
Abstract
RNA modifications are emerging as critical regulators in cancer biology, thanks to their ability to influence gene expression and the predominant protein isoforms expressed during cell proliferation, migration, and other pro-oncogenic properties. The reversibility and dynamic nature of post-transcriptional RNA modifications allow cells to quickly adapt to microenvironmental changes. Recent literature has revealed that the deregulation of RNA modifications can promote a plethora of developmental diseases, including tumorigenesis. In this review, we will focus on four key post-transcriptional RNA modifications which have been identified as contributors to the pathogenesis of brain tumors: m6A, alternative polyadenylation, alternative splicing and adenosine to inosine modifications. In addition to the role of RNA modifications in brain tumor progression, we will also discuss potential opportunities to target these processes to improve the dismal prognosis for brain tumors.
Similar content being viewed by others

Introduction
Of the approximately 25,000 people diagnosed annually with primary malignant brain tumors in the USA, 80% are gliomas, one of the most lethal types of cancer [89, 101, 131]. Amongst gliomas, glioblastoma (GBM) is the most aggressive, characterized by a median patient survival of less than 15 months following surgical resection and concurrent radiotherapy and chemotherapy with temozolomide (TMZ) [67, 111]. Most of the literature on gliomas has historically focused on transcriptional control of gene expression [81, 124]. However, the role of post-transcriptional RNA modifications in cellular function and glioma progression has recently begun to surface, mostly due to advances in next generation sequencing (NGS) [46, 115, 145]. The insights gained from these advances highlight the importance of post-transcriptional control of gene expression in the development and progression of brain tumors as well as neurological disorders such as autism, Alzheimer’s disease, and Parkinson’s disease [21, 22, 51, 62].
Before messenger RNA (mRNA) translation and protein synthesis can occur, nascent mRNA transcripts require processing and nucleic acid modifications. These include splicing out of introns, the non-coding sections of the transcripts, as well as methylation of certain bases. RNA modifications modulate most steps of gene expression, from indirectly controlling DNA transcription, by regulating expression of mRNAs encoding transcription factors, to directly affecting mRNA translation [23, 144]. While many RNA modifications were originally discovered decades ago [24, 92], studies of many of these modifications, such as N6-methyladenosine (m6A), were previously limited by the inability to distinguish between certain nucleotides during reverse transcription [20, 96]. However, with the recent advances in NGS, over one hundred different types of RNA modifications have now been described [8, 11, 65, 69]. Other RNA modifications include N1-methyladenosine (m1A) and 5-methylcytosine (m5C), which are not found only in mRNA: m5C can also be found in transfer RNA (tRNA) and m1A in tRNAs, ribosomal RNAs (rRNAs) and long non-coding RNAs (lncRNAs) [25, 96]. Within rRNAs, the most abundant modification are 2′-O-methylations, which are known to play a significant role in ribosome function [30]. These modifications are not further discussed here and are reviewed elsewhere [96], as their roles in brain tumorigenesis are not well established. For the purpose of this review, we will focus on the most common RNA modifications that have been characterized in glioma: m6A, alternative polyadenylation (APA), alternative splicing, and adenosine to inosine (A-to-I) modifications. RNA modifications are a highly conserved mechanism utilized by eukaryotes throughout phylogeny to enhance biologic complexity [7]. RNA modifications are typically both reversible and dynamic, allowing for rapid cellular adaptation to changes in the microenvironment, thus limiting the size of the genome necessary to encode adaptive molecules [6, 28]. This mechanism is particularly beneficial for cancer cells to adapt to acute microenvironmental changes [6, 28]. In contrast to the relatively long half-life of mRNA in mammalian cells (median of 9 h) through protracted transcriptional changes, dynamic RNA alterations can be completed in < 30 s [12]. This allows for rapid cellular adaptation to harsh environments, most commonly induced in cancer by microenvironmental stresses, such as hypoxia, or toxic therapy [23, 100]. While several informative publications have underscored the pivotal role of RNA modifications in glioma progression [79, 84], a comprehensive overview on the topic is currently lacking. We believe that a better understanding of the molecular mechanisms behind glioma progression is critical for the development of novel therapeutic approaches that could ultimately improve the outcome of patients with glioma.
N6-methyladenosine (m6A) modifications
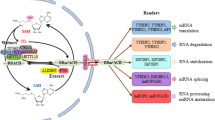
Of the known RNA modifications, the methylation of adenosine at the nitrogen-6 position to create N6-methyladenosine (m6A) makes up the majority of the internal mRNA modifications in eukaryotes, and has emerged as a critical regulator in many aspects of RNA biology, including pre-mRNA splicing, polyadenylation, localization, and mRNA translation [23, 27, 95]. The addition, removal and recognition of m6A is catalyzed by methyltransferases, demethylases, and binding proteins, otherwise known as “writers,” “erasers” and “readers”, respectively [82]. M6A generally occurs within long exons, around stop codons, and in 3′ untranslated regions (3′-UTRs) [140, 141]. However, approximately 30% of target sites for m6A writers are also located in intronic RNA regions [69], indicating that m6A methylation may occur co-transcriptionally, before or during splicing. In addition, mRNA splicing factor precursors co-localize with m6A methyltransferases in nuclear speckles, suggesting the involvement of intronic m6A residues in alternative splicing [61, 69].
The relative ease by which m6A can be added or removed facilitates rapid changes in gene expression, as m6A modifications are reported to promote mRNA decay through binding of specific degradative protein complexes [83, 140]. This is in contrast to the historical notion that RNA molecules remain largely unchanged after initial covalent modifications [33]. To catalyze m6A mRNA methylation, the multi-subunit writer complex comprises a catalytic subunit, known as methyltransferase-like 3 (METTL3), a second augmenting methyltransferase subunit (METTL14) utilized in substrate recognition, as well as the Wilms’ tumor 1-associating protein (WTAP) [9, 82, 127]. Lacking the methyltransferase activity of the other subunits, WTAP is instead likely to be involved in m6A modifications by promoting the recruitment of the METTL3-METTL14 complex to target mRNAs, in addition to inducing the translocation of the complex to nuclear speckles [68, 69]. WTAP overexpression promotes the migratory and invasive capabilities of GBM cells by epidermal growth factor receptor (EGFR) stimulation, although no further mechanistic insights were provided in this study [54]. WTAP mutations are extremely rare in cancer, occurring in only 0.5% of gliomas [13]. Instead, utilizing Quaking gene isoform 6 (QKI-6) knockout and QKI-6 mutant studies in glioma U87 and U251 cell lines, and tissues derived from GBM, Xi et al. found that WTAP is regulated by QKI-6. WTAP mRNAs contain a specific sequence known as a QKI response element (QRE) in its 3′ UTR region whereby QKI-6 induces WTAP expression [134]. Moreover, QKI-6 is directly controlled by microRNAs (miRNAs), with miR-29a overexpression leading to reduced QKI-6 activity and decreased glioma tumor growth and increased survival [134]. While further studies are required to fully elucidate the mechanism behind WTAP function in GBM pathogenesis, it is reasonable to postulate that WTAP’s activity of recruiting methyltransferases to specific unidentified targets facilitates GBM progression, and the utilization of miRNA-based therapies could prove beneficial for glioma treatment.
Proteins often referred to as “m6A readers” selectively bind to mRNAs modified with m6A. The specific type of reader protein regulates different functions: binding of YTH domain containing family protein (YTHDC1) to m6A induces mRNA splicing by recruiting splicing factor SRSF3 [135], whereas binding of YTHDF2 targets the transcripts for degradation by recruiting them to cytoplasmic processing (P) bodies within mammalian cells [29, 50, 128]. In contrast, transcript binding by YTHDF1 and YTHDF3 enhances their translation [70, 128, 129]. To facilitate transcript binding, a hydrophobic pocket within the YTH domain interacts with the methyl group exposed in m6A [64, 70, 117, 136]. While YTHDF1 and YTHDF2 mutations only occur in 0.9 and 0.5% of glioma cases respectively [13], several published datasets, including from The Cancer Genome Atlas (TCGA), show that YTHDF1 and YTHDF2 mRNA expression levels are positively correlated with malignancy of gliomas, with significant increases in higher grade gliomas, suggesting a role for these m6A readers in glioma progression [13, 15, 112]. While this seems counterintuitive at first glance, given the different effects on mRNAs by binding these proteins, one possible explanation is provided by Wang et al. [129]. Using photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) and RNA immunoprecipiation (RIP-seq), the authors identified 1260 and 1276 mRNA targets for YTHDF1 and YTHDF2, respectively [129]. While these proteins share 622 mRNA targets, YTHDF1 and YTHDF2 also bind to ~ 650 unique mRNA targets each [129]. Although this experiment was performed in human cervical cancer HeLa cells, unique regulation of separate mRNAs by YTHDF1 versus YTHDF2 in gliomas could provide an intriguing explanation for overexpression of both genes in high grade gliomas. For example, YTHDF1 could drive translation of pro-oncogenic transcripts, while YTHDF2 might drive degradation of tumor suppressor encoding transcripts. While YTHDF1 and YTHDF2 expression promote pancreatic and lung cancer cell proliferation, no equivalent research has to date determined a causal relationship between YTHDF1 or YTHDF2 expression in gliomagenesis [17, 104, 105]. It also remains to be determined if YTHDF1 and/or YTHDF3 are upregulated epigenetically in gliomas.
The reversal of m6A methylation is catalyzed by demethylases known as fat mass and obesity-associated protein (FTO), and ALKBH5 [125, 146], both acting as so-called “erasers” for m6A modifications [53, 146]. In glioma, mutations occur only in 0.1% of cases for ALKBH5 and no mutations have been reported in FTO [13]. However, as mentioned earlier, high expression of ALKBH5, which could occur through the induction of hypoxia-inducible factors (HIFs), as seen in breast cancer [142], is linked to worse GBM patient outcome [139, 143]. To further investigate the mechanism, Zhang et al. immunoprecipitated RNAs using m6A primary antibodies and performed microarray analysis. This approach identified ALKBH5 mRNAs targets, such as the proto-oncogene FOXM1. Demethylation of m6A residues in the 3′-UTR of the FOXM1 pre-mRNA results in increased transcript stability and enhanced FOXM1 protein expression [143]. This leads to downstream STAT3 activation and thus increased GBM proliferation, invasion and metastasis [39]. Demethylation of mRNA increases binding of the RNA stabilizer protein Hu-antigen R (HUR), and thus leads to increased stability of the targeted mRNA [83].
According to the TCGA, genetic amplifications at the METTL3 locus arise in ~ 1% of gliomas [13]. In addition, several studies have shown that METTL3 mRNA and m6A levels are elevated in glioma compared to normal brain [19, 112, 125], therefore leading multiple researchers to investigate the effects of upregulating and suppressing METTL3 on glioma growth. Early research indicated that short hairpin RNA (shRNA) mediated silencing of METTL3 in several glioma cell lines, both in vitro as well as in in vivo orthotopic models, resulted in enhanced GBM growth [19]. As a possible explanation of this phenotype, the authors found by RNA-seq that oncogenes such as ADAM19 and KLF4 were upregulated by METTL3 silencing and tumor suppressors such as CDKN2A and BRCA2 were downregulated. However, more recent work suggests a different scenario. Visvanathan et al., utilizing methylated RNA immunoprecipitation-seq (MeRIP-seq) on glioma cell line MGG8, followed by gene set enrichment (GSEA) and Gene Ontology (GO) analyses, proposed that METTL3 silencing may in fact disrupt tumorigenic pathways that facilitate glioma progression, such as NOTCH, c-Myc and NFκB [63, 125]. This concept was further supported by Li et al., who found that both genetic knockout and knockdown of METTL3 significantly decreased proliferation of GBM cell lines U251 and U87MG in cell viability assays [63]. In vivo, xenograft tumor size was reduced compared to controls after inoculation of shMETTL3 GBM cells into mice [63]. While it is difficult to speculate on these inconsistencies, it is possible that some of the observed differences could be due to intertumor heterogeneity and the use of different cell lines [125]. The METTL3-METTL14 complex shares similar structures with other DNA and protein methyltransferases, including disrupter of telomeric silencing 1-like (DOT1L), as both contain Rossmann fold structural motifs [99, 108]. Notably, small-molecule inhibitors of DOT1L are currently undergoing clinical trials to treat acute myeloid leukemia (AML), suggesting the potential to develop novel drug therapies targeting against this family of proteins in brain tumors [110].
Additional m6A methylation regulators are also emerging as critical components of GBM tumorigenesis [19, 126]. Chai et al. reported that many of the main regulators of m6A modifications are differentially expressed between different glioma grades. Specifically, there is positive correlation between WHO grade and expression of WTAP, YT521-B homology (YTH) domain containing family (YTHDF) and AlkB homolog 5 (ALKBH5), whereas there is a negative correlation between FTO and WHO grade [15]. These results suggest a potential interplay among these regulatory elements and glioma malignancy [15]. A possible mechanism of action for these enzymes in the context of glioma progression was recently proposed by Li et al. The authors suggest that METTL3 is involved in decreasing nonsense-mediated mRNA decay (NMD) of transcripts encoding for splicing factors by m6A deposition around the start codon of serine and arginine rich splicing factor (SRSF) mRNAs. The methylation around the start codon then prevents NMD of SRSF mRNAs, thus resulting in increased alternative splicing and isoform switching in glioma [63].
Alternative 3′ polyadenylation (APA)
APA is a mechanism that allows a single gene to encode multiple mRNAs, and represents a critical post-transcriptional regulator of gene expression [38]. For the mRNA transcript to undergo APA, a two-step endonucleolytic cleavage of the pre-mRNA occurs at its 3′-UTR, or in some cases within exons and introns of the transcript, followed by addition of repetitive adenosine monophosphate nucleotide units to the end, creating the poly(A) tail [38, 77]. With over 50% of human genes associated with APA, transcripts can be diversified while limiting the size of the genome [38, 78, 119]. These isoform variations are dependent on the location of the alternative poly(A) site (PAS), as some sites can be located within introns or exons, a process known as coding region alternative polyadenylation (CR-APA) (Fig. 1a). This can result in decreased binding of miRNAs to the transcript, as a result of the entire 3′-UTR being cleaved out, in addition to the generation of additional protein isoforms due to the exclusion of exons from the transcript [38, 78, 119]. Another form of APA, termed untranslated region alternative polyadenylation (UTR-APA), occurs when alternative PASs are located in different regions of the 3′-UTR. This results in different 3′-UTR lengths but the same protein isoform, as the coding region remains unaffected (Fig. 1b) [38]. Moreover, previous studies reported that 3′-UTR shortening by APA, by preventing the suppressive effects of miRNAs and other RNA binding proteins, induces the activation of proto-oncogenes [1, 79, 80, 113].

Different variants of APA. a In CR-APA the PAS is located within the coding region, which after polyadenylation can result in variations in the coding region at the C-terminal end, resulting in different protein isoforms. Protein isoforms could potentially have different functions within the cell to either lead to or hinder cell proliferation and tumor progression. b In UTR-APA, the PAS can be located within the 3′-UTR. Therefore, depending on the location of the PAS, the length of the 3′-UTR could be altered, thus generating new mRNA isoforms whilst not affecting the protein produced. However, the alteration of the 3′-UTR could affect accessibility to regulatory sites, such as for miRNA binding, which would affect expression level of the protein. While not generating new protein isoforms after UTR-APA, oncogenic proteins can be either over or under-expressed as a result of this process
The process of polyadenylation is facilitated by a multimeric protein complex comprised of four primary subunits: the cleavage and polyadenylation specificity factor (CPSF), cleavage stimulation factor (CSTF), mammalian cleavage factor I (CFIm) and cleavage factor II (CFIIm) [18, 40, 120]. CFIm performs a crucial regulatory role in polyA site (PAS) selection by acting as an enhancer-dependent activator [10, 18, 147].
One of the subunits of the CFIm complex, CFIm25, is believed to directly facilitate the recognition of certain PAS sequences, especially those rich in UGUA sequences [79]. Depletion of CFIm25 in GBM leads to 3′-UTR shortening and increased stability of specific transcripts, in turn leading to increased production of oncogenic proteins such as Pak1 and Pak2, key components of the Ras signalling pathway [18]. Activation of the Ras signalling pathway then results in increased cell proliferation and increased GBM aggressiveness [18].
O6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair enzyme that acts to convert methylguanine back to guanine by removing the methyl or akyl group from the O6 position of guanine, without causing breaks in the DNA. Promoter methylation of the MGMT gene in GBM, present in over 40% of cases, results in improved survival in patients treated with TMZ in addition to radiotherapy [45]. APA has been recently identified as an additional mechanism by which MGMT is repressed in GBM. Through the usage of an alternate, distally located PAS of the MGMT transcript, APA results in a transcript variant with an elongated 3′-UTR [60]. This longer 3′-UTR contains miRNA binding sites that act as targets for several miRNAs, including miR-181d, miR-34a, and miR-648, which act to induce degradation of the MGMT transcript [52]. Importantly, this type of promoter-independent silencing of MGMT has also been shown to confer tumor sensitivity to alkylating agents [52, 60]. This concept has important clinical implications, as it should support the use of techniques aiming to identify MGMT protein (e.g. by immunohistochemistry), rather than promoter methylation, to determine MGMT status. However, additional studies are warranted to further elucidate mechanisms of APA in GBM.
Alternative splicing
As mentioned, following transcription, processing of the pre-mRNA transcript precedes downstream translation and protein synthesis. In addition to the modifications discussed above, splicing results in the formation of multiple mRNA and protein isoforms from one gene (Fig. 2). Alternative splicing is performed by excising introns out of a given transcript using a large molecular complex known as the spliceosome, allowing for the synthesis of multiple protein isoforms from one gene [76, 86]. With > 80% of human genes being affected by alternative splicing, the proteome is greatly diversified as a result of the generation of two or more distinct mature mRNA transcripts from each pre-mRNA [47, 116]. By controlling the splice isoforms produced, cells can dynamically change gene expression and favor certain mRNA and protein isoforms to overcome stresses within the microenvironment [85, 91]. The process of splicing is composed of two major steps: the assembly of the spliceosome complex and the actual splicing of the pre-mRNA. The spliceosome is comprised of U1, U2, U4, U5, and U6 small nuclear ribonucleic proteins (snRNPs) and, in the case of the major human spliceosome, it includes over 300 additional proteins [47, 130]. The spliceosome complex is assembled on each target transcript and is directed by specific sequence elements contained within the pre-mRNA, such as the 5′ splice site, the branch point sequence and the 3′ splice site [47, 130]. The mechanistic details behind the cleavage and removal of the spliced out regions of the mRNA have been discussed elsewhere and are beyond the scope of this review [47]. For effective cell adaptation to rapid microenvironmental changes, splicing needs to be as efficient and as precise as possible [76]. However, in reality, pre-mRNA splicing can take up to several hours to be completed, mostly due to varying intron lengths [76]. The precision of splicing is also critical, as a shift in the reading frame and consequent irregularities in splicing can promote the development and progression of different diseases, including cystic fibrosis and glioma [31, 36, 90, 123].

Schematic representation of protein isoforms that can be generated from alternative splicing of pre-mRNA transcript. After removal of the intronic segments of the nascent mRNA transcript by the spliceosome complex (not shown), exons can be ligated to form mature RNA forms that are translated into different protein variants. The different splice variants are implicated in glioma pathogenesis
The ubiquitous presence of alternative splicing within cells underscores its relevance for the pathogenesis of tumors, including GBM. Glioma, as described for many other tumors, display a high degree of intertumor heterogeneity [16, 124]. Among the GBM subtypes that can be identified by RNA expression profiling, the mesenchymal (MES) subtype is the most aggressive variant, with higher rates of proliferation in vitro and in vivo and increased radiation resistance [55, 75, 102]. Guardia et al. reported important splicing differences (4934 splicing events affecting 3243 genes) between the MES subtype and the proneural (PN) subtype [43], suggesting a contribution of these events to glioma heterogeneity and plasticity. A large body of literature describes the effects that alternative splicing has on uncontrolled cell proliferation in GBM. The generation of different protein isoforms through alternative splicing promotes increased proliferation and evasion of apoptosis in GBM [118]. Tiek et al. found that different splice variants of Estrogen-related receptor β (ERR-β) influence GBM progression [121]. ERR-β is an orphan nuclear receptor expressed in the brain, where alternative splicing of the 3′ of the pre-mRNA transcript leads to 3 different isoforms: the ERR-β short form (ERR-βsf), ERR-β2, and ERR-β with exon 10 deleted. ERR-β2 drives G2/M cell cycle arrest and induces apoptosis [44]. Exploring ERR-β2 function in GBM, these authors found that by favoring expression of ERR-β2 over other splice variants and by inhibiting the splicing regulatory cdc2-like kinases (CLKs), they could suppress GBM cell migration and proliferation, in combination with an ERR-β2 agonist [121].
The MAPK interacting kinase (Mnks) family of proteins include MNK1 and MNK2 and are the kinases responsible for phosphorylation of eukaryotic translation initiation factor 4E (eIF4E) on Ser-209 [56]. While genetic alterations of the MKNK1 and MKNK2 genes are rare in cancer, being present in 0.5 and 2.4% of cases respectively in glioma [13], both proteins undergo alternative splicing to create distinct protein isoforms [84]. Previous studies have suggested that MNK1 positively regulates the expression of TGFβ, known to regulate proliferation, invasion and immune evasion [4, 41, 42]. A recent study by Garcia-Recio et al. reported that the MNK1b isoform (the spliced variant of MNK1a lacking the 89 C-terminal amino acids [42]) can act as a marker for prognosis in breast cancer patients [37]. Unfortunately, the clinical outcomes resulting from the formation of Mnk1a and Mnk1b have not been analyzed in detail with regard to brain tumors. Mnk2b, acts as an oncogenic protein by inducing eIF4E phosphorylation and not activating p38-MAPK, leading to enhanced translation of mRNAs encoding factors implicated in tumor formation, such as c-MYC and cyclin D1 [94]. On the other hand, one of the MNK2 isoforms, Mnk2a, acts as a tumor suppressor by co-localizing with and activating the p38-MAPK pathway to induce apoptosis and suppressing Ras-induced transformation [73]. Typically, the p38-MAPK pathway is activated in response to environmental changes and affects transcription, gene expression and efficacy of drug therapies [5, 59, 66]. The mechanism of action of many anticancer drugs has been linked to induction of the p38-MAPK pathway. Thus, one potential therapeutic strategy could rely on utilizing drugs to favor the Mnk2a isoform, which can activate the p38-MAPK stress response more effectively and promote apoptosis [103, 133]. As a specific pre-clinical example in GBM, Mogilevsky et al. recently used splice switching oligonucleotides (SSOs) that bind to Mnkb2 splice sites on the MKNK2 pre-mRNA to disrupt normal splicing by blocking interactions between the pre-mRNA and the spliceosome. This resulted in suppression of Mnk2b production and GBM growth in-vivo, and re-sensitized cells to chemotherapy [75]. Finally, a recent article reported non-coding mutations in the 5′ splice site binding region in U1 spliceosomal small nuclear RNAs (snRNAs), resulting in increased 5′ cryptic splicing events and aberrant RNA splicing in Sonic hedgehog (SHH) medulloblastoma, compared to control cells [114]. This results in the inactivation of tumor suppressing genes such as PTCH1 while activating oncogenic genes such as GLI2 and CCND2 [114]. While no literature to date has investigated snRNA mutations in GBM, these studies emphasize how controlling the regulation of alternative splicing to favour the production of tumor suppressive isoforms has potential for developing novel therapeutic approaches for GBM.
Adenosine to inosine modifications
A-to-I modifications comprise the irreversible, hydrolytic deamination of adenosine nucleoside bases by adenosine deaminases acting on dsRNA (ADARs), converting the adenosine to inosine in RNAs [3, 87, 98, 106]. Inosines are then recognized by the translation machinery as guanosines rather than adenosines, due to preferential base pairing of inosine with cytidine (Fig. 3) [26, 97, 122]. This substitution can lead to codon changes, modify the amino acid sequence of proteins, change the secondary structure of RNA, and result in the addition or removal of splice sites to expand the proteome [2, 87, 93]. In mammals, three enzymes that catalyze A-to-I modifications have been identified to date: ADAR1, ADAR2 and ADAR3 [88]. A-to-I modifications can also affect processes such as the binding of miRNAs to 3′-UTRs through editing of the miRNA seed sequence (the miRNA recognition site in RNAs), affecting target specificity of the mature miRNA [32, 57, 137]. A-to-I modifications are even responsible for directing the alternative splicing of its own pre-mRNA with the ADAR2 enzyme, resulting in multiple isoforms with different catalytic functions each [34].

The hydrolytic deamination of adenosine is catalyzed by ADAR enzymes, resulting in the formation of inosine. Due to structural similarities between inosine and guanosine, the translational machinery reads the nucleoside as the latter, potentially resulting in changes to codon sequences or the addition/removal of splice sites. The A-to-I modification can also serve to direct alternative splicing to create oncogenic protein isoforms that promote glioma progression
While found in all human tissues, A-to-I editing is most prevalent in the brain, and aberrant editing is linked to the development of various brain pathologies including amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, epilepsy and glioma [14, 34, 93, 107]. A-to-I editing is extremely prevalent in healthy brains, with almost 100% of some miRNA strands being edited, such as in the case of miR-589-3p, resulting in inhibition of aberrant cell proliferation [14]. In GBM, while mutations or deletions of the ADARB1 gene occur only in 0.6% of cases [13], ADAR2 activity is decreased, leading to significant hypo-editing of miRNAs, resulting in miRNA target switching by changing the miRNA seed sequence [14]. In the case of miR-589-3p, this is retargeted from tumor-suppressor Protocadherin 9 (PCDH9) to Disintegrin and Metalloproteinase 12 (ADAM12), facilitating GBM invasion by supporting cell adhesion [14, 58, 74].
GBM is known to utilize glutamate to promote proliferation and migration [49], a process mostly mediated by calcium permeable AMPA-type glutamate receptor signaling. One of the subunits, glutamate receptor subunit B (GluR-B), encoded by GRIA2, was one of the first known ADAR targets [71, 138]. By editing a single adenosine on the GRIA2 transcript, known as the glutamine/arginine (Q/R) site targeted by ADAR2 [72], the originally encoded Q codon is converted to positively charged R encoding codon, allowing for the incorporation of the GluR-B subunit into the AMPA receptor, rendering it impermeable to positively charged calcium ions [132]. If the transcript is left unedited due to a reduction in ADAR activity, such as in GBM, AMPA remains permeable to calcium. This results in increased activity of AMPA-type glutamate receptors independently from glutamate activation, leading to excitotoxicity and epileptic seizures, typically associated with glioma [48, 72, 88]. However, observed changes in ADAR2 activity are not due to decreased expression of ADAR2, but rather to inhibition of self-editing leading to decreased alternative splicing of the ADAR2 pre-mRNA transcript [72]. Q/R site editing of GRIA2 is edited in virtually 100% of healthy mammalian brain tissue [109]. In contrast, Maas et al. found that Q/R editing decreases to 90% in lower grade astrocytoma and decreases further to 69–88% in GBM [72]. Unfortunately, the cellular mechanisms that induce these changes and regulate ADAR2 activity in brain tumors remain unclear [72]. Thus, an emphasis on targeting the self-editing site to further increase editing at that location, in combination with increasing expression of ADAR2, should be the focus of further pharmacological research.
In other studies, the activity of ADAR2 was deemed essential to prevent glioma proliferation and growth through the editing of the CDC14B pre-mRNA transcript involved in the Skp2/p21/p27 pathway [35]. As ADAR2 activity decreases in gliomas, CDC14B pre-mRNA modification is decreased, resulting in overexpression of Skp2 and downregulation of the known tumor suppressors p21 and p27 [35]. This eventually induces glioma cells to bypass the G1/S checkpoint, promoting increased cell proliferation [35]. Oakes et al. found that as a negative regulator of ADAR2 activity, ADAR3, which is genetically amplified in ~ 2% of glioma, inhibits the binding of ADAR2 to the GRIA2 pre-mRNA transcript, preventing RNA editing [88], although the exact mechanism by which ADAR3 performs this function remains unclear. Without the deaminase activity of ADAR1 and ADAR2 in vitro or in vivo, it is unlikely that ADAR3 can directly edit pre-mRNA transcripts. However, as ADAR3 is known to bind to dsRNA despite not being able to perform A-to-I modifications, it has been hypothesized that ADAR3 could act as a direct physical block, preventing ADAR2 from mRNA binding and subsequent editing [88]. An alternative scheme is that ADAR3 facilitates the alternative splicing of the GRIA2 pre-mRNA transcript, hence providing another method of preventing ADAR2 editing to promote glioma proliferation and malignant progression [88]. Sequestering ADAR3 or upregulating ADAR2 would serve as potential therapeutic strategies that could increase pre-mRNA transcript editing, decreasing GBM progression.
Conclusions
The studies outlined in this review highlight the importance of RNA modifications in brain tumor progression, specifically in glioma. The flexibility conferred by post-transcriptional control adds another dimension by which gene expression can be regulated beyond what is directly coded from DNA. The addition of multiple modifications on the same transcript could thus increase the complexity of multiple regulatory networks. This plasticity is particularly relevant for cancer cells to adapt to unexpected microenvironmental changes. However, as showcased throughout this review, deregulation of the RNA modification machinery and altered gene expression are associated with many of the hallmarks of cancer, such as apoptosis evasion and uncontrolled proliferation. Given the significant contribution to brain tumor malignancy, greater emphasis on clarifying the role of RNA regulation and modifications in glioma progression is needed. Targeting the regulatory enzymes controlling the post-transcriptional modifications discussed in this paper warrants further detailed investigation, as this remains an unexplored strategy that could ultimately improve the prognosis of brain tumor patients.
Availability of data and materials
Not applicable.
Abbreviations
- 3′- UTR:
-
3′ - Untranslated region
- AML:
-
Acute myeloid leukemia
- ADAM12:
-
Disintegrin and Metalloproteinase 12
- ADAR:
-
Adenosine deaminases acting on dsRNA
- ALKBH5:
-
AlkB Homolog 5
- APA:
-
Alternative polyadenylation
- CFIIm:
-
Mammalian cleavage factor II
- CFIm:
-
Mammalian cleavage factor I
- CLK:
-
Cdc2-like kinases
- CPSF:
-
Cleavage and polyadenylation specificity factor
- CSTF:
-
Cleavage stimulation factor
- DOT1L:
-
Disrupter of telomeric silencing 1-like
- EGFR:
-
Epidermal growth factor receptor
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- ERR-β:
-
Estrogen-related receptor β
- ERR-βsf:
-
ERR-β short form
- FTO:
-
Fat mass and obesity-associated protein
- GBM:
-
Glioblastoma
- GO:
-
Gene Ontology
- GSEA:
-
Gene set enrichment analysis
- GluR:
-
Glutamate receptor
- HIF:
-
Hypoxia-inducible factors
- m6A:
-
N6-methyladenosine
- MeRIP-seq:
-
Methylated RNA immunoprecipitation-seq
- MES:
-
Mesenchymal
- METTL14:
-
Methyltransferase-like 14
- METTL3:
-
Methyltransferase-like 3
- MGMT:
-
O6-methylguanine-DNA-methyltransferase
- miRNA:
-
Micro RNA
- NGS :
-
Next generation sequencing
- NMD:
-
Nonsense-mediated mRNA decay
- PAS:
-
Poly(A) site
- PAR-CLIP:
-
Photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation
- PCDH9:
-
Protocadherin 9
- PN:
-
Proneural
- QKI:
-
Quaking I
- RIP-seq:
-
RNA immunoprecipitation sequencing
- SgRNA:
-
Guide RNA
- ShRNA:
-
Short hairpin RNA
- snRNA:
-
Small nuclear RNA
- SRSF:
-
Serine and arginine rich splicing factor
- SSO:
-
Splice switching oligonucleotides
- TCGA:
-
The Cancer Genome Atlas
- TMZ:
-
Temozolomide
- WTAP:
-
Wilms’ tumor 1-associating protein
- YTH:
-
YT521-B homology
- YTHDC:
-
YTH domain containing
- YTHDF:
-
YTH N6-methyladenosine RNA binding protein
References
Akman BH, Can T, Elif Erson-Bensan A (2012) Estrogen-induced upregulation and 3′-UTR shortening of CDC6. Nucleic Acids Res 40:10679–10688. https://doi.org/10.1093/nar/gks855
Barbieri I, Kouzarides T (2020) Role of RNA modifications in cancer. Nat Rev Cancer. https://doi.org/10.1038/s41568-020-0253-2
Bass BL, Weintraub H (1988) An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. https://doi.org/10.1016/0092-8674(88)90253-X
Beier CP, Kumar P, Meyer K, Leukel P, Bruttel V, Aschenbrenner I et al (2012) The Cancer stem cell subtype determines immune infiltration of Glioblastoma. Stem Cells Dev 21:2753–2761. https://doi.org/10.1089/scd.2011.0660
Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S, Lee JC, Haegeman G et al (1996) The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis response to tumor necrosis factor. EMBO J 15:1914–1923. https://doi.org/10.1002/j.1460-2075.1996.tb00542.x
Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J et al (2016) Stem cell function and stress response are controlled by protein synthesis. Nature. https://doi.org/10.1038/nature18282
Bludau I, Aebersold R (2020) Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat Rev Mol Cell Biol. https://doi.org/10.1038/s41580-020-0231-2
Boccaletto P, MacHnicka MA, Purta E, Pitkowski P, Baginski B, Wirecki TK et al (2018) MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1030
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F (1994) Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei: internal mRNA methylation requires a multisubunit complex. J Biol Chem 269:17697–17704
Brown KM, Gilmartin GM (2003) A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol Cell. https://doi.org/10.1016/S1097-2765(03)00453-2
Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X et al (2011) The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. https://doi.org/10.1093/nar/gkq1028
Carmo-Fonseca M, Kirchhausen T (2014) The timing of pre-mRNA splicing visualized in real-time. Nucleus 5:11–14. https://doi.org/10.4161/nucl.28056
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse Glioma. Cell 164:550–563. https://doi.org/10.1016/j.cell.2015.12.028
Cesarini V, Silvestris DA, Tassinari V, Tomaselli S, Alon S, Eisenberg E et al (2018) ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1257
Chai RC, Wu F, Wang Q-X, Zhang S, Zhang KN, Liu YQ et al (2019) m 6 A RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging (Albany NY). https://doi.org/10.18632/aging.101829
Chandran UR, Luthra S, Santana-Santos L, Mao P, Kim S-H, Minata M et al (2015) Gene expression profiling distinguishes proneural glioma stem cells from mesenchymal glioma stem cells. Genomics Data 5:333–336. https://doi.org/10.1016/j.gdata.2015.07.007
Chen J, Sun Y, Xu X, Wang D, He J, Zhou H et al (2017) YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. https://doi.org/10.1080/15384101.2017.1380125
Chu Y, Elrod N, Wang C, Li L, Chen T, Routh A et al (2019) Nudt21 regulates the alternative polyadenylation of Pak1 and is predictive in the prognosis of glioblastoma patients. Oncogene. https://doi.org/10.1038/s41388-019-0714-9
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G et al (2017) m6A RNA methylation regulates the self-renewal and tumorigenesis of Glioblastoma stem cells. Cell Rep. https://doi.org/10.1016/j.celrep.2017.02.059
Dai Q, Fong R, Saikia M, Stephenson D, Yu YT, Pan T et al (2007) Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N6-methyladenosine. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm657
Delaidelli A, Jan A, Herms J, Sorensen PH (2019) Translational control in brain pathologies: biological significance and therapeutic opportunities. Acta Neuropathol 137:535–555
Delaidelli A, Negri GL, Jan A, Jansonius B, El-Naggar A, Lim JKM et al (2017) MYCN amplified neuroblastoma requires the mRNA translation regulator eEF2 kinase to adapt to nutrient deprivation. Cell Death Differ. https://doi.org/10.1038/cdd.2017.79
Delaunay S, Frye M (2019) RNA modifications regulating cell fate in cancer. Nat Cell Biol 21:552–559
Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.71.10.3971
Dinescu S, Ignat S, Lazar A, Constantin C, Neagu M, Costache M (2019) Epitranscriptomic signatures in lncRNAs and their possible roles in Cancer. Genes (Basel) 10:52. https://doi.org/10.3390/genes10010052
Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G (2011) Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis 32:1569–1577
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. https://doi.org/10.1038/nature11112
Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS et al (2016) The dynamic N1 -methyladenosine methylome in eukaryotic messenger RNA. Nature. https://doi.org/10.1038/nature16998
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M et al (2016) YTHDF2 destabilizes m 6 A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. https://doi.org/10.1038/ncomms12626
Erales J, Marchand V, Panthu B, Gillot S, Belin S, Ghayad SE et al (2017) Evidence for rRNA 2′-O-methylation plasticity: control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1707674114
Faustino NA (2003) Pre-mRNA splicing and human disease. Genes Dev 17:419–437. https://doi.org/10.1101/gad.1048803
Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T (2016) RNA modifications: what have we learned and where are we headed? Nat Rev Genet 17:365–372
Fu Y, Dominissini D, Rechavi G, He C (2014) Gene expression regulation mediated through reversible m 6 A RNA methylation. Nat Rev Genet. 15:293–306
Fu Y, Zhao X, Li Z, Wei J, Tian Y (2016) Splicing variants of ADAR2 and ADAR2-mediated RNA editing in glioma. Oncol Lett 12:788–792
Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M et al (2013) ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene. https://doi.org/10.1038/onc.2012.125
Garcia-Blanco MA, Baraniak AP, Lasda EL (2004) Alternative splicing in disease and therapy. Nat Biotechnol 22:535–546. https://doi.org/10.1038/nbt964
García-Recio EM, Pinto-Díez C, Pérez-Morgado MI, García-Hernández M, Fernández G, Martín ME et al (2016) Characterization of MNK1b DNA Aptamers that inhibit proliferation in MDA-MB231 breast cancer cells. Mol Ther - Nucleic Acids 5. https://doi.org/10.1038/mtna.2015.50
Di Giammartino DC, Nishida K, Manley JL (2011) Mechanisms and consequences of alternative Polyadenylation. Mol Cell 43:853–866. https://doi.org/10.1016/j.molcel.2011.08.017
Gong AH, Wei P, Zhang S, Yao J, Yuan Y, Zhou AD et al (2015) FoxM1 drives a feed-forward STAT3-activation signaling loop that promotes the self-renewal and tumorigenicity of glioblastoma stem-like cells. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-14-2800
Gruber AR, Martin G, Keller W, Zavolan M (2012) Cleavage factor I m is a key regulator of 3′ UTR length. RNA Biol. https://doi.org/10.4161/rna.22570
Grzmil M, Huber RM, Hess D, Frank S, Hynx D, Moncayo G et al (2014) MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J Clin Invest. https://doi.org/10.1172/JCI70198
Grzmil M, Morin P, Lino MM, Merlo A, Frank S, Wang Y et al (2011) MAP kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β signaling pathway in human glioblastoma. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-10-3112
Guardia GDA, Correa BR, Araujo PR, Qiao M, Burns S, Penalva LOF et al (2020) Proneural and mesenchymal glioma stem cells display major differences in splicing and lncRNA profiles. Npj genomic med 5:2. https://doi.org/10.1038/s41525-019-0108-5
Heckler MM, Zeleke TZ, Divekar SD, Fernandez AI, Tiek DM, Woodrick J et al (2016) Antimitotic activity of DY131 and the estrogen-related receptor beta 2 (ERRβ2) splice variant in breast cancer. Oncotarget. https://doi.org/10.18632/oncotarget.9719
Hegi ME, Diserens AC, Gorlia T, Hamou MF, De Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. https://doi.org/10.1056/NEJMoa043331
Helm M, Motorin Y (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18:275–291. https://doi.org/10.1038/nrg.2016.169
Hoskins AA, Moore MJ (2012) The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci 37:179–188. https://doi.org/10.1016/j.tibs.2012.02.009
Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H et al (2002) Blockage of Ca 2+ − permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. https://doi.org/10.1038/nm746
Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T et al (2007) Ca 2+ −permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. https://doi.org/10.1523/JNEUROSCI.2180-07.2007
Ivanova I, Much C, Di Giacomo M, Azzi C, Morgan M, Moreira PN et al (2017) The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal Transcriptome and oocyte competence. Mol Cell. https://doi.org/10.1016/j.molcel.2017.08.003
Jan A, Jansonius B, Delaidelli A, Bhanshali F, An YA, Ferreira N et al (2018) Activity of translation regulator eukaryotic elongation factor-2 kinase is increased in Parkinson disease brain and its inhibition reduces alpha synuclein toxicity. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-018-0554-9
Jesionek-Kupnicka D, Braun M, Trąbska-Kluch B, Czech J, Szybka M, Szymańska B et al (2019) MiR-21, miR-34a, miR-125b, miR-181d and miR-648 levels inversely correlate with MGMT and TP53 expression in primary glioblastoma patients. Arch Med Sci. https://doi.org/10.5114/aoms.2017.69374
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y et al (2011) N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. https://doi.org/10.1038/nchembio.687
Jin D-I, Lee SW, Han M-E, Kim H-J, Seo S-A, Hur G-Y et al (2012) Expression and roles of Wilms’ tumor 1-associating protein in glioblastoma. Cancer Sci 103:2102–2109. https://doi.org/10.1111/cas.12022
Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T et al (2017) Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med 23:1352–1361. https://doi.org/10.1038/nm.4415
Joshi S (2014) Mnk kinase pathway: cellular functions and biological outcomes. World J Biol Chem. https://doi.org/10.4331/wjbc.v5.i3.321
Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K (2007) Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science (80). https://doi.org/10.1126/science.1138050
Kodama T, Ikeda E, Okada A, Ohtsuka T, Shimoda M, Shiomi T et al (2004) ADAM12 is selectively overexpressed in human glioblastomas and is associated with glioblastoma cell proliferation and shedding of heparin-binding epidermal growth factor. Am J Pathol. https://doi.org/10.1016/S0002-9440(10)63429-3
Koul HK, Pal M, Koul S (2013) Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 4:342–359. https://doi.org/10.1177/1947601913507951
Kreth S, Limbeck E, Hinske LC, Schütz SV, Thon N, Hoefig K et al (2013) In human glioblastomas transcript elongation by alternative polyadenylation and miRNA targeting is a potent mechanism of MGMT silencing. Acta Neuropathol. https://doi.org/10.1007/s00401-013-1081-1
Lamond AI, Spector DL (2003) Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4:605–612. https://doi.org/10.1038/nrm1172
Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo ARF, Kool M et al (2013) XThe eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. https://doi.org/10.1016/j.cell.2013.04.055
Li F, Yi Y, Miao Y, Long W, Long T, Chen S et al (2019) N6-methyladenosine modulates nonsense-mediated mRNA decay in human Glioblastoma. Cancer Res. https://doi.org/10.1158/0008-5472.can-18-2868
Li F, Zhao D, Wu J, Shi Y (2014) Structure of the YTH domain of human YTHDF2 in complex with an m6A mononucleotide reveals an aromatic cage for m6A recognition. Cell Res 24:1490–1492. https://doi.org/10.1038/cr.2014.153
Li Z, Zhao P, Xia Q (2019) Epigenetic methylations on N6-adenine and N6-adenosine with the same input but different output. Int J Mol Sci 20:2931. https://doi.org/10.3390/ijms20122931
Lin J, Chang S-Y, Hsieh D-S, Lee C-F, Yu D-S (2005) Modulation of mitogen-activated protein kinase cascades by differentiation-1 protein: acquired drug resistance of hormone independent prostate cancer cells. J Urol 174:2022–2026. https://doi.org/10.1097/01.ju.0000176476.14572.39
Liu C-A, Chang C-Y, Hsueh K-W, Su H-L, Chiou T-W, Lin S-Z et al (2018) Migration/invasion of malignant Gliomas and implications for therapeutic treatment. Int J Mol Sci. https://doi.org/10.3390/ijms19041115
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L et al (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. https://doi.org/10.1038/nchembio.1432
Liu N, Pan T (2016) N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol 23:98–102. https://doi.org/10.1038/nsmb.3162
Luo S, Tong L (2014) Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1412742111
Maas S, Melcher T, Herb A, Seeburg PH, Keller W, Krause S et al (1996) Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J Biol Chem. https://doi.org/10.1074/jbc.271.21.12221
Maas S, Patt S, Schrey M, Rich A (2001) Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.251531398
Maimon A, Mogilevsky M, Shilo A, Golan-Gerstl R, Obiedat A, Ben-Hur V et al (2014) Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. https://doi.org/10.1016/j.celrep.2014.03.041
Manini I, Caponnetto F, Bartolini A, Ius T, Mariuzzi L, Di Loreto C et al (2018) Role of microenvironment in Glioma invasion: what we learned from in vitro models. Int J Mol Sci 19:147. https://doi.org/10.3390/ijms19010147
Mao P, Joshi K, Li J, Kim S-H, Li P, Santana-Santos L et al (2013) Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci 110:8644–8649. https://doi.org/10.1073/pnas.1221478110
El Marabti E, Younis I (2018) The Cancer Spliceome: reprograming of alternative splicing in Cancer. Front Mol Biosci 5. https://doi.org/10.3389/fmolb.2018.00080
Marsollier A-C, Joubert R, Mariot V, Dumonceaux J (2018) Targeting the Polyadenylation signal of pre-mRNA: A new gene silencing approach for Facioscapulohumeral dystrophy. Int J Mol Sci 19:1347. https://doi.org/10.3390/ijms19051347
Masamha CP, Wagner EJ (2018) The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis 39:2–10. https://doi.org/10.1093/carcin/bgx096
Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu A Bin, et al. (2014) CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. https://doi.org/10.1038/nature13261
Mayr C, Bartel DP (2009) Widespread shortening of 3′UTRs by alternative cleavage and Polyadenylation activates oncogenes in Cancer cells. Cell. https://doi.org/10.1016/j.cell.2009.06.016
McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM et al (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. https://doi.org/10.1038/nature07385
Meyer KD, Jaffrey SR (2014) The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15:313–326. https://doi.org/10.1038/nrm3785
Minton K (2014) Stabilizing stemness through m6A. Nat Rev Mol Cell Biol 15:77–77. https://doi.org/10.1038/nrm3745
Mogilevsky M, Shimshon O, Kumar S, Mogilevsky A, Keshet E, Yavin E et al (2018) Modulation of MKNK2 alternative splicing by splice-switching oligonucleotides as a novel approach for glioblastoma treatment. Nucleic Acids Res. https://doi.org/10.1093/nar/gky921
Nakayama K, Kataoka N (2019) Regulation of gene expression under hypoxic conditions. Int J Mol Sci 20. https://doi.org/10.3390/ijms20133278
Nilsen TW, Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463:457–463. https://doi.org/10.1038/nature08909
Nishikura K (2010) Functions and regulation of RNA editing by ADAR Deaminases. Annu Rev Biochem. https://doi.org/10.1146/annurev-biochem-060208-105251
Oakes E, Anderson A, Cohen-Gadol A, Hundley HA (2017) Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem. https://doi.org/10.1074/jbc.%20M117.779868
Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C et al (2019) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro-Oncology. https://doi.org/10.1093/neuonc/noz150
Pagani F, Baralle FE (2004) Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 5:389–396. https://doi.org/10.1038/nrg1327
Pagliarini V, Naro C, Sette C (2015) Splicing regulation: A molecular device to enhance Cancer cell adaptation. Biomed Res Int 2015:1–13. https://doi.org/10.1155/2015/543067
Perry RP, Kelley DE (1974) Existence of methylated messenger RNA in mouse L cells. Cell. https://doi.org/10.1016/0092-8674(74)90153-6
Picardi E, Manzari C, Mastropasqua F, Aiello I, D’Erchia AM, Pesole G (2015) Profiling RNA editing in human tissues: towards the inosinome atlas. Sci Rep. https://doi.org/10.1038/srep14941
Robichaud N, Hsu BE, Istomine R, Alvarez F, Blagih J, Ma EH et al (2018) Translational control in the tumor microenvironment promotes lung metastasis: phosphorylation of eIF4E in neutrophils. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1717439115
Rottman F, Shatkin AJ, Perry RP (1974) Sequences containing methylated nucleotides at the 5′ termini of messenger RNAs: possible implications for processing. Cell 3:197–199. https://doi.org/10.1016/0092-8674(74)90131-7
Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA modifications in gene expression regulation. Cell 169:1187–1200. https://doi.org/10.1016/j.cell.2017.05.045
Rueter SM, Dawson TR, Emeson RB (1999) Regulation of alternative splicing by RNA editing. Nature. https://doi.org/10.1038/19992
Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T (2010) Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol. https://doi.org/10.1038/nchembio.434
Schapira M (2016) Chemical inhibition of protein Methyltransferases. Cell Chem Biol 23:1067–1076. https://doi.org/10.1016/j.chembiol.2016.07.014
Schiera G, Di Liegro C, Di Liegro I (2017) Molecular determinants of malignant brain cancers: from intracellular alterations to invasion mediated by extracellular vesicles. Int J Mol Sci 18:2774. https://doi.org/10.3390/ijms18122774
Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M (2006) Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol 2:494–503. https://doi.org/10.1038/ncpneuro0289
Segerman A, Niklasson M, Haglund C, Bergström T, Jarvius M, Xie Y et al (2016) Clonal variation in drug and radiation response among Glioma-initiating cells is linked to proneural-Mesenchymal transition. Cell Rep 17:2994–3009. https://doi.org/10.1016/j.celrep.2016.11.056
Seufferlein T, Seckl MJ, Schwarz E, Beil M, Wichert GV, Baust H et al (2002) Mechanisms of nordihydroguaiaretic acid-induced growth inhibition and apoptosis in human cancer cells. Br J Cancer 86:1188–1196. https://doi.org/10.1038/sj.bjc.6600186
Sheng H, Li Z, Su S, Sun W, Zhang X, Li L et al (2019) YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis. https://doi.org/10.1093/carcin/bgz152
Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L et al (2019) YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. https://doi.org/10.1038/s41467-019-12801-6
Silvestris DA, Picardi E, Cesarini V, Fosso B, Mangraviti N, Massimi L et al (2019) Dynamic inosinome profiles reveal novel patient stratification and gender-specific differences in glioblastoma. Genome Biol. https://doi.org/10.1186/s13059-019-1647-x
Singh M (2013) Dysregulated A to I RNA editing and non-coding RNAs in neurodegeneration. Front Genet:3. https://doi.org/10.3389/fgene.2012.00326
Śledź P, Jinek M (2016) Structural insights into the molecular mechanism of the m6A writer complex. Elife 5. https://doi.org/10.7554/eLife.18434
Sommer B, Köhler M, Sprengel R, Seeburg PH (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67:11–19. https://doi.org/10.1016/0092-8674(91)90568-J
Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR et al (2018) The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131:2661–2669. https://doi.org/10.1182/blood-2017-12-818948
Stupp R, Hegi ME, Mason WP, van den Bent M, Taphoorn M, Janzer RC et al (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10:459–466. https://doi.org/10.1016/S1470-2045(09)70025-7
Sun L, Hui A-M, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S et al (2006) Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9:287–300. https://doi.org/10.1016/j.ccr.2006.03.003
Sun M, Ding J, Li D, Yang G, Cheng Z, Zhu Q (2017) NUDT21 regulates 3′-UTR length and microRNA-mediated gene silencing in hepatocellular carcinoma. Cancer Lett. https://doi.org/10.1016/j.canlet.2017.09.026
Suzuki H, Kumar SA, Shuai S, Diaz-Navarro A, Gutierrez-Fernandez A, De Antonellis P et al (2019) Recurrent non-coding U1-snRNA mutations drive cryptic splicing in Shh medulloblastoma. Nature. https://doi.org/10.1038/s41586-019-1650-0
Synhaeve NE, van den Bent MJ, French PJ, Dinjens WNM, Atmodimedjo PN, Kros JM et al (2018) Clinical evaluation of a dedicated next generation sequencing panel for routine glioma diagnostics. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-018-0633-y
Tazi J, Bakkour N, Stamm S (2009) Alternative splicing and disease : an overview. Biochim Biophys Acta - Rev Cancer 1792:14–26. https://doi.org/10.1016/j.bbadis.2008.09.017.Alternative
Theler D, Dominguez C, Blatter M, Boudet J, Allain FHT (2014) Solution structure of the YTH domain in complex with N6-methyladenosine RNA: A reader of methylated RNA. Nucleic Acids Res. https://doi.org/10.1093/nar/gku1116
Thorne AH, Cavenee WK, Furnari FB (2015) Alternative RNA splicing in the pathogenesis of GBM. Med Res Arch. https://doi.org/10.18103/mra.v0i1.11
Tian B, Hu J, Zhang H, Lutz CS (2005) A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. https://doi.org/10.1093/nar/gki158
Tian B, Manley JL (2017) Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 18:18–30. https://doi.org/10.1038/nrm.2016.116
Tiek DM, Khatib SA, Trepicchio CJ, Heckler MM, Divekar SD, Sarkaria JN et al (2019) Estrogen-related receptor β activation and isoform shifting by cdc2-like kinase inhibition restricts migration and intracranial tumor growth in glioblastoma. FASEB J. https://doi.org/10.1096/fj.201901075r
Tomaselli S, Locatelli F, Gallo A (2014) The RNA editing enzymes ADARs: mechanism of action and human disease. Cell Tissue Res 356:527–532. https://doi.org/10.1007/s00441-014-1863-3
Venables JP (2004) Aberrant and alternative splicing in Cancer. Cancer Res 64:7647–7654. https://doi.org/10.1158/0008-5472.CAN-04-1910
Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of Glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Visvanathan A, Patil V, Abdulla S, Hoheisel JD, Somasundaram K (2019) N 6 -Methyladenosine landscape of glioma stem-like cells: METTL3 is essential for the expression of actively transcribed genes and sustenance of the oncogenic signaling. Genes (Basel). https://doi.org/10.3390/genes10020141
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V et al (2018) Essential role of METTL3-mediated m 6 A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. https://doi.org/10.1038/onc.2017.351
Wang P, Doxtader KA, Nam Y (2016) Structural basis for cooperative function of Mettl3 and Mettl14 Methyltransferases. Mol Cell. https://doi.org/10.1016/j.molcel.2016.05.041
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D et al (2014) N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature. https://doi.org/10.1038/nature12730
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H et al (2015) N6-methyladenosine modulates messenger RNA translation efficiency. Cell. https://doi.org/10.1016/j.cell.2015.05.014
Wang Y, Ji L, Huang B, Xu Y-M, Li J, Huang L-F et al (2015) Mechanism of alternative splicing and its regulation. Biomed Reports. https://doi.org/10.3892/br.2014.407
Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM et al (2015) Glioma. Nat Rev Dis Prim 1:15017. https://doi.org/10.1038/nrdp.2015.17
Wright A, Vissel B (2012) The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci 5. https://doi.org/10.3389/fnmol.2012.00034
Wu J, Suzuki H, Akhand AA, Zhou YW, Hossain K, Nakashima I (2002) Modes of activation of mitogen-activated protein kinases and their roles in cepharanthine-induced apoptosis in human leukemia cells. Cell Signal 14:509–515. https://doi.org/10.1016/S0898-6568(01)00278-9
Xi Z, Wang P, Xue Y, Shang C, Liu X, Ma J et al (2017) Overexpression of miR-29a reduces the oncogenic properties of glioblastoma stem cells by downregulating quaking gene isoform 6. Oncotarget. https://doi.org/10.18632/oncotarget.15327
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF et al (2016) Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. https://doi.org/10.1016/j.molcel.2016.01.012
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y et al (2014) Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. https://doi.org/10.1038/nchembio.1654
Yablonovitch AL, Deng P, Jacobson D, Li JB (2017) The evolution and adaptation of A-to-I RNA editing. PLoS Genet 13:e1007064. https://doi.org/10.1371/journal.pgen.1007064
Yang JH, Sklar P, Axel R, Maniatis T (1997) Purification and characterization of a human RNA adenosine deaminase for glutamate receptor B pre-mRNA editing. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.94.9.4354
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S et al (2017) MicroRNA-145 modulates N6-methyladenosine levels by targeting the 3′-untranslated mRNA region of the N6-Methyladenosine binding YTH domain family 2 protein. J Biol Chem. https://doi.org/10.1074/jbc.%20M116.749689
Yue Y, Liu J, He C (2015) RNA N 6 -methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 29:1343–1355. https://doi.org/10.1101/gad.262766.115
Zhang C, Fu J, Zhou Y (2019) A review in research Progress concerning m6A methylation and Immunoregulation. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.00922
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I et al (2016) Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m 6 A-demethylation of NANOG mRNA. Proc Natl Acad Sci 113:E2047–E2056. https://doi.org/10.1073/pnas.1602883113
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z et al (2017) m6A Demethylase ALKBH5 maintains Tumorigenicity of Glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. https://doi.org/10.1016/j.ccell.2017.02.013
Zhao BS, Roundtree IA, He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18:31–42. https://doi.org/10.1038/nrm.2016.132
Zhao Z, Meng F, Wang W, Wang Z, Zhang C, Jiang T (2017) Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci Data. https://doi.org/10.1038/sdata.2017.24
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ et al (2013) ALKBH5 is a mammalian RNA Demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. https://doi.org/10.1016/j.molcel.2012.10.015
Zhu Y, Wang X, Forouzmand E, Jeong J, Qiao F, Sowd GA et al (2018) Molecular mechanisms for CFIm-mediated regulation of mRNA alternative Polyadenylation. Mol Cell. https://doi.org/10.1016/j.molcel.2017.11.031
Acknowledgements
Data generated by the TCGA Research Network were used: https://www.cancer.gov/tcga.
Funding
This work was partially supported by Canadian Cancer Society Research Institute (CCSRI) Impact Grant (Grant #703205; to PHS), and CIHR Foundation Grant FDN-143280 (to PHS). AD is supported by a 4 Year Fellowship and a Killam Doctoral Scholarship from the University of British Columbia, BC, Canada.
Author information
Authors and Affiliations
Contributions
AH and AD conceptualized the manuscript. AH carried out the literature searches, drafted the original manuscript and generated the figures. PHS was responsible for acquisition of funding and all authors were involved in the review, revision and approval of the manuscript before submission.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, A.Z., Delaidelli, A. & Sorensen, P.H. RNA modifications in brain tumorigenesis. acta neuropathol commun 8, 64 (2020). https://doi.org/10.1186/s40478-020-00941-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-00941-6