
Abstract
Congenital myopathies (CM) form a genetically heterogeneous group of disorders characterized by perinatal muscle weakness. Here, we report an 11-year old male offspring of consanguineous parents of Lebanese origin. He presented with proximal weakness including Gower’s sign, and skeletal muscle biopsy revealed myopathic changes with core-like structures. Whole exome sequencing of this index patient lead to the discovery of a novel genetically defined CM subtype based on bi-allelic mutations in the uncoordinated mutant number-45 myosin chaperone B (UNC45B) NM_173167:c.2261G > A, p.Arg754Gln. The mutation is conserved in evolution and co-segregates within the pedigree with the phenotype, and located in the myosin binding armadillo repeat domain 3 (ARM3), and has a CADD Score of 35. On a multimeric level, UNC45B aggregates to a chain which serves as an assembly line and functions as a “template” defining the geometry, regularity, and periodicity of myosin arranged into muscle thick filaments. Our discovery is in line with the previously described myopathological phenotypes in C. elegans and in vertebrate mutants and knockdown–models. In conclusion, we here report for the first time a patient with an UNC45B mutation causing a novel genetically defined congenital myopathy disease entity.
Similar content being viewed by others


Text
Congenital myopathies (CM) form a genetically heterogeneous group of disorders characterized by perinatal muscle weakness [18, 20]. Here, we report an 11-year old male offspring of consanguineous parents of Lebanese origin. He presented with proximal weakness including Gower’s sign, and skeletal muscle biopsy revealed myopathic changes with core-like structures. Genomic investigation of this index patient lead to the discovery of a novel genetically defined CM subtype based on bi-allelic mutations in the uncoordinated mutant number-45 myosin chaperone B (UNC45B) gene. Regarding medical history, the mother reported reduced fetal movements during pregnancy. After birth, the patient presented as a floppy infant with feeding difficulties, improving after the first year of life. He was able to sit and walk independently at 10 and 20 months of age, respectively. Currently, his Gower’s time is >10s (Fig. 1b), he is unable to run, shows a Trendelenburg sign (Additional file 1: Figure S1h), he is overweight and has a static disease course. He talks with a nasal voice without chewing or swallowing difficulties. Facial weakness and ophthalmoplegia are absent. While he had a reduced forced vital capacity of 70%, his echocardiogram showed a normal heart function. Serum creatinine kinase levels were not elevated. For further clinical details, please see Additional file 2: Supplementary Material.

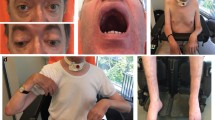
Phenotypic, myopathological, and electron microscopical findings in our patient with an overview of the UNC45B variant identified in this study. a Front and side view of our patient, showing hyperlordosis and obesity. b Gower’s sign in our patient. c Increased fiber size caliber spectrum with internalized nuclei predominantly in hypertrophic muscle fibers (arrows) and atrophic muscle fibers (arrowheads). H&E staining, original magnification × 400; scale bar 20 μm. d Disorganization of myofibrillary architecture evidenced by pale centers of muscle fibers presenting as core-like structures. Enzyme histochemistry with NADH; original magnification × 400; scale bar 20 μm. e Focal myofibrillary disintegration (arrows) and occasional non-subsarcolemmal muscle fiber nuclei. Semithin section, toluidine blue staining; scale bar 20 μm. f Subsarcolemmal core-like structure and Z-band streaming in electron microscopy (EM). g Pedigree and chromatograms of the index patient, healthy sisters, and healthy mother. The healthy father’s blood sample was unavailable for dideoxy sequencing. h UNC45B multiple sequence alignment made with Jalview shows high evolutionary conservation at amino acid residue p.Arg754 (NP_775259.1 Homo sapiens, the mutated sequence from our patient c.2261G > A p.Arg754Gln, XP_001174363.2 P. troglodytes, XP_0011113905.2 m. mulatta, XP_005624856.1 C. lupus, XP_002695676.1 B. taurus, NP_848795.3 m. musculus, NP_001100498.1 R. norvegicus, XP_004946569.1 g. gallus, NP_705959.1 D. rerio, NP_001172057.1 x. tropicalis, NP_524796.1 D. melanogaster, XP_310258.5 A. gambiae, and NP_497205.1 C. elegans). i Variant in the UNC45B gene (NM_173167.3, 20 exons) identified in our patients and concomitant position in the j. UNC45B protein structure (Q8IWX7) based on 931-aa isoform (ENST00000268876.9, NP_775259.1); pictogram with protein domains: Tetratricopeptide repeats (TPR, red) and Armadillo/beta-catenin-like repeats (ARM, green), N-terminal region of protein in blue, central region (131–506) in white, UCS region (Unc45−/Cro1p−/She4p-related protein) in red (507–931). Gene and protein sequences are drawn with the IBS Biocuckoo web server [14]
At the age of 10 years, left femoral quadriceps muscle biopsy showed myopathic changes, i.e., fiber size variability including hypertrophic and atrophic fibers and central nuclei (Fig. 1c) with core-like lesions mainly in the center of muscle fibers (Fig. 1d). Fiber type distribution was altered as type-2 fibers were virtually absent (Fig. 1d, Additional file 1: Figure S1a). Small neonatal myosin positive fibers indicated regeneration (Additional file 1: Figure S1b). Electron microscopy unraveled numerous core-like alterations of myofibrillary architecture with Z-band streaming (Fig. 1f). Some mitochondria showed prominent matrix granula, globoidal inclusions, and even paracristalline intramitochondrial inclusions (Additional file 1: Figure S1f). We also observed subsarcolemmal accumulations of organelles (Additional file 1: Figure S1e).
To uncover underlying disease-causing mutations, we performed whole-exome sequencing (WES) (Additional file 2: Table S1 and Supplementary Material) [3]. By stringent filtering for various inheritance models (Additional file 2: Table S2), the most likely autosomal-recessive model in a consanguineous family let us to the solution. Based on skeletal muscle expression levels (Additional file 1: Figure S1 g) and previously reported animal models [4, 9, 11, 13, 21], we consider a strictly conserved homozygous base pair exchange in UNC45B (NM_173167:c.2261G > A, p.Arg754Gln, Fig. 1g-j) in a homozygous region as pathogenic. The variant leads to an amino acid substitution with a change in polarity and mass (dissimilarity 43 in Sneath’s index) in the armadillo repeat domain 3 (ARM3), and is reported with a CADD Score of 35. UNC45B is highly conserved and constrained against loss-of-function variants in the gnomAD population database (Additional file 2: Table S3). UNC-45 proteins show a three-domain configuration, with an N-terminal tetratricopeptide repeat (TPR) domain, poorly conserved central domain, and a C-terminal UCS domain (Unc45−/Cro1p−/She4p-related protein) [13]. Three consensus TPR repeats participate in protein-protein interaction especially with Hsp70 and Hsp90 [17]. The C-terminal UCS domain has been shown to form a putative myosin-binding groove, largely stabilized by electrostatic interactions [6]. Our patient’s missense mutation is located in the third ARM domain at residue p.Arg754 at the C-terminal UCS region, which might abrogate the interaction between UNC45B and myosin heavy chain [19], thus impairing myofibrillogenesis (Additional file 1: Figure S2d). Indeed, in-silico modeling and docking studies of the human UNC45B protein showed that the binding groove in the UCS domain is a negatively-charged surface at the R18R19 helices of UNC45B and serves as “place-holder” for the charged loop-U and β-sheets residues of myosin (MYH7) [6]. The p.Arg754Gln mutation is actually found directly in R18H1 [6, 12]. We calculated a change to a decreased isoelectric point and lower net charge from wildtype to p.Arg754Gln mutant in the R18-R19 residues at pH 7.4 by using the Prot-pi tool (Additional file 1: Figure S2c).
Notably, all three isoforms of UNC45B are highly expressed in skeletal muscle and the p.Arg754Gln mutation affects all isoforms (Additional file 1: Figure S2a,b), only one of the three isoforms is highly expressed in cardiac muscle. In a D. melanogaster model, an Unc-45 knockdown showed a severe cardiac phenotype with dilated cardiomyopathy and reduced muscle contractility [15]. Unc-45b knockdown in zebrafish and also the steif/unc-45b mutants resulted in paralysis and cardiac dysfunction based on severely disrupted myofibrillogenesis [4, 21]. Therefore, our patient might develop cardiomyopathy at later ages. Knockdown of unc-45b severely affected sarcomere organization including M- and Z-lines of skeletal muscles of embryos [2].
From the essential physiological function of UNC45B in muscle development, it can be deducted that deleterious genetic variants may lead to myopathy. On a multimeric level, UNC45B aggregates to a chain which serves as an assembly line for beta (β)-myosin heavy chain, encoded by MYH7 [1, 5], and functions as a “template” defining the geometry, regularity, and periodicity of myosin arranged into muscle thick filaments [16]. After myosin incorporates into thick filaments, Unc45b and Hsp90 dissociate from myosin ensuring the proper myosin filament assembly [15]. Once disassociated, UNC45B binds to a VCP cofactor protein UFD-2 and an E3 ligase CHIP which leads to poly-ubiquitylation of UNC45B and its subsequent proteasomal degradation [10] (Additional file 1: Figure S2d).
Therefore, we hypothesize that our patient’s mutation in UNC45B in the UCS domain might directly lead to myofibrillogenesis failure (Additional file 1: Figure S2d). Of note, a heterozygous missense variant in UNC45B (c.2413C > T, p.Arg805Trp) has been reported to cause a dominant form of glaucoma without further confirmation since publication [8]. Noteworthy, there is a high heterozygous allele carrier status of 18/272310 in gnomAD of this c.2413C > T variant in healthy adults.
In conclusion, we here report for the first time a patient with an UNC45B mutation causing a novel genetically defined congenital myopathy disease entity. Our discovery is in line with the previously described myopathological phenotypes in C. elegans and in vertebrate mutants and knockdown–models [4, 7, 9, 11, 19].
Availability of data and materials
The next generation sequencing datasets generated and analyzed during the current study are not publicly available because of patient confidentiality and since we do not have informed consent for that.
References
Ao W, Pilgrim D (2000) Caenorhabditis elegans UNC-45 is a component of muscle thick filaments and colocalizes with myosin heavy chain B, but not myosin heavy chain a. J Cell Biol 148:375–384. https://doi.org/10.1083/jcb.148.2.375
Bernick EP, Zhang PJ, Du S (2010) Knockdown and overexpression of Unc-45b result in defective myofibril organization in skeletal muscles of zebrafish embryos. BMC Cell Biol 11:70. https://doi.org/10.1186/1471-2121-11-70
Dafsari HS, Sprute R, Wunderlich G, Daimaguler HS, Karaca E, Contreras A, Becker K, Schulze-Rhonhof M, Kiening K, Karakulak T, Kloss M, Horn A, Pauls A, Nurnberg P, Altmuller J, Thiele H, Assmann B, Koy A, Cirak S (2019) Novel mutations in KMT2B offer pathophysiological insights into childhood-onset progressive dystonia. J Hum Genet 64:803–813. https://doi.org/10.1038/s10038-019-0625-1
Etard C, Behra M, Fischer N, Hutcheson D, Geisler R, Strahle U (2007) The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis. Dev Biol 308:133–143. https://doi.org/10.1016/j.ydbio.2007.05.014
Etard C, Roostalu U, Strahle U (2008) Shuttling of the chaperones Unc45b and Hsp90a between the a band and the Z line of the myofibril. J Cell Biol 180:1163–1175. https://doi.org/10.1083/jcb.200709128
Fratev F, Osk Jonsdottir S, Pajeva I (2013) Structural insight into the UNC-45-myosin complex. Proteins 81:1212–1221. https://doi.org/10.1002/prot.24270
Geach TJ, Zimmerman LB (2010) Paralysis and delayed Z-disc formation in the Xenopus tropicalis unc45b mutant dicky ticker. BMC Dev Biol 10:75. https://doi.org/10.1186/1471-213x-10-75
Hansen L, Comyn S, Mang Y, Lind-Thomsen A, Myhre L, Jean F, Eiberg H, Tommerup N, Rosenberg T, Pilgrim D (2014) The myosin chaperone UNC45B is involved in lens development and autosomal dominant juvenile cataract. Eur J Hum Genet 22:1290–1297. https://doi.org/10.1038/ejhg.2014.21
Hoppe T, Cassata G, Barral JM, Springer W, Hutagalung AH, Epstein HF, Baumeister R (2004) Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell 118:337–349. https://doi.org/10.1016/j.cell.2004.07.014
Janiesch PC, Kim J, Mouysset J, Barikbin R, Lochmuller H, Cassata G, Krause S, Hoppe T (2007) The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat Cell Biol 9:379–390. https://doi.org/10.1038/ncb1554
Landsverk ML, Li S, Hutagalung AH, Najafov A, Hoppe T, Barral JM, Epstein HF (2007) The UNC-45 chaperone mediates sarcomere assembly through myosin degradation in Caenorhabditis elegans. J Cell Biol 177:205–210. https://doi.org/10.1083/jcb.200607084
Lee CF, Hauenstein AV, Fleming JK, Gasper WC, Engelke V, Sankaran B, Bernstein SI, Huxford T (2011) X-ray crystal structure of the UCS domain-containing UNC-45 myosin chaperone from Drosophila melanogaster. Structure 19:397–408. https://doi.org/10.1016/j.str.2011.01.002
Lee CF, Melkani GC, Bernstein SI (2014) The UNC-45 myosin chaperone: from worms to flies to vertebrates. Int Rev Cell Mol Biol 313:103–144. https://doi.org/10.1016/b978-0-12-800177-6.00004-9
Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, Lahrmann U, Zhao Q, Zheng Y, Zhao Y, Xue Y, Ren J (2015) IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics (Oxford, England) 31:3359–3361. https://doi.org/10.1093/bioinformatics/btv362
Melkani GC, Bodmer R, Ocorr K, Bernstein SI (2011) The UNC-45 chaperone is critical for establishing myosin-based myofibrillar organization and cardiac contractility in the drosophila heart model. PLoS One 6:e22579. https://doi.org/10.1371/journal.pone.0022579
Pokrzywa W, Hoppe T (2013) Chaperoning myosin assembly in muscle formation and aging. Worm 2:e25644–e25644. https://doi.org/10.4161/worm.25644
Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210. https://doi.org/10.1016/s0092-8674(00)80830-2
Scoto M, Cullup T, Cirak S, Yau S, Manzur AY, Feng L, Jacques TS, Anderson G, Abbs S, Sewry C, Jungbluth H, Muntoni F (2013) Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur J Hum Genet 21:1249–1252. https://doi.org/10.1038/ejhg.2013.31
Srikakulam R, Liu L, Winkelmann DA (2008) Unc45b forms a cytosolic complex with Hsp90 and targets the unfolded myosin motor domain. PLoS One 3:e2137. https://doi.org/10.1371/journal.pone.0002137
Wang H, Schanzer A, Kampschulte B, Daimaguler HS, Logeswaran T, Schlierbach H, Petzinger J, Ehrhardt H, Hahn A, Cirak S (2018) A novel SPEG mutation causes non-compaction cardiomyopathy and neuropathy in a floppy infant with centronuclear myopathy. Acta Neuropathol Commun 6:83. https://doi.org/10.1186/s40478-018-0589-y
Wohlgemuth SL, Crawford BD, Pilgrim DB (2007) The myosin co-chaperone UNC-45 is required for skeletal and cardiac muscle function in zebrafish. Dev Biol 303:483–492. https://doi.org/10.1016/j.ydbio.2006.11.027
Acknowledgments
We furthermore thank the Cologne Center for Genomics (CCG) at the University of Cologne for performing Whole Exome Sequencing of the patient and providing the sequencing data. We also thank the Regional Computing Center of the University of Cologne (RRZK) for providing computing time for the bioinformatics analyses on the DFG-funded High-Performance Computing (HPC) system CHEOPS.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft, Germany grants (CI 218/1–1) to Dr. Sebahattin Cirak. Dr. Hormos Salimi Dafsari was supported by the Gerok program of the Faculty of Medicine, University of Cologne.
Author information
Authors and Affiliations
Contributions
Manuscript writing, genetic data and bioinformatic analysis, and sequencing were performed by HSD. NMK contributed to manuscript writing and figure generation. H-SD contributed to the genomic analysis and manuscript writing. Myopathological and electron microscopy examinations were performed and interpreted by AB, JW, and MD. Patient recruitment and examination, analysis of WES data, obtainment of funding, study design, manuscript writing, and supervision of the study were contributed by SC. All authors (HSD, NMK, H-SD, AB, JD, JW, MD, SC) have critically reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee (ethics committee of the Medical Faculty, University Hospital Cologne, University of Cologne [17–096]) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from the legal guardian/parent of the index case.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Further myopathological, electron microscopical and phenotypic findings in our patient with UNC45B variant. Figure S2. Gene and isoform expression of UNC45B in various tissues and a possible disease model scheme.
Additional file 2: Table S1.
Detailed metrics of Whole Exome Sequencing in our patient with coverage (1x, 2x, 10x, 20x, 30x, 100x, mean). Table S2. Results of the variant filtering and the specific criteria we applied on the dataset. Table S3. Prediction of pathogenicity for our patient’s UNC45B variant via multiple scoring tools.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dafsari, H.S., Kocaturk, N.M., Daimagüler, HS. et al. Bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B are a cause for congenital myopathy. acta neuropathol commun 7, 211 (2019). https://doi.org/10.1186/s40478-019-0869-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0869-1