
Abstract
To identify pathogenetic markers and potential drivers of different lesion types in the white matter (WM) of patients with progressive multiple sclerosis (PMS), we sequenced RNA from 73 different WM areas. Compared to 25 WM controls, 6713 out of 18,609 genes were significantly differentially expressed in MS tissues (FDR < 0.05). A computational systems medicine analysis was performed to describe the MS lesion endophenotypes. The cellular source of specific molecules was examined by RNAscope, immunohistochemistry, and immunofluorescence. To examine common lesion specific mechanisms, we performed de novo network enrichment based on shared differentially expressed genes (DEGs), and found TGFβ-R2 as a central hub. RNAscope revealed astrocytes as the cellular source of TGFβ-R2 in remyelinating lesions. Since lesion-specific unique DEGs were more common than shared signatures, we examined lesion-specific pathways and de novo networks enriched with unique DEGs. Such network analysis indicated classic inflammatory responses in active lesions; catabolic and heat shock protein responses in inactive lesions; neuronal/axonal specific processes in chronic active lesions. In remyelinating lesions, de novo analyses identified axonal transport responses and adaptive immune markers, which was also supported by the most heterogeneous immunoglobulin gene expression. The signature of the normal-appearing white matter (NAWM) was more similar to control WM than to lesions: only 465 DEGs differentiated NAWM from controls, and 16 were unique. The upregulated marker CD26/DPP4 was expressed by microglia in the NAWM but by mononuclear cells in active lesions, which may indicate a special subset of microglia before the lesion develops, but also emphasizes that omics related to MS lesions should be interpreted in the context of different lesions types. While chronic active lesions were the most distinct from control WM based on the highest number of unique DEGs (n = 2213), remyelinating lesions had the highest gene expression levels, and the most different molecular map from chronic active lesions. This may suggest that these two lesion types represent two ends of the spectrum of lesion evolution in PMS. The profound changes in chronic active lesions, the predominance of synaptic/neural/axonal signatures coupled with minor inflammation may indicate end-stage irreversible molecular events responsible for this less treatable phase.
Similar content being viewed by others
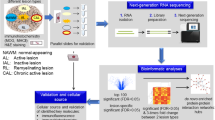


Introduction
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating and neurodegenerative disease of the CNS. Without treatment, a secondary progressive course (SPMS) develops in about half of the patients [65]. Neuroimaging, treatment responses and pathology all show differences between the early and late phase of MS, indicating that disease mechanisms change during the natural course [31]. Therefore, modern systems medicine approaches may help to increase our understanding of MS progression and to find novel, mechanistic treatment targets.
Inflammatory demyelination affects osmotic homeostasis, energy coupling with oligodendrocytes, and contributes to glutamate excitotoxicity, axonal damage and fibrillary gliosis that may inhibit remyelination [23, 49]. Key elements of the degenerative process are chronic oxidative injury [29], accumulation of mitochondrial damage resulting in chronic cell stress and imbalance of ionic homeostasis [9, 60], microglia activation, and age-related iron accumulation in the brain [61]. As the disease progresses, diffuse changes can be observed in the normal appearing white and grey matter (NAWM, NAGM), and B cell follicle-like cellular aggregates in the meninges contribute to subpial cortical lesions [48, 59, 72].
WM lesions are inherent characteristics of MS from the early phase, and both quantitative and qualitative changes in the WM can be observed as the disease progresses: microglia activation in the NAWM [22], increasing number of chronic active lesions, and decreasing number of remyelinating lesions [16, 69]. B cells are also present in active WM lesions in progressive MS, and the number of plasma cells is higher in lesions from progressive MS compared to acute MS [24, 58, 54, 75].
The lesion evolution and fate in the WM can be classified into distinct groups based on the distribution and density of inflammatory cells and myelin loss [72]. During lesion evolution, active lesions develop from the NAWM and are characterized by myelin breakdown and massive infiltration by macrophages and activated microglia. Lesions may remyelinate [56], and partially remyelinated axons and activated microglia are seen [72]. Lesions can develop into inactive lesions with sharply demarcated hypocellular areas of demyelination and axonal degeneration with little to no inflammatory activity [25, 72]. As the disease progresses, the number of chronic active (smoldering, slowly expanding, mixed active/inactive) lesions with a hypocellular demyelinated core and a rim of activated glia increases [25, 46, 56]. The number of chronic active lesions inversely correlates with the proportion of remyelinating lesions, and patients with more severe disease have a higher proportion of such lesions [56].
The molecular mechanisms driving the development and evolution of the different cellular MS endophenotypes are largely unknown. To identify dominant pathways of lesion genesis, unbiased omics investigation of precisely defined and microdissected lesions at these different stages of lesion formation and their comparison to controls is required. We addressed this need by generating and analyzing the first tissue map of the transcriptional landscape of lesion evolution and fate in progressive MS brain by deep next-generation RNA sequencing to identify key pathways, molecules and their cellular source (Fig. 1). Two recent studies have performed nuclei-RNA sequencing on MS WM tissue; however, their focus have been on specific cell types, i.e. neurons and oligodendrocytes [39, 74] We performed bulk RNA sequencing that neglects the cell type but major strengths are high coverage from a high number of samples, and analysis of nuclear, cytoplasmic and extracellular RNA both coding and non-coding [19]. Here, we have re-analyzed the data from our original study, since we discovered that a bug in the analysis scripts resulted in incorrect label annotation files for 10 out of the 100 samples [20]. With our comprehensive transcriptomics data, we have been able to extract mechanistic signatures that differentiate between lesions. We identified lesion-specific protein complex networks by using de novo network enrichment. We further validated the differential expression of key molecules and examined their cellular source by RNAscope, immunohistochemistry, and by immunofluorescence. This specific selection and validation of mechanistic signatures in different lesion types emphasize the value of precision in the characterization of the diverse phenotype of lesions, when understanding the complex and heterogeneous pathogenesis of MS.

Outline of the systems medicine approach to identify mechanistic drivers of different MS lesion types. Using RNAseq we analysed the transcriptome of normal-appearing white matter (NAWM), and lesion evolution/fate (active, inactive, chronic active, remyelinating) in the white matter (WM) of patients with progressive MS. We performed a comprehensive computational data analysis – from differential expression to de novo network enrichment – and examined selected molecules of interest by a combination of RNAscope, immunohistochemistry and immunofluorescence to confirm their cellular source and protein expression levels
Materials and methods
Human postmortem brain tissue
MS and control tissue samples were supplied by the UK Multiple Sclerosis Society Tissue Bank (UK Multicentre Research Ethics Committee, MREC/02/2/39), funded by the Multiple Sclerosis Society of Great Britain and Northern Ireland (registered charity 207,495). A total of 73 snap-frozen tissue blocks from ten progressive MS patients and 25 blocks from five donors without neurological disease were chosen. The death-tissue preservation interval was between 8 and 30 h. Clinical data are summarized in Additional file 4: Table S1.
Lesion classification and immunohistochemistry/immunofluorescence
Snap frozen tissue was sectioned (10-μm), fixed (4% PFA), blocked in PBS with 10% normal horse serum (NHS) and stained for myelin oligodendrocyte glycoprotein (MOG) (R. Reynolds, Imperial College, UK) and HLA-DR (Dako UK Ltd) followed by biotinylated secondary antibody (Jackson Immunoresearch Laboratories, Cambridgeshire, UK), avidin/biotin staining (Vector Laboratories, Burlingame, CA) and DAB staining (Vector Laboratories, Burlingame, CA). All sections were also stained with haematoxylin-eosin (H&E) (R. Reynolds, Imperial College, UK). The five pathological areas (NAWM, active, chronic active, inactive and repairing/early remyelinating lesions) were characterized based on MOG+ staining showing myelin integrity and HLA-DR+ staining showing the inflammatory state, and each lesion type was defined as described (Reynolds et al., 2011). All antibodies and concentration are listed in Additional file 1. Quantification of CD20+ cells were performed in four active and three remyelinating lesions from seven different patients. Ten pictures depending on size per lesion type were taken at an objective lens magnification of 20x. The CD20+ cells were manually counted, and the average number of cells per lesion from each patient was compared by using Mann-Whitney Test performed in Graphpad Prism.
RNAscope
The RNAscope 2.5 Duplex Assay (ACD Biosystems) was performed according to the ACD protocol for fresh-frozen tissue. Brains sections from one patient with both chronic active and remyelinating lesions and from two patients with either chronic active or remyelinating lesions were hybridized with two mRNA probes per experiment. Hs-GFAP (Cat No. 311801) was used as the astrocyte marker and Hs-AIF1/IBA1 (Cat No. 433121) was used as the microglial marker together with Hs-TGFBR2 (Cat No. 407941). Additionally, the Duplex Negative Control Probes (Cat. No. 320751) was used on one section per slide to confirm signal specificity, and the Duplex Positive Control Probes to confirm sensitivity (Additional file 3: Figure S1). The probes were amplified according to manufacturer’s instructions and labeled with the following red or green color for each experiment. The target probes were also combined with immunohistochemistry (anti-GFAP and anti-MHCII) as described above.
RNA extraction from specific histological brain areas
The brain fields of interest were manually microdissected under a magnifying glass in a cryostat. The amount of collected tissue ranged between 10 and 100 mg/sample depending on the lesion size and thickness. A total of 25 WM control areas, 19 NAWM, 6 remyelinating, 18 active, 13 inactive and 17 chronic active lesions were harvested. Total RNA was isolated from the frozen brain samples according to the manufacturer’s instruction (miRNeasy Mini Kit, Qiagen) including DNAse I treatment. RNA concentration was measured using NanoDrop spectrophotometer ND-1000 (Thermo Scientific), and the integrity of RNA (RIN) was measured by using the Bioanalyzer 2100 (Agilent Technologies). RNA integrity was good quality (RIN 6 ± 1.7) among the samples, therefore the fragmentation time and cleanup steps during library preparation have been adapted for each sample based on the RIN value.
Next-generation sequencing
One μg of RNA per sample was processed to remove ribosomal RNA followed by library preparation for RNA sequencing using TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Human/Mouse/Rat Set (Illumina). Pooled indexed libraries were loaded into the flow cell followed by 2 × 80 bp paired-end sequencing on an Illumina NextSeq550.
Raw data analysis and quality control
Demultiplexing was carried out with Casava software (Illumina) configured to allow one mismatch during the identification of the indexes. Data were filtered with Trimmomatic [6] (TRIM:2:30:10 LEADING:20 TRAILING:20 SLIDING:4:20 TRAILING:20 MIN:17). Filtered transcripts were aligned against the human reference genome from UCSC [42] (GRCh38/hg38) with STAR 2.5.3a [14] using default mode/parameters and counted using HTSeq-count [4] using strict mode.
Statistical analysis
Differentially Expressed Genes (DEGs) between different lesion types vs. control WM were identified with the edgeR package (3.8) [73]. The generalized linear model used for our analysis adjusted for library size and biological replicates (same lesion type//same sample//from same patient). Furthermore, we corrected for age and sex of the patients. Genes that had very low expression were excluded following the edgeR userGuide. Therefore, genes were expected to be presented with more than two counts per million (CPM) in at least as many samples as present in the smallest lesion group. Adjusted P value filtering using the procedure of Benjamini and Hochberg was used to identify genes significantly differently expressed between MS brain areas and control brain areas.
Volcano plots, heatmaps and pathways
Volcano plots and heatmaps were created in R studio, and Venn diagrams were produced using an online tool at http://bioinformatics.psb.ugent.be/webtools/Venn/. Predefined pathways were identified by importing the DEGs of selected gene sets to different enrichment tools using Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla) [17] WebGestalt [86] and FunRich [67]. Charts were produced using meta-chart.com. KeyPathwayMiner [1, 2] was used to conduct de-novo network enrichment analyses. The biological network was selected and downloaded from the Integrated Interactions Database (IID) [44] restricted to only brain specific interactions based on evidence type: experimental detection, orthology or prediction. The network and the gene lists were uploaded to the web-interface of KeyPathwayMiner and further processed and analysed in the cytoscape app. Hubs were selected based on the highest betweenness centrality value.
Data availability
All data is deposited and can be post-analyzed online at “msatlas.dk”. Raw data are available upon special request and will be also publicly available in GEO (ID GSE138614). The analysis script is in Additional file 2.
Results
Comparison of the WM transcriptome between MS and control
First, we compared the transcriptome of the global MS tissue (NAWM and lesions) to control WM tissue: out of 18,609 detected genes, 6713 were DEGs (FDR < 0.05 compared to control WM) (Additional file 5: Table S2 and Fig.2A). More than 3000 DEGs were detected for each lesion type, respectively. In the NAWM, only 465 DEGs were present, and the highest number of DEGs was found in chronic active lesions (Fig. 2b). More DEGs with fold change in expression level (log2FC > 1/<− 1, FDR < 0.05) were upregulated (n = 750) than downregulated (n = 206) in the global MS WM transcriptome landscape (Fig. 2a).

Change in gene expression profile during the evolution and fate of WM lesions in progressive MS. a. Visualization of the transcriptional landscape of genes (n = 18,722) detected between MS and non-MS (dots in graph); DEGs are indicated in bright red and orange, where orange indicates log2FC > 1 or < − 1. b. Total number of up- and downregulated DEGs when comparing each lesion type or NAWM to control WM. c. The Venn diagram represents the number of overlapping and lesion-specific DEGs between WM lesion types (active, inactive, remyelinating, chronic active) and NAWM compared to control WM tissue. FDR: false discovery rate; FC: fold change; WM: white matter; NAWM: normal-appearing WM; DEGs: differentially expressed genes (FDR < 0.05)
To identify common and uniquely expressed genes, we compared DEGs between each lesion type (Fig. 2c). We identified 282 common DEGs: among them genes encoding for proinflammatory cytokines, chemokines and complement factors (e.g. IL16, TNFDF14, TNFAIP8, TNFRSF10A, CXCL12, C7, CFH, CFI). In contrast, the number of unique lesion specific DEGs was much higher, 4034 (Fig. 2c, Additional file 6: Table S3).

TGFβ-R2 as a major hub in the common MS lesion-specific de novo network. a. De-novo enriched protein-protein network of the 282 DEGs that were expressed in all MS WM tissue 84 nodes, 169 protein connections). b. Genes of TGFβ receptors and ligands in different lesion types. Asterisks indicate significant changes (*FDR < 0.05, **FDR < 0.01, ***FDR < 0.001). The number indicate the fold change (FC). c. TGFβ-R2 protein expression in remyelinating lesions by immunohistochemistry. d. TGFBR2 (green) RNA co-localizes with GFAP (red) RNA in remyelinating lesions by RNAscope. e. TGFBR2 (green) RNA do not co-localize with IBA1 RNA by RNAscope in remyelinating lesions. FDR: false discovery rate; DEGs: differentially expressed genes (FDR < 0.05)
The common MS lesion-specific de novo network and TGFβ-R2 as a major hub
We extracted the identified 282 common lesion DEGs (Fig. 2c) and examined their de novo enriched network based on protein-protein interactions (Fig. 3a). The biggest de novo network contained 84 proteins of DEGs with 169 connections. The biggest central hub based on betweenness centrality was TGFΒR2 beside other major hubs of STAT6, COL3A1, CASP1, NFATC2, YWHAQ, A2M and CASP7 (Fig. 3a). Beside TGFBR2, five out of six ligands and one additional receptor were also significant DEGs in at least one lesion type (Fig. 3b). To check cellular source, we stained for TGFβ-R2 in remyelinating lesions that had the highest expression level (Log2FC = 1.55, FDR = 0.0001), and the cell morphology of positive cells indicated astrocytes (Fig. 3c). By using RNAscope, we found GFAP and TGFBR2 mRNA co-expressed in remyelinating lesions (Fig. 3d). Microglia did not express TGFBR2 in this lesion type, as IBA1 and TGFBR2 were expressed in different cells far from each other (Fig. 3e).
Additional hubs in the de novo enriched network of shared DEGs were also linked to inflammation, such as STAT6 (IL4 induced transcription factor), CASP1 (part of the inflammasome) and NFAC2 (nuclear factor of activated T cells). The major impact of inflammation as a common mechanism behind lesions was also supported by connected DEGs in the network, e.g. IL16, CXCL12, MERTK, CASP4, C7, CD37, or CASP7 (Fig. 3a).
Transcriptome changes among lesion types
To identify transcriptome changes and generate the molecular signatures of different WM lesion types in progressive MS, we first selected the DEGs for all lesion types compared to control (FDR < 0.05) (Fig. 4a). For each of these selected DEGs, we then calculated the fold-changes between the different lesions types and chose those that were > 2 times (1.5 < log2FC) differentially regulated between at least two lesion types for generation of heatmaps (Fig. 4b). By applying the fold change threshold between lesion types, the heatmap consisted of 28 DEGs, where the Long Intergenic Non-Protein Coding RNA 326 (LINC00326) was the only DEG that was downregulated (Fig. 4b). Remyelinating lesions had the most upregulated DEGs in both analyses. It differed the most from all the other lesion types, when comparing all the DEGs (Fig. 4a). A few genes were much higher expressed in remyelinating lesions compared to the rest; either involved in axonal assembly (SPAG17, DNAH11, DCDC1 DNAAF1) or unknown (FHAD1, TTC34). The metabolic gene (ADAMST18), chemokine receptor (CXCR4), plasma protease inhibitor (SERPNA4) and lincRNA (RP1-111D6.3) were particularly less upregulated in chronic active lesions (Fig. 4b). The pattern of DEGs were more similar between active and remyelinating lesions, while chronic active DEGs pattern resembled more to inactive lesions (Fig. 4b).

Transcriptome changes among lesions in progressive MS. a. Heatmap showing the 1487 DEGs in all WM lesion types compared to WM. b. Heatmap showing 28 DEGs in different WM lesion types compared to control WM with a selection of the DEGs with highest fold changes (log2FC > 1.5 for at least one pair of lesions types). FC: fold change; FDR: false discovery rate; WM: white matter; NAWM: normal appearing WM; DEGs: differentially expressed genes (FDR < 0.05)
Cellular processes and cellular locations of lesion specific DEGs
To examine molecular processes of unique DEGs for each lesion type, we used GOrilla. We uploaded the unique DEGs (290 for active, 447 for remyelinating, 1068 for inactive, and 2213 for chronic active, Additional file 6: Table S3) to Gorilla, and extracted pathways (0 for remyelinating, 3 for active, 2 for chronic active, 11 for inactive) and molecular functions (0 for remyelinating and active, 24 for chronic active, 13 for inactive) (Additional file 7: Table S4).). In active lesions, the three identified pathways were “Immune System”, “Innate Immune System” and “Neutrophil degranulation”. For chronic active lesions, the two pathways identified were the “Histamine H1 receptor signaling” and the “Wnt signaling pathway”; the 11 identified pathways in inactive lesion were mainly related to cellular responses to stress and heat shock proteins, metabolism and the “Neutrophil degranulation” (Additional file 7: Table S4).
We classified the biological processes in six groups: “immune-related”, “cell activation and extracellular transduction”, “protein modifications”, “metabolic processes”, “extracellular secretion and exocytosis”, and “brain (neuron) specific” (Fig. 5a). Active lesions were mostly enriched in immune-related biological processes (54%); inactive lesions were enriched in metabolic processes (56%); and chronic active lesions were highly enriched in both cell activation/intracellular transduction signaling and brain (neuron) specific biological processes (73%). No specific known biological process was identified in remyelinating lesion.

Lesion/specific biological processes and cellular locations. a. The bar chart shows the number of biological processes in the different lesion types (active, inactive, chronic active, remyelinating) based on the lesion-specific DEGs. We have classified them in six groups: “immune-related”, “cell activation and extracellular transduction”, “protein modifications”, “metabolic processes”, “extracellular secretion and exocytosis”, and “brain (neuron) specific”. b. The circle diagram for each lesion type (active, inactive, chronic active) represents the percentage of cellular components identified from the lesion-specific DEGs and grouped in “granules”, “vesicles”, “extracellular space”, “cytoplasm”, “mitochondria” and “brain (neuron) specific”. No predefined cellular component was predicted for remyelination. FDR: false discovery rate; DEGs: differentially expressed genes (FDR < 0.05)
We grouped the cellular components identified from the lesion-specific DEGs in “granules”, “vesicles”, “extracellular space”, “cytoplasm”, “mitochondria” and “brain (neuron) specific” (Fig. 5b). In active lesions, the DEGs of cellular components were related to vesicles, extracellular space, cytoplasm and granules. In inactive lesions, cytoplasm was the dominant area of localization besides mitochondria, granules, vesicles, and extracellular space. In chronic active lesions, 60% of the cellular components were brain/neuron specific, and the rest of unique DEGs were related to cytoplasm and extracellular space. No specific known cellular components were identified in remyelinating lesions.
Unique de novo protein-protein networks of different lesion types
Beside the known predetermined pathways, we examined de novo pathways based on the lesion stage-specific gene expression. By using KeyPathwayMiner, we mapped each of the highly significant lesion-specific DEGs (FDR < 0.001: active = 164, remyelinating = 235, inactive = 484, chronic active = 853) (Fig. 6) to a brain-specific protein-protein network, and retrieved the biggest de novo subnetwork with hubs for each lesion type (Fig. 6). We selected the ten major hubs in each lesion type based on the magnitude of betweenness centrality. In the active lesion-specific biggest network (n = 43 with 60 connections), the ten major hubs were SH2B3, RAC2, RAB23, ANXA2, SMURF1, TGFB1L1, TRIM38, TNFAIP3, CSF2RB and PRKCZ reflecting processes involved in autoimmunity risk and immune responses (Fig. 6a). The remyelinating lesion-specific network contained 50 DEGs with 66 connections, and the ten major hubs were KLK6, APP, PLAU, CTGF, EFEMP1, RRAS, CCL5 ROR2 and CD8A indicating ongoing adaptive immune responses and upregulated growth factor genes; additional well known immune related genes were also present in the network, such as ILGR2, TNFs, NCAM1 (Fig. 6b). However, none of the genes of tissue-resident CD8+ T cells were significantly changed in this lesion (CXCR6, GPR56, CD49a, CD44, PD-1, CD103, CD69), and the genes of cytotoxic molecules granzyme B and GPR56 were not changed either (www.msatlas.dk). The inactive lesion-specific network consisted of 147 DEGs with 346 connections. The ten major hubs were SMOC1, SEMA6D, FLNA, HSPD1, HSPA4, NXF1, XPO1, RAC1, GABARAPL2 and BAG3 pointing to oxidative stress and protein modification (Fig. 6c). The chronic active lesion-specific network included 315 DEGs with 795 connections, and the ten major hubs were SLC39A10, EGFR, ABL1, SLIT2, PRKCB, TAB2, EPHA7, RGS14, HERC2 and COPS5; in general, this lesion-specific network was focused on neuronal and axonal changes (Fig. 6d).

Lesion specific signatures and their biggest de novo network enrichment. By using KeyPathwayMiner, we retrieved the biggest de novo brain-specific protein-protein network from each of the lesion-specific gene list ((FDR < 0.001: active = 164, remyelinating = 235, inactive = 484, chronic active = 853) without any exception nodes. a. Forty-three nodes with 60 connections in active lesions. b. Fifty nodes with 66 connections in remyelinating lesions. c. One hundred forty-seven nodes with 346 connections in inactive lesions. d. One hundred fifteen nodes with 795 connections in chronic active lesions. Red color indicates upregulation, while blue color indicates downregulation of the nodes. The size of the node indicates higher betweenness centrality. FDR: false discovery rate
Transcriptome signature of the NAWM and CD26 a lesion marker in diverse cell types
To address changes before the evolution of lesions, we examined all DEGs (n = 465) in NAWM. We detected 305 upregulated and 160 downregulated DEGs (Fig. 2b). Of the ten upregulated genes, microglia/macrophages/immune related DEGs (GPNMB, CD163, HLA-DRB5, F13A1, IGHG1), mitochondrial humanins (MTRNR2L8, MTRNR2L12) and brain specific genes (POR7A5, NPIPA5) were present. The downregulated DEGs were not related to specific cell types or pathogenic mechanisms (U2AF1L5, SLC25A48, CAMPD8), or belonged to the long-intronic-non-coding (linc) RNA class (LNC0706, RNU5D-1, RAB6C-AS1, RP1-74D7.1) (Fig. 7a). The 16 unique NAWM DEGs (Figs. 2c and 7b) also belonged to unknown/unspecific functional group (ERP29, TTC23) or non-protein-coding RNAs (SNAPc1, RNU5D-1, FCF1P2) besides regulation of proliferation (EIF3C, PROM1/CD133, DDIT4L), metabolic (CYP3A4, P2RY1), axonal specific (DRAXIN), and cellular interaction (ADGRE4P) DEGs.

Molecular signature of NAWM and CD26/DPP4 expression. a. The top ten up- and downregulated DEGs in NAWM compared to control WM. b. The 16 uniquely expressed DEGs in NAWM. c. The RNA expression level of CD26/DPP4 in the NAWM, chronic active remyelinating, active and inactive lesions. d. The protein expression of CD26/DPP4 in the NAWM and absence in control WM. e. The co-localization of CD26/DPP4 with IBA1 in the NAWM. f. Expression of CD26/DPP4 at a vessel in an active lesion. FDR: false discovery rate; DEGs: differentially expressed genes (FDR < 0.05); WM: white matter; NAWM: normal appearing WM
Among the common DEGs, we found CD26/DPP4 encoding for dipeptidylpeptidase 4 that was also identified in the NAWM in a previous study by RNA-seq and DNA methylation analysis [36] (Fig. 7c). We confirmed the protein expression of CD26 in the NAWM, and its absence in control WM by immunohistochemistry (Fig. 7d). The morphology of cells expressing CD26 in NAWM indicated microglia, and CD26 co-localized with IBA1 (Fig. 7e). In the active lesions, CD26 was expressed by mononuclear cells in the vicinity of vessels rather than by microglia (Fig. 7f).
Immunoglobulin signatures in the different WM lesion types
We noticed that immunoglobulin genes were present among the top 10 upregulated DEGs in the global WM tissue of MS (Fig. 8a). To examine their distribution in the different lesion types, we produced a heatmap with all significant (FDR < 0.05) immunoglobulin transcripts (Fig. 8b). IGHG1 encoding for heavy chain IgG1was highly expressed in all lesions and in the NAWM; IGKC encoding for the constant region of light kappa chain was highly expressed in all lesions; genes encoding for IgG2, IgG3 and IgM heavy chains (IGHG2, IGHG3, IGHM), and variable region of light kappa chain (IGKV4–1) were only present in remyelinating lesions. To examine, if such high expression of immunoglobulin genes was due to a higher number of B cells, we investigated the presence of B cells by quantifying CD20+ cells in active (n = 3) and remyelinating (n = 4) lesions each from different patients (n = 7). We found that CD20+ cells were mostly present in active lesions, but remyelinating lesions had the most heterogenous upregulated transcripts (Fig. 8c).

Immunoglobulin signatures and B cells in WM lesions of patients with progressive MS. a. Immunoglobulin transcripts among the top 10 upregulated DEGs in the global MS WM tissue compared to control WM tissue. b. Heatmap of immunoglobulin transcripts in the different lesion types and NAWM. c. Number of CD20+ cells in active (n = 3) and remyelinating (n = 4) lesions, each lesion from different brains. The same brains and lesions were used for generating transcriptome signatures. Around 10 pictures per lesion in each patient were taken at magnification of 20x and the average cell number per lesion were calculated. Statistical difference between the CD20+ cells was calculated using the Mann-Whitney test. Immunohistochemistry shows CD20+ cells at small vessels within an active and remyelinating MS lesion, respectively. FDR: false discovery rate; DEGs: differentially expressed genes (FDR < 0.05); WM: white matter; NAWM: normal appearing WM
Discussion
We introduce the first mechanistic investigation of transcriptome signatures of lesion evolution and fate in the WM of patients with progressive MS across all major WM lesion types: NAWM, active, inactive, chronic active and remyelinating lesions (compared to control WM). One study applied next generation RNA sequencing to examine gene expression in the NAWM [37], and a very recent work examined oligodendrocyte nuclei signatures in MS lesions [39]. We controlled for confounders using generalized mixed effect linear models considering age, sex and multiple samples of the same patient. We corrected all results for multiple testing with a target FDR value < 0.05 to use a conservative statistical estimation of gene expression changes. We detected a high number of differently expressed genes (DEGs) in different lesion types (compared to the control samples) using an FDR-corrected p-value threshold of 0.05. Most of these DEGs with high fold change were upregulated in MS tissue. We then, for the first time, interrogated the human interactome for sub-networks that putatively drive MS lesion evolution mechanistically.
Common DEGs in different lesion types
The central hub in the de novo network based on shared DEGs was TGFβ-R2 that was most upregulated in remyelinating lesions (Fig 3). By immunohistochemistry and RNAscope, we found that TGFβ-R2 was expressed by astrocytes in remyelinating lesions. The gene expression of one of its major ligands, TGFβ1 was significantly expressed in active and remyelinating lesions only, in contrast to genes of TGFβ2 and TGFβ3. TGFβ1 has been associated with reparatory function in the CNS [15]. A recent study on microarray data from spinal cord periplaque vs. NAWM identified TGFβ1 in the context of astrocytosis and remodeling [64]. Astrocyte targeted overexpression of TGFβ1 resulted in earlier and more severe experimental autoimmune encephalomyelitis [57, 89], while systemic administration inhibited disease [47]. A previous study suggested that TGFβ activity leads to the formation of chronic MS lesions [13]. Indeed, we found that TGFΒ2 was a DEG in chronic active lesions in addition to active and remyelinating ones. But it may have a dual role in by both regulating the immune responses and promoting neuronal survival [27], and is an important player of neural stem/precursor cells (NPCs) immunomodulation [12]. Since we observed unique neuron/axon specific activities in chronic active lesions (Fig. 5), the highly significant expression of TGFΒ2 may be also related to this. Our data indicates that the effect of TGFβ ligands and their receptors in the MS-CNS may depend on the lesion types with different inflammatory and cellular environment [15].
Transcriptome changes specific to different lesion types
In order to investigate unique transcriptional changes at different stages of lesion evolution and fate, we applied a comprehensive approach: (i) we identified DEGs that were differentially regulated at least among two lesion types visualized in a global transcriptome heatmap (Fig. 4); (ii) we investigated predefined pathways based on DEGs that were present only in one lesion type (Fig. 5); (iii) we extracted unique significant (FDR < 0.001) up- and downregulated genes in each lesion type, and created de novo enriched protein interaction networks with major hubs for these DEGs (Fig. 6).
Active lesions
Based on all the different analyses, the molecular signature of active lesions indicated the classic inflammatory MS lesion governed by immune responses (Figs. 5 and 6, Additional file 6: Table S3): over 50% of the biological processes were immune-related and were distributed to granules, vesicles, extracellular space and the cytoplasm, suggesting extracellular effects on the microenvironment. The ten major hubs in the de novo network enrichment analysis have all been related to MS or autoimmune/brain diseases (Fig. 6): MS susceptibility genes (SH2B3 [3], RAB23 [50]), potential biomarkers (CSF2RB [70], ANXA2 [38]), or potential roles in inflammatory/brain diseases or MS pathogenesis (RAC2 [78], Smurf1 [62], TGFB1I1 [53], TRIM38 [35], TNFAIP3 [33], PRKCZ [51]). Active lesions exhibited the least unique DEGs (n = 290, FDR < 0.05), which could suggest that active lesion is the primary step for lesion evolution, and the source of all other lesion types.
Remyelinating lesions
The signature of active lesions was more similar to remyelinating lesions than to inactive and chronic active lesions, and remyelinating lesions had the second least unique DEGs (n = 447, FDR < 0.05). Remyelinating lesion type differed the most from all the others in the context of DEGs present in all lesion types (Fig. 4a). However, the most different molecular signature was found between chronic active and remyelinating lesions when setting a threshold between the difference in expression level between at least two lesion types (Fig. 4b). We did not detect any known predefined pathways, biological processes, cellular component enrichments or molecular function with Gorilla, WeGestalt or FunEnrich in remyelinating lesions. That contrasted with the highest expression level of shared lesion DEGs (Fig. 4b). Some of these were related to dynein-dependent axonal transport during brain development (Spag17, Dnah11, DNAAF1, DCDC1), which could suggest neuronal response to stress [84].
In the de novo network enrichment analysis of unique DEGs, well known immune-related genes were present such as TNF, CCL5, NCAM1, PLAU, CD8 and ILGR2. This may indicate an overlap between active and remyelinating lesions, and may be related to early remyelinating events also indicated by partial remyelination of these lesions. The genes for molecules characterizing tissue-resident CD8+ cells (Trm cells) were not significantly changed, and the genes for cytotoxic molecules granzyme B and GPR56 were also not upregulated.
Unique downregulation of two hubs supported protective events: KLK6, which has been indicated as a marker for disease worsening in EAE and MS [5], and FA2H that has been linked to WM neurodegeneration [45]. Two additional upregulated hubs supported regenerative processes: CTGF, a central mediator of tissue remodeling [52] and EFEMP1 promoter of cell growth often implicated in cancer [91]. However, CTGF, has also been found to inhibit myelination [21]. Despite the observed remyelination, myelin genes (MBP, MOG, MAG) were not upregulated. This was characteristic of all lesion types (data not shown) similar to recent data by microarray [92]. A recent work that examined oligodendrocyte heterogeneity by single nuclei sequencing and found that there was a reduction of OPCs in MS and NAWM compared to control, and the subclusters of mature oligodendrocytes were skewed between MS and control tissue [40]. The number of OPCs and oligodendrocytes are reduced in MS lesions [7, 39, 55], which may also be responsible for the observed absence of changes in myelin gene expression.
Chronic active lesions
In contrast to remyelinating lesions, chronic active lesions were characterized by a high number of differentially regulated predefined biological processes; the highest number of DEGs (n = 5739) and the highest number of unique DEGs (n = 2213). This distinctive nature of chronic active lesions was also reflected by the biggest de novo network of unique DEGs. The heatmap also suggested that chronic active lesions were the most different from remyelinating lesions (Fig. 4b).
We observed a dominance of neuron/synapse specific biological processes in white matter lesions (Fig. 5, Additional file 7: Table S4) that was somewhat unexpected. Recent evidence has localized mRNA translation in subcellular regions of neurons (dendrites, axons, synapses, somas) [76]. It is also well known that OPCs do express many synaptic markers and are shown to form synapses in the WM as well [28]. It may be RNA in vesicles transported through the axons or maybe some genes that also play a role in glia cells. In contrast to active lesions, intracellular transduction events were more common, such as binding reactions with metal ions, anions, ATP, and cytoskeleton proteins (Additional file 7: Table S4). Synaptic and axonal events were also supported by the increased enrichment of lesion-specific DEGs belonging to the cadherin (CDH) family, potassium family (KCN and KCTD), the ephrin (ECH) receptors and GABA genes (Additional file 6: Table S3). Five of the ten biggest hubs in the de novo network enrichment analysis were all also neuron-related: SLC39A10 [63], SLIT2 [88], EPHA7 [10], RGS14 [79], HERC2 [11], COPS5 [87]. DEGs suggested lower inflammatory response in chronic active lesions compared to the other lesion types. The only identified immune related process was” response to wound healing”, which could suggest a more post-inflammatory reaction (Additional file 7: Table S4). One of the major hubs, SLIT2, mentioned above, is released from neurons and also inhibits leukocyte chemotaxis migration [88].
Altogether, such neuron/synaptic gene activity in chronic active lesions indicates intrinsic irreversible pathogenic events less coupled with inflammatory reactions, and could also explain why immune-related treatments work less in progressive MS, where this lesion type increases [56].
Inactive lesions
Inactive lesions had over twice as many unique DEGs as active lesions (n = 1068). These suggested protein modifications, cellular stress, heat shock protein responses, metabolic events, catabolic and “breaking down” of components (Fig. 5, Additional file 6: Table S3, Additional file 7: Table S4). This was further supported by major hubs in the de novo network enrichment analysis (HSPD1, HSPA4) (Fig. 6). Two nuclei exporter proteins (NXF1, XPO1) were also among the ten major hubs, and they are known to be upregulated after neuronal damage to prevent repeated neurotoxicity [30, 43, 82]. The export, folding/unfolding of proteins plus the catalytic and oxidoreductase reactions takes place in the cytoplasm, which was also predicted as the dominant cellular location (Fig. 5b) including different organelle compartments, vesicles and mitochondria (Additional file 7: Table S4).
NAWM and Dipeptidylpeptidase IV (CD26/DPP4)
We found 465 DEGs in NAWM (Figs. 2 and 7). Most of the upregulated genes were microglia/macrophage/immune related, which may suggest an altered phenotype of diffusely activated microglia throughout the NAWM as indicated recently [85]. Mitochondrial and brain specific genes were also altered, suggesting other intrinsic events either before the development of active lesions or indicating changes as consequence of pathological events and lesion development at other sites. Mitochondrial humanins (MTRNR2L12, MTRNR2L8) that may protect cells from oxidative stress [90] were significantly upregulated in both NAWM and all lesions.
The lower number of DEGs also contrasted the thousands of DEGs in lesions: while NAWM differed from control WM only by 465 DEGs, it differed from lesions by 3894 DEGs. The unique-specific DEGs in NAWM were enriched by unspecified or non-protein-coding genes, which may suggest that more unconventional approaches may be needed to understand mechanistic changes before lesion evolution.
We also found CD26 in the NAWM and all lesion types (Fig. 7). A recent study also detected significant expression of CD26 in both DNA methylation and RNA seq data in the NAWM tissue [37]. CD26/DPP4 is a membrane-associated exopeptidase that by engaging inhibitory ligands may limit autoimmunity in mice by regulating Th1 responses [68, 80], and by hydrolyzing substrates CXCL12 and CCL5 [8]. By using immunohistochemistry and immunofluorescence, we found that CD26 was expressed by microglia in the NAWM. In contrast, in active lesions, the CD26+ cells had mononuclear morphology.. These data suggested that CD26 may be related to an altered microglia function/phenotype in the NAWM and continue to be significantly expressed in lesion types. The recent report of protection against cuprizone-induced demyelination by an inhibitory ligand of CD26 [18] also suggests regulation of microglia function, since the role of T cells in this model is probably minor [32, 71]. The different cellular source of CD26 in NAWM and active lesions also indicate that differential regulation of a gene should be addressed in the context of lesion type and cellular source to interpret potentially different functional outcomes.
Immunoglobulins and B cells
We noticed that immunoglobulin genes were among the top 10 upregulated genes in WM MS tissue vs. control WM (Fig. 8). The highly significant expression of immunoglobulin genes among the total MS-WM can be explained by presence of plasma cells, or by increased transcription of rearranged B cell receptors secreted also as antibodies. There is an increasing recognition of B cells and plasma cells also in the WM lesions of PMS besides the B cell-rich aggregates [57, 61, 79, 80]. Here, we found B cells in WM lesions mostly located around the vessels. All lesions had IGHG1 and IGKC highly upregulated. A proteomic study of CSF also found the protein of Ig gamma-1 chain (IGHG1) more abundant in fulminant MS samples compared to control [26]. Another study also confirmed the expression of IGKC and IGHG1 in NAGM and GM lesions with both microarray and qPCR [83]. The transcribed immunoglobulin genes we detected, may be secreted because among the top 10 upregulated is J-CHAIN, which serve to link immunoglobulins in dimer (IgA) or pentamer (IgM) as secretory components [40]. The dominance of immunoglobulin genes among the top upregulated DEGs was disproportional to the number of B cells (Fig. 8), indicating either a restricted B cell clonality, or high secretion of immunoglobulins.
IGKV4–1, IGHM, IGHG2, IGHG3 were uniquely upregulated in remyelinating lesions. The IGHV4 transcript was also most frequently found in B cell receptor transcriptome of the CSF and paired brain-draining lymph node tissue [41, 66, 81], and maybe related to rare T cell exposed motifs [34]. The specific presence of the variable regions in the remyelinating lesions may indicate a more heterogeneous B cell phenotype with paratopes to a wider range of epitopes. The heterogenous upregulated transcripts for immunoglobulins of other groups (IGHG2, IGHG3) in remyelinating lesions could be due to another minor overlooked mature B cell subpopulation. The presence of B cells in remyelinating lesions were also emphasized by de novo pathway analysis where both T- and B cell markers were present (Fig. 6). A recent work also emphasized the presence of plasma cells in lesions of patients with progressive MS [58]. Altogether, these data argue for important role of B cells even in the WM of progressive MS. Whether the heterogeneous immunoglobulin genes in remyelinating lesions could reflect some special subset of B cells is not clear; we were not able to detect IL10 transcripts in remyelinating lesions that may be related to regulatory B cells [77], but we did find the IL10 receptor (IL10RA) highly upregulated in the lesions, mostly in remyelinating ones (log2FC 2,3 and FDR 4.2 × 10− 5).
Conclusion
In conclusion, by next-generation RNA sequencing and a comprehensive computational systems medicine approach, we identified the first mechanistic transcriptome signature of lesion evolution and fate in the progressive MS brain WM.
The major de novo network of shared DEGs among the different lesions emphasize inflammation as a common mechanism, and support the view also provided by GWAS that MS is an inflammatory disease, even at the later progressive stage.
We found that each lesion type had a huge complexity of molecular pathways, and although we tried to categorize them to simplify for better understanding, many pathways were unexpected and overlapping indicating a dynamic process of lesion evolution. TGFβR2 was major hub of shared DEGs by all lesions and NAWM, and it was expressed by astrocytes in remyelinating lesions. Our data also suggest that chronic active lesions that are more frequent during the late progressive phase have more complex molecular mechanisms and changes in multiple pathways. This lesion type was profoundly different from all other lesion types, and also from control WM. NAWM was more similar to control WM than to any other lesion types. This indicates that major gene expression changes occur both at early lesion genesis, and in lesions most characteristic as the late progressive phase develops. Besides unique sub-networks mechanistically evolving different lesions stages, some molecules were specifically regulated: CD26/DPP4 by microglia in the NAWM and by mononuclear cells in active lesions. The uniqueness of lesion types also indicates that omics approaches should consider lesion stages, when expression and regulation of different molecules are addressed. Although this study indicates the extreme diverse molecular events on transcriptome level at different lesion stages, our comprehensive unbiased search across subsets of multiple lesions provided a discovery of specific molecular mechanistic signatures validated by different approaches.
A limitation of our study is the absence of controls with other neurological disease, and the lack of separation of rim and core in the chronic active and inactive lesions. Nevertheless, the combination of different bioinformatics methods and validation by immunohistochemistry supported our conclusions, and overcome these limitation for the interpretation of changes in transcriptome signatures during lesion evolution and fate in the WM of progressive MS brain.
Availability of data and materials
The datasets generated and/or analysed during the current study are available as interactive online database linked to bioinformatics approaches at „msatlas.dk”.
References
Alcaraz N, Kücük H, Weile J, Wipat A, Baumbach J (2011) KeyPathwayMiner: detecting case-specific biological pathways using expression data. Internet Math 7:299–313. https://doi.org/10.1080/15427951.2011.604548
Alcaraz N, Pauling J, Batra R, Barbosa E, Junge A, Christensen AGL, Azevedo V, Ditzel HJ, Baumbach J (2014) KeyPathwayMiner 4.0: condition-specific pathway analysis by combining multiple omics studies and networks with Cytoscape. BMC Syst Biol 8:99. https://doi.org/10.1186/s12918-014-0099-x
Alcina A, Vandenbroeck K, Otaegui D, Saiz A, Gonzalez JR, Fernandez O, Cavanillas ML, Cénit MC, Arroyo R, Alloza I, García-Barcina M, Antigüedad A, Leyva L, Izquierdo G, Lucas M, Fedetz M, Pinto-Medel MJ, Olascoaga J, Blanco Y, Comabella M, Montalban X, Urcelay E, Matesanz F (2010) The autoimmune disease-associated KIF5A, CD226 and SH2B3 gene variants confer susceptibility for multiple sclerosis. Genes Immun 11:439–445. https://doi.org/10.1038/gene.2010.30
Anders S, Pyl PT, Huber W (2015) HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. https://doi.org/10.1093/bioinformatics/btu638
Bando Y, Hagiwara Y, Suzuki Y, Yoshida K, Aburakawa Y, Kimura T, Murakami C, Ono M, Tanaka T, Jiang Y-P, Mitrovi B, Bochimoto H, Yahara O, Yoshida S (2018) Kallikrein 6 secreted by oligodendrocytes regulates the progression of experimental autoimmune encephalomyelitis. Glia 66:359–378. https://doi.org/10.1002/glia.23249
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Boyd A, Zhang H, Williams A (2013) Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 125:841–859. https://doi.org/10.1007/s00401-013-1112-y
Busso N, Wagtmann N, Herling C, Chobaz-Péclat V, Bischof-Delaloye A, So A, Grouzmann E (2005) Circulating CD26 is negatively associated with inflammation in human and experimental arthritis. Am J Pathol 166:433–442. https://doi.org/10.1016/S0002-9440(10)62266-3
Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ (2011) Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 69:481–492. https://doi.org/10.1002/ana.22109
Clifford MA, Athar W, Leonard CE, Russo A, Sampognaro PJ, Van der Goes M-S, Burton DA, Zhao X, Lalchandani RR, Sahin M, Vicini S, Donoghue MJ (2014) EphA7 signaling guides cortical dendritic development and spine maturation. Proc Natl Acad Sci 111:4994–4999. https://doi.org/10.1073/pnas.1323793111
Cubillos-Rojas M, Schneider T, Hadjebi O, Pedrazza L, de Oliveira JR, Langa F, Guénet J-L, Duran J, de Anta JM, Alcántara S, Ruiz R, Pérez-Villegas EM, Aguilar-Montilla FJ, Carrión ÁM, Armengol JA, Baple E, Crosby AH, Bartrons R, Ventura F, Rosa JL (2016) The HERC2 ubiquitin ligase is essential for embryonic development and regulates motor coordination. Oncotarget 7:56083–56106. https://doi.org/10.18632/oncotarget.11270
De Feo D, Merlini A, Brambilla E, Ottoboni L, Laterza C, Menon R, Srinivasan S, Farina C, Garcia Manteiga JM, Butti E, Bacigaluppi M, Comi G, Greter M, Martino G (2017) Neural precursor cell-secreted TGF-β2 redirects inflammatory monocyte-derived cells in CNS autoimmunity. J Clin Invest 127:3937–3953. https://doi.org/10.1172/JCI92387
De Groot CJ, Montagne L, Barten AD, Sminia P, Van Der Valk P (1999) Expression of transforming growth factor (TGF)-beta1, −beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J Neuropathol Exp Neurol 58:174–187
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Dobolyi A, Vincze C, Pál G, Lovas G (2012) The Neuroprotective functions of transforming growth factor Beta proteins. Int J Mol Sci 13:8219–8258. https://doi.org/10.3390/ijms13078219
Dutta R, Trapp BD (2014) Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol 27:271–278. https://doi.org/10.1097/WCO.0000000000000094
Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48. https://doi.org/10.1186/1471-2105-10-48
Elbaz EM, Senousy MA, El-Tanbouly DM, Sayed RH (2018) Neuroprotective effect of linagliptin against cuprizone-induced demyelination and behavioural dysfunction in mice: a pivotal role of AMPK/SIRT1 and JAK2/STAT3/NF-κB signalling pathway modulation. Toxicol Appl Pharmacol 352:153–161. https://doi.org/10.1016/j.taap.2018.05.035
Elkjaer ML, Frisch T, Reynolds R, Kacprowski T, Burton M, Kruse TA, Thomassen M, Baumbach J, Illes Z (2019) RETRACTED ARTICLE: unique RNA signature of different lesion types in the brain white matter in progressive multiple sclerosis. Acta Neuropathol Commun 7:58. https://doi.org/10.1186/s40478-019-0709-3
Elkjaer ML, Frisch T, Reynolds R, Kacprowski T, Burton M, Kruse TA, Thomassen M, Baumbach J, Illes Z (2019) Retraction note: unique RNA signature of different lesion types in the brain white matter in progressive multiple sclerosis. Acta Neuropathol Commun 7:136. https://doi.org/10.1186/s40478-019-0790-7
Ercan E, Han JM, Di Nardo A, Winden K, Han M-J, Hoyo L, Saffari A, Leask A, Geschwind DH, Sahin M (2017) Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J Exp Med 214:681–697. https://doi.org/10.1084/jem.20160446
Felts PA, Woolston A-M, Fernando HB, Asquith S, Gregson NA, Mizzi OJ, Smith KJ (2005) Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain 128:1649–1666. https://doi.org/10.1093/brain/awh516
Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3):393–399
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132:1175–1189. https://doi.org/10.1093/brain/awp070
Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, Mandrekar J, Bramow S, Metz I, Brück W, Lassmann H, Lucchinetti CF (2015) Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 78:710–721. https://doi.org/10.1002/ana.24497
Füvesi J, Hanrieder J, Bencsik K, Rajda C, Kovács SK, Kaizer L, Beniczky S, Vécsei L, Bergquist J (2012) Proteomic analysis of cerebrospinal fluid in a fulminant case of multiple sclerosis. Int J Mol Sci 13:7676–7693. https://doi.org/10.3390/ijms13067676
Goris A, Williams-Gray CH, Foltynie T, Brown J, Maranian M, Walton A, Compston DAS, Barker RA, Sawcer SJ (2007) Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson’s disease. J Neurol 254:846–848. https://doi.org/10.1007/s00415-006-0414-6
Habermacher C, Angulo MC, Benamer N (2019) Glutamate versus GABA in neuron–oligodendroglia communication. Glia 67:2092–2106. https://doi.org/10.1002/glia.23618
Haider L, Zrzavy T, Hametner S, Höftberger R, Bagnato F, Grabner G, Trattnig S, Pfeifenbring S, Brück W, Lassmann H (2016) The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 139:awv398. https://doi.org/10.1093/brain/awv398
Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez-Martinez A, Cooper-Knock J, Higginbottom A, Lin Y-H, Bauer CS, Dodd JE, Myszczynska MA, Alam SM, Garneret P, Chandran JS, Karyka E, Stopford MJ, Smith EF, Kirby J, Meyer K, Kaspar BK, Isaacs AM, El-Khamisy SF, De Vos KJ, Ning K, Azzouz M, Whitworth AJ, Shaw PJ (2017) SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8:16063. https://doi.org/10.1038/ncomms16063
Hemmer B, Kerschensteiner M, Korn T (2015) Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 14:406–419. https://doi.org/10.1016/S1474-4422(14)70305-9
Hiremath MM, Chen VS, Suzuki K, Ting JPY, Matsushima GK (2008) MHC class II exacerbates demyelination in vivo independently of T cells. J Neuroimmunol 203:23–32. https://doi.org/10.1016/j.jneuroim.2008.06.034
Hoffjan S, Okur A, Epplen JT, Wieczorek S, Chan A, Akkad DA (2015) Association of TNFAIP3 and TNFRSF1A variation with multiple sclerosis in a German case-control cohort. Int J Immunogenet 42:106–110. https://doi.org/10.1111/iji.12183
Høglund RA, Lossius A, Johansen JN, Homan J, Benth JŠ, Robins H, Bogen B, Bremel RD, Holmøy T (2017) In Silico prediction analysis of Idiotope-driven T-B cell collaboration in multiple sclerosis. Front Immunol 8:1255. https://doi.org/10.3389/fimmu.2017.01255
Hu M-M, Shu H-B (2017) Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cell Mol Immunol 14:331–338. https://doi.org/10.1038/cmi.2016.66
Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, Haroutunian V, Zhu J, Donovan MJ, Sharp AJ, Casaccia P (2013) Epigenome-wide differences in pathology-free regions of multiple sclerosis–affected brains. Nat Neurosci 17:121–130. https://doi.org/10.1038/nn.3588
Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, Haroutunian V, Zhu J, Donovan MJ, Sharp AJ, Casaccia P (2014) Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat Neurosci 17:121–130. https://doi.org/10.1038/nn.3588
Iparraguirre L, Muñoz-Culla M, Prada-Luengo I, Castillo-Triviño T, Olascoaga J, Otaegui D (2017) Circular RNA profiling reveals that circular RNAs from ANXA2 can be used as new biomarkers for multiple sclerosis. Hum Mol Genet 26:3564–3572. https://doi.org/10.1093/hmg/ddx243
Jäkel S, Agirre E, Mendanha Falcão A, van Bruggen D, Lee KW, Knuesel I, Malhotra D, Ffrench-Constant C, Williams A, Castelo-Branco G (2019) Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566:543–547. https://doi.org/10.1038/s41586-019-0903-2
Johansen FE, Braathen R, Brandtzaeg P (2000) Role of J chain in secretory immunoglobulin formation. Scand J Immunol 52:240–248. https://doi.org/10.1046/j.1365-3083.2000.00790.x
Johansen JN, Vartdal F, Desmarais C, Tutturen AEVV, de Souza GA, Lossius A, Holmøy T (2015) Intrathecal BCR transcriptome in multiple sclerosis versus other neuroinflammation: equally diverse and compartmentalized, but more mutated, biased and overlapping with the proteome. Clin Immunol 160:211–225. https://doi.org/10.1016/j.clim.2015.06.001
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12:996–1006. https://doi.org/10.1101/gr.229102 Article published online before print in may 2002
Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, Casaccia P (2010) HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci 13:180–189. https://doi.org/10.1038/nn.2471
Kotlyar M, Pastrello C, Sheahan N, Jurisica I (2016) Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res 44:D536–D541. https://doi.org/10.1093/nar/gkv1115
Kruer MC, Paisán-Ruiz C, Boddaert N, Yoon MY, Hama H, Gregory A, Malandrini A, Woltjer RL, Munnich A, Gobin S, Polster BJ, Palmeri S, Edvardson S, Hardy J, Houlden H, Hayflick SJ (2010) Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 68:611–618. https://doi.org/10.1002/ana.22122
Kuhlmann T, Lassmann H, Brück W (2008) Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol 115:275–287. https://doi.org/10.1007/s00401-007-0320-8
Kuruvilla AP, Shah R, Hochwald GM, Liggitt HD, Palladino MA, Thorbecke GJ (1991) Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc Natl Acad Sci U S A 88:2918–2921
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128:2705–2712. https://doi.org/10.1093/brain/awh641
Lassmann H (2014) Multiple sclerosis: lessons from molecular neuropathology. Exp Neurol 262(Pt A):2–7. https://doi.org/10.1016/j.expneurol.2013.12.003
Lim YS, Tang BL (2013) The Evi5 family in cellular physiology and pathology. FEBS Lett 587:1703–1710. https://doi.org/10.1016/j.febslet.2013.04.036
Lin R, Taylor BV, Simpson S, Charlesworth J, Ponsonby A-L, Pittas F, Dwyer T, van der Mei IAF (2014) Novel modulating effects of PKC family genes on the relationship between serum vitamin D and relapse in multiple sclerosis. J Neurol Neurosurg Psychiatry 85:399–404. https://doi.org/10.1136/jnnp-2013-305245
Lipson KE, Wong C, Teng Y, Spong S (2012) CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 5:S24. https://doi.org/10.1186/1755-1536-5-S1-S24
Liu Y, Hu H, Wang K, Zhang C, Wang Y, Yao K, Yang P, Han L, Kang C, Zhang W, Jiang T (2014) Multidimensional analysis of gene expression reveals TGFB1I1-induced EMT contributes to malignant progression of astrocytomas. Oncotarget 5:12593–12606. https://doi.org/10.18632/oncotarget.2518
Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, Reynolds R, O’Connor KC, Hafler DA, O’Connor KC, Hafler DA (2011) Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 134:534–541. https://doi.org/10.1093/brain/awq350
Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (1999) A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain 122:2279–2295. https://doi.org/10.1093/brain/122.12.2279
Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, Huitinga I (2018) Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol 135:511–528. https://doi.org/10.1007/s00401-018-1818-y
Luo J, Ho PP, Buckwalter MS, Hsu T, Lee LY, Zhang H, Kim D-K, Kim S-J, Gambhir SS, Steinman L, Wyss-Coray T (2007) Glia-dependent TGF-beta signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J Clin Invest 117:3306–3315. https://doi.org/10.1172/JCI31763
Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, Bien CG, Bauer J, Lassmann H (2018) The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 141:2066–2082. https://doi.org/10.1093/brain/awy151
Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, Aloisi F, Reynolds R (2010) A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 68:477–493. https://doi.org/10.1002/ana.22230
Mahad D, Lassmann H, Turnbull D (2008) Review: mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol 34:577–589. https://doi.org/10.1111/j.1365-2990.2008.00987.x
Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183–193. https://doi.org/10.1016/S1474-4422(14)70256-X
Makioka K, Yamazaki T, Takatama M, Ikeda M, Okamoto K (2014) Immunolocalization of Smurf1 in Hirano bodies. J Neurol Sci 336:24–28. https://doi.org/10.1016/j.jns.2013.09.028
McClintick JN, Xuei X, Tischfield JA, Goate A, Foroud T, Wetherill L, Ehringer MA, Edenberg HJ (2013) Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 47:505–515. https://doi.org/10.1016/j.alcohol.2013.07.002
Nataf S, Barritault M, Pays L (2017) A unique TGFB1-driven genomic program links Astrocytosis, low-grade inflammation and partial demyelination in spinal cord Periplaques from progressive multiple sclerosis patients. Int J Mol Sci 18:2097. https://doi.org/10.3390/ijms18102097
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000) Multiple Sclerosis. N Engl J Med 343:938–952. https://doi.org/10.1056/NEJM200009283431307
Obermeier B, Mentele R, Malotka J, Kellermann J, Kümpfel T, Wekerle H, Lottspeich F, Hohlfeld R, Dornmair K (2008) Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med 14:688–693. https://doi.org/10.1038/nm1714
Pathan M, Keerthikumar S, Ang C-S, Gangoda L, Quek CYJ, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, Bacic A, Hill AF, Stroud DA, Ryan MT, Agbinya JI, Mariadason JM, Burgess AW, Mathivanan S (2015) FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15:2597–2601. https://doi.org/10.1002/pmic.201400515
Preller V, Gerber A, Wrenger S, Togni M, Marguet D, Tadje J, Lendeckel U, Röcken C, Faust J, Neubert K, Schraven B, Martin R, Ansorge S, Brocke S, Reinhold D (2007) TGF-beta1-mediated control of central nervous system inflammation and autoimmunity through the inhibitory receptor CD26. J Immunol 178:4632–4640. https://doi.org/10.4049/jimmunol.178.7.4632
Prineas JW, Kwon EE, Cho E-S, Sharer LR, Barnett MH, Oleszak EL, Hoffman B, Morgan BP (2001) Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol 50:646–657. https://doi.org/10.1002/ana.1255
Ragnedda G, Disanto G, Giovannoni G, Ebers GC, Sotgiu S, Ramagopalan SV (2012) Protein-protein interaction analysis highlights additional loci of interest for multiple sclerosis. PLoS One 7:e46730. https://doi.org/10.1371/journal.pone.0046730
Remington LT, Babcock AA, Zehntner SP, Owens T (2007) Microglial recruitment, activation, and proliferation in response to primary demyelination. Am J Pathol 170:1713–1724. https://doi.org/10.2353/ajpath.2007.060783
Reynolds R, Roncaroli F, Nicholas R, Radotra B, Gveric D, Howell O Djordje @bullet, @bullet G, Howell O(2011) The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol 122:155–170. https://doi.org/10.1007/s00401-011-0840-0
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Schirmer L, Velmeshev D, Holmqvist S, Kaufmann M, Werneburg S, Jung D, Vistnes S, Stockley JH, Young A, Steindel M, Tung B, Goyal N, Bhaduri A, Mayer S, Engler JB, Bayraktar OA, Franklin RJM, Haeussler M, Reynolds R, Schafer DP, Friese MA, Shiow LR, Kriegstein AR, Rowitch DH (2019) Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573:75–82. https://doi.org/10.1038/s41586-019-1404-z
Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, Andreoni L, Trivedi P, Salvetti M, Faggioni A, Aloisi F (2007) Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med PubMed – NCBI. http://www.ncbi.nlm.nih.gov/pubmed/17984305. Accessed 14 Jan 2016
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F (2004) Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14:164–174. https://doi.org/10.1111/j.1750-3639.2004.tb00049.x
Shen P, Fillatreau S (2015) Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol 15:441–451. https://doi.org/10.1038/nri3857
Sironi M, Guerini FR, Agliardi C, Biasin M, Cagliani R, Fumagalli M, Caputo D, Cassinotti A, Ardizzone S, Zanzottera M, Bolognesi E, Riva S, Kanari Y, Miyazawa M, Clerici M (2011) An evolutionary analysis of RAC2 identifies haplotypes associated with human autoimmune diseases. Mol Biol Evol 28:3319–3329. https://doi.org/10.1093/molbev/msr164
Squires KE, Gerber KJ, Pare J-F, Branch MR, Smith Y, Hepler JR (2018) Regulator of G protein signaling 14 (RGS14) is expressed pre- and postsynaptically in neurons of hippocampus, basal ganglia, and amygdala of monkey and human brain. Brain Struct Funct 223:233–253. https://doi.org/10.1007/s00429-017-1487-y
Steinbrecher A, Reinhold D, Quigley L, Gado A, Tresser N, Izikson L, Born I, Faust J, Neubert K, Martin R, Ansorge S, Brocke S (2001) Targeting dipeptidyl peptidase IV (CD26) suppresses autoimmune encephalomyelitis and up-regulates TGF-beta 1 secretion in vivo. J Immunol 166:2041–2048
Stern JNH, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, Huttner AJ, Laman JD, Nagra RM, Nylander A, Pitt D, Ramanan S, Siddiqui BA, Vigneault F, Kleinstein SH, Hafler DA, O’connor KC, Vander HJA, Church G, Donahue WF, Hintzen RQ, Huttner AJ, Laman JD, Nagra RM, Nylander A, Pitt D, Ramanan S, Siddiqui BA, Vigneault F, Kleinstein SH, Hafler DA, O’connor KC, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, Huttner AJ, Laman JD, Nagra RM, Nylander A, Pitt D, Ramanan S, Siddiqui BA, Vigneault F, Kleinstein SH, Hafler DA, O’connor KC (2014) B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 6:248ra107. https://doi.org/10.1126/scitranslmed.3008879
Tajiri N, De La Peña I, Acosta SA, Kaneko Y, Tamir S, Landesman Y, Carlson R, Shacham S, Borlongan CV (2016) A nuclear attack on traumatic brain injury: sequestration of cell death in the nucleus. CNS Neurosci Ther 22:306–315. https://doi.org/10.1111/cns.12501
Torkildsen Ø, Stansberg C, Angelskår SM, Kooi E-J, Geurts JJG, van der Valk P, Myhr K-M, Steen VM, Bø L (2010) Upregulation of immunoglobulin-related genes in cortical sections from multiple sclerosis patients. Brain Pathol 20:720–729. https://doi.org/10.1111/j.1750-3639.2009.00343.x
Tuck E, Cavalli V (2010) Roles of membrane trafficking in nerve repair and regeneration. Commun Integr Biol 3:209–214. https://doi.org/10.4161/cib.3.3.11555
van der Poel M, Ulas T, Mizee MR, Hsiao C-C, Miedema SSM, Adelia SKG, Helder B, Tas SW, Schultze JL, Hamann J, Huitinga I (2019) Transcriptional profiling of human microglia reveals grey–white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun 10:1139. https://doi.org/10.1038/s41467-019-08976-7
Wang J, Vasaikar S, Shi Z, Greer M, Zhang B (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45:W130–W137. https://doi.org/10.1093/nar/gkx356
Wang R, Wang H, Carrera I, Xu S, Lakshmana MK (2015) COPS5 protein overexpression increases amyloid plaque burden, decreases Spinophilin-immunoreactive Puncta, and exacerbates learning and memory deficits in the mouse brain. J Biol Chem 290:9299–9309. https://doi.org/10.1074/jbc.M114.595926
Wu JY, Feng L, Park HT, Havlioglu N, Wen L, Tang H, Bacon KB, Jiang Zh Z, Zhang Xc X, Rao Y (2001) The neuronal repellent slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature 410:948–952. https://doi.org/10.1038/35073616
Wyss-Coray T, Borrow P, Brooker MJ, Mucke L (1997) Astroglial overproduction of TGF-beta 1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol 77:45–50
Yen K, Lee C, Mehta H, Cohen P (2013) The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol 50:R11–R19. https://doi.org/10.1530/JME-12-0203
Yin X, Fang S, Wang M, Wang Q, Fang R, Chen J (2016) EFEMP1 promotes ovarian cancer cell growth, invasion and metastasis via activated the AKT pathway. Oncotarget 7:47938–47953. https://doi.org/10.18632/oncotarget.10296
Zeis T, Howell OW, Reynolds R, Schaeren-Wiemers N (2018) Molecular pathology of multiple sclerosis lesions reveals a heterogeneous expression pattern of genes involved in oligodendrogliogenesis. Exp Neurol 305:76–88. https://doi.org/10.1016/j.expneurol.2018.03.012
Acknowledgements
We are grateful for the technical assistance of Carmen Picon Munoz PhD and Cornelius K. Donat PhD (Division of Brain Science, Imperial College, London, UK).
Funding
Lundbeckfonden R118-A11472 (to ZI), Lundbeckfonden R260–2017-1247 and R296–2018-2502 (to MLE), Scleroseforeningen R458-A31829-B15690 and R487-A33600-B15690 (to ZI), Region of Southern Denmark 14/24200 (to ZI), Jascha Fonden 5589 (to ZI), Direktør Ejnar Jonasson kaldet Johnsen og hustrus mindelegat 5609 (to ZI), Odense University Hospital (OUH) 29A-1501 (to ZI, Sanofi-Genzyme REG-NOBA-COMPL-SD-017 (to ZI), and FIKP 2 theme 20765/3/2018/FEKUTSTRAT (to ZI). JB and TB are grateful for financial support from Braumbach’s VILLUM Young Investigator grant nr. 13154. JB’s work was also funded by H2020 project nr. 777111 (REPOTRIAL).
Author information
Authors and Affiliations
Contributions
MLE contributed to concept and design, obtaining research grants, acquisition, analysis and interpretation of data, drafting of the manuscript, revision of the manuscript, and critical revision of the manuscript for important intellectual content. TF contributed to acquisition and analysis of data, drafting of the manuscript, revision of the manuscript, and statistical analysis. RR contributed to acquisition of data, analysis and interpretation of data, and revision of the manuscript. TK contributed to analysis of data, drafting of the manuscript, revision of the manuscript, and critical revision of the manuscript for important intellectual content. MB contributed to analysis of data, and revision of the manuscript. TAK contributed to concept and design, acquisition, analysis of data, and revision of the manuscript. MT contributed to concept and design, acquisition, analysis of data, and revision of the manuscript. JB contributed to concept and design, obtaining research grants, acquisition, analysis and interpretation of data, drafting of the manuscript, revision of the manuscript, and critical revision of the manuscript for important intellectual content. ZI contributed to concept and design, obtaining research grants, acquisition, analysis and interpretation of data, drafting of the manuscript, revision of the manuscript, and critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
MS and control tissue samples were supplied by the UK Multiple Sclerosis Tissue Bank (UK Multicentre Research Ethics Committee, MREC/02/2/39), funded by the Multiple Sclerosis Society of Great Britain and Northern Ireland (registered charity 207,495).
Consent for publication
Not applicable.
Competing interests
Dr. Illes reports personal fees from Biogen, personal fees from Sanofi-Genzyme, personal fees from Merck, personal fees from Novartis, outside the submitted work.
The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Supplementary Methods
Additional file 2.
The R script for analyzing differentially expressed genes
Additional file 3:
Figure S1. RNAscope : Negative (red and green targeting two bacterial genes)and postive (PPIB /POLR2A POLR2A ) controls
Additional file 4:
Table S1. Clinical and demographic data of the MS patients and non-neurological disease controls
Additional file 5:
Table S2. Number of samples, detected genes and significant genes in the post-mortem human brain samples
Additional file 6:
Table S3. Lesion specific genes
Additional file 7. Table S4.
Pathways of cellular processes in different lesion types based on unique differentially expressed genes.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Elkjaer, M.L., Frisch, T., Reynolds, R. et al. Molecular signature of different lesion types in the brain white matter of patients with progressive multiple sclerosis. acta neuropathol commun 7, 205 (2019). https://doi.org/10.1186/s40478-019-0855-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0855-7