
Abstract
The protein α-synuclein (αsyn) forms pathologic aggregates in a number of neurodegenerative diseases including Lewy body dementia (LBD) and Parkinson’s disease (PD). It is unclear why diseases such as LBD may develop widespread αsyn pathology, while in Alzheimer’s disease with amygdala restricted Lewy bodies (AD/ALB) the αsyn aggregates remain localized. The amygdala contains αsyn aggregates in both LBD and in AD/ALB; to understand why αsyn pathology continues to progress in LBD but not in AD/ALB, tissue from the amygdala and other regions were obtained from 14 cases of LBD, 9 cases of AD/ALB, and 4 controls for immunohistochemical and biochemical characterization. Utilizing a panel of previously characterized αsyn antibodies, numerous unique pathologies differentiating LBD and AD/ALB were revealed; particularly the presence of dense neuropil αsyn aggregates, astrocytic αsyn, and αsyn-containing dystrophic neurites within senile plaques. Within LBD, these unique pathologies were predominantly present within the amygdala. Biochemically, the amygdala in LBD prominently contained specific carboxy-truncated forms of αsyn which are highly prone to aggregate, suggesting that the amygdala may be prone to initiate development of αsyn pathology. Similar to carboxy-truncated αsyn, it was demonstrated herein that the presence of aggregation prone A53T αsyn is sufficient to drive misfolding of wild-type αsyn in human disease. Overall, this study identifies within the amygdala in LBD the presence of unique strain-like variation in αsyn pathology that may be a determinant of disease progression.
Similar content being viewed by others


Introduction
Aggregates comprised of the pre-synaptic neuronal protein, α-synuclein (αsyn), are the major component in Lewy body (LB) inclusions that pathologically define Parkinson’s disease (PD) and Lewy body dementia (LBD) [52, 96]; additionally 40–60% of Alzheimer’s disease (AD) cases display LBs that are most commonly localized to the amygdala [3, 37, 73, 84, 102]. αsyn aggregates are not merely associated with these diseases but in fact can have etiologic roles, whereby aggregation promoting point mutations (A53T in particular) in the SNCA gene encoding αsyn have been discovered to cause familial PD/LBD [15, 20, 83]. It is unclear what factors prompt physiologic αsyn to misfold and form pathologic inclusions, however once formed these aggregates are key to disease progression as they can likely spread between cells and induce further pathology along with resultant cellular toxicity in a prion-like fashion [21, 54, 103, 110]. Prominent in synucleinopathies is the occurrence of post-translational modifications of αsyn which may influence the tendency of the protein to misfold and aggregate; in disease, 90% or more of αsyn becomes phosphorylated at Ser129 and 10–20% may become carboxy (C)-terminally truncated within LB enriched extracts [2, 4, 53, 60, 62]. C-terminal truncation of αsyn in particular may be crucial, as these species aggregate even more readily than disease-causal mutant forms of αsyn [16, 40, 41, 71, 72, 90, 93]. Another important modulator of αsyn pathology in LBD and AD is concurrent AD pathologic changes such as tau neurofibrillary tangles and Aβ plaques which are present at a moderate to severe stage in the majority of LBD cases and worsen clinical outcomes [43, 44, 97, 100]. Tau and Aβ purportedly harbor prion-like properties similarly to αsyn and have in-vitro demonstrated the capacity to cross-seed αsyn aggregation [32, 38, 77, 95] which may be evidenced in human disease by lesions containing both misfolded tau and αsyn within the same cell; these co-localized aggregates are often within the medial temporal lobe (MTL) of LBD patients [33, 46, 49, 88].
The prion-like spread model of αsyn pathology is complicated due to the presence of atypical synucleinopathies that do not conform to typical staging schema of caudal to rostral spread [11, 39, 48, 85, 100]; in particular, AD with amygdala predominant LB pathology (AD/ALB) is especially confounding as extensive αsyn aggregates may be predominantly present within just one brain region and lack evidence of initial pathology elsewhere or continued spread to other regions [37, 100, 102]. Furthermore, different synucleinopathies appear to have separate patterns of regional initiation and progression of pathology; αsyn aggregation in the brain purportedly begins within autonomic medullary neurons in PD temporally succeeded by mesencephalic and telencephalic regions which contrasts with LBD in which olfactory and limbic structures may display extensive pathology with minimal midbrain involvement [10, 48, 49, 52, 100, 112]. In addition to heterogeneity in pathologic progression, αsyn aggregates can appear in a number of different morphologies in the same brain including Lewy neurites (LNs), cortical LBs, classical brainstem LBs with a clearly defined core and halo, and other forms often distinct across anatomic regions [24, 36, 49, 105]. Separate types of Lewy pathology may correlate with symptomatic progression differentially; for example, it has been suggested that small, neuritic aggregates of αsyn present in the cortex predict symptomatic severity superiorly to the presence of cortical LBs alone [14, 59, 89]. Similarly, total burden of Lewy pathology within the SNpc to include smaller neuropil aggregates predicted the degree of striatal dopaminergic deficit whereas counts of only brainstem type LBs did not correlate with severity of dopaminergic loss [57]. The diversity of pathologic presentations of synucleinopathies both between and within disease types has been noted extensively in support of the notion that different “strains” of pathologic αsyn may occur [23, 79, 81].
If the prion-like hypothesis of αsyn aggregation holds true, then therapeutic approaches should focus on brain regions not only in which aggregates initially form, but as well those in which more fulminant “strains” occur which may be evidenced by biochemically and histochemically unique properties of αsyn pathologies [23, 26, 80]. The amygdala is an optimal brain region to study in relation to factors governing the transition of LB pathology from an incidental and localized finding as is the case in AD/ALB [74, 86, 102] and possibly incidental Lewy body disease (iLBD) [25, 30, 64] versus a toxic, seemingly prion-like neurodegenerative disorder in LBD and PD. Furthermore, the amygdala is uniquely vulnerable in a number of neurodegenerative diseases as it is afflicted early on in multiple disorders and prone to develop concurrent pathologies [73]. This study aims to define the biochemical and immunohistochemical differences in the intrinsic nature of Lewy related pathology (LRP) within the amygdala in LBD compared with AD/ALB to reveal the molecular alterations in pathologic αsyn aggregates associated with pathology that remains localized in AD/ALB but not LBD. Additionally, the LBD amygdala was compared with other brain regions in LBD to probe for differences in pathology that may underlie the early involvement of the amygdala compared to the midbrain and higher cortical regions.
Materials and methods
Autopsy case material
Human brain tissue was obtained through the University of Florida Neuromedicine Human Brain Tissue Bank in accordance with institutional review board approval. Post-mortem pathological staging and diagnoses were made according to respective neuropathological criteria for AD and LBD [42, 70]. Apolipoprotein E (APOE) genotypes were determined by genotyping SNPs rs7412 and rs429358 using TaqMan SNP genotyping assay (ThermoFisher). For immunohistochemical studies, sections from the cingulate cortex, amygdala, and midbrain of 9 formalin fixed cases of diffuse LBD and 9 formalin fixed cases of AD/ALB were used. Some studies included midbrain and hippocampus sections from a previously described patient with the SNCA A53T mutation [27]. For biochemical fractionation and comparison, frozen brain tissues from the medial temporal lobe, amygdala, or cingulate cortex grey matter from 8 cases of diffuse LBD, 2 cases of AD/ALB, and 4 controls were used. In total, the case selection includes 14 sporadic LBD, 1 familial PD/LBD, 9 AD/ALB, and 4 controls without synucleinopathy (Table 1).
Transgenic mouse tissue
Hemizygous M83 transgenic mice overexpress human αsyn harboring the A53T mutation and when intra-muscularly seeded with pre-formed αsyn fibrils they accumulate pathologic inclusions that spread throughout most of the neuro-axis [94]. Hemizygous M20 mice overexpress wild type (WT) human αsyn and when intra-cerebrally injected with preformed αsyn fibrils develop extensive αsyn pathology [91]. Paraffin embedded sections from the brains of the M20 and M83 mouse lines, induced to develop pathology, were obtained in order to confirm the specificity of antibody 3H11.
Antibodies
Anti-phosphorylated Ser129 (pSer129) αsyn rabbit monoclonal antibody EP1536Y was obtained from Abcam (Cambridge, MA). Antibodies 9C10 and 94-3A10 are mouse monoclonal antibodies specific for N-terminal (2–21) or C-terminal (130–140) residues of αsyn respectively [22]. Antibody 3H11 is a mouse monoclonal antibody raised against central residues (43–63) of human αsyn that does not react with αsyn harboring the A53T mutation [23, 92]. Antibody 5G4, a mouse monoclonal antibody raised against central residues (44–57) with high affinity for oligomeric αsyn, was obtained from MilliporeSigma (Burlington, MA, USA) [56, 58]. Antibody 7F2 is a mouse monoclonal antibody generated against the AT8 epitope specific for phosphorylated tau particularly at pT205 [98]. Antibody 33.1.1 is a mouse monoclonal antibody raised against Aβ residues 1–16 that detects Aβ plaques [69]. Other antibodies utilized include polyclonal rabbit anti-glial fibrillary acidic protein (GFAP) from Dako (Santa Clara, CA, USA) and polyclonal rabbit anti-vimentin (C-20) from Santa Cruz (Dallas, TX, USA).
Expression and purification of recombinant αsyn proteins
Recombinant WT or A53T human αsyn were expressed from the pRK172 plasmid containing the cDNA for the SNCA gene as described previously [34, 107]. Constructs were expressed in E.coli BL21 (DE3) and purified as previously described utilizing size exclusion and Mono Q anion exchange chromatography [34]. Recombinant proteins were diluted in pH 7.4 sterile phosphate buffered saline (PBS)(Invitrogen), and concentrations were determined using the bicinchoninic acid assay (BCA) from Pierce (Waltham, MA, USA) with bovine serum albumin (BSA) as the standard.
Biochemical fractionation
Unfixed frozen human tissue was thawed and samples were retrieved from grey matter of the hippocampus and surrounding MTL structures; when identified, the amygdala was separately processed. For all cases, ~ 250 mg of MTL or amygdala was homogenized within 3 mL/g tissue high-salt (HS) buffer (50 mM Tris–HCl, pH 7.5, 0.75 M NaCl, 2 mM EDTA, 50 mM NaF with a cocktail of protease inhibitors) and sedimented at 100,000 x g for 30 min at 4 °C. The HS supernatant was collected (HS fraction) and insoluble material was re-suspended in 2 mL/g tissue HS buffer/1% Triton X-100. Re-suspended material was again sedimented at 100,000 x g for 30 min at 4 °C and the supernatant was collected (HS/T fraction). Pellets were homogenized in 3 mL/g tissue HS buffer/1 M sucrose and sedimented at 100,000 x g for 30 min at 4 °C in order to remove myelin which was discarded in the supernatant. Pellets were subsequently re-suspended in 2 mL/g tissue radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) and sedimented at 100,000 x g for 30 min at 4 °C; supernatants were collected as the RIPA fractions. Lastly, remaining insoluble material was homogenized in 1 mL/g tissue SDS/Urea buffer (4 M urea, 2% SDS, 25 mM Tris–HCl pH 7.6), probe sonicated, and then stored as the SDS/Urea fraction. The concentration of each fraction was determined using the BCA assay with BSA as the standard. SDS containing sample buffer was added to sequential fractions and all samples were further boiled for 10 min except for the SDS/Urea fraction. Cingulate grey matter was previously obtained and fractionated similarly [23]. Fractions were stored at − 80 °C until western blot analysis.
Immunohistochemistry
Immunostaining of the sections was performed using established methods [26]. Paraffinized sections were rehydrated and subsequent antigen retrieval was performed in a steam bath for 60 min in a solution of modified citrate buffer (Target Retrieval Solution Citrate pH 6; Agilent, Santa Clara, CA). If indicated, additional antigen retrieval was performed by exposing sections to 70% formic acid for 20 min at room temperature. Endogenous peroxidase was quenched by incubation of sections in 1.5% hydrogen peroxide/0.005% Triton-X-100/PBS solution for 20 min. Non-specific antibody binding was minimized with a 2% fetal bovine serum (FBS)/0.1 M Tris, pH 7.6 block solution; primary antibodies were diluted in block solution and applied to tissue sections at 4 °C overnight. A mixture of biotinylated secondary antibody (Vector Laboratories; Burlingame, CA) and Impress polymer secondary antibody (Vector Laboratories; Burlingame, CA) were similarly diluted in block solution and applied to sections for 1 h at room temperature. An avidin-biotin complex (ABC) system (Vectastain ABC Elite kit; Vector Laboratories, Burlingame, CA) was used to enhance detection of the immunocomplexes, which were visualized using the chromogen 3,3′-diaminobenzidine (DAB kit; KPL, Gaithersburg, MD). Tissue sections were counterstained with hematoxylin. Slides were digitally scanned using an Aperio ScanScope CS instrument (40× magnification; Aperio Technologies Inc., Vista, CA), and images of representative areas of pathology were captured using the ImageScope software (40× magnification; Aperio Technologies Inc.). Semi quantitative assessment of LRP was performed by two independent observers.
Immunofluorescence
Deparaffinization and antigen retrieval procedures were identical to those used for immunohistochemistry. Sections were blocked using a solution of 5% milk/0.1 M Tris (pH 7.6) to prevent non-specific staining. Thereafter, sections were incubated overnight (4 °C) using combinations of primary antibodies diluted in 5% FBS/0.1 M Tris (pH 7.6) followed with subsequent incubation for 1 h at room temperature using secondary antibodies (diluted in 5% FBS/0.1 M Tris, pH 7.6) conjugated to Alexa 647, 594, or Alexa 488 (Invitrogen). Nonspecific fluorescence was quenched using 0.3% Sudan Black/70% ethanol. Sections were stained with 5 μg/mL 4′,6-diamindino-2-phenylindole. Sections were cover-slipped with Fluoromount-G (SouthernBiotech) and visualized using either an Olympus BX51 microscope mounted with a DP71 Olympus digital camera to capture images at 20x magnification or an Olympus IX81-DSU spinning disk confocal microscope equipped with a cooled FluoView II charge coupled device (CCD) digital monochrome camera.
Western blot analysis
For biochemical characterization of fractionated human brain tissue, 20 μg of lysate from either the HS or SDS/urea fractions were loaded onto 15% polyacrylamide gels and resolved by SDS-PAGE, followed by electrophoretic transfer onto 0.2 μm pore size nitrocellulose membranes (Bio-Rad, Hercules, CA), in carbonate transfer buffer (10 mM NaHCO3, 3 mM Na2CO3, pH 9.9) [29]. For analysis of recombinant protein, 200 ng of WT or A53T human αsyn was analyzed. Membranes were blocked in 5% dry milk/Tris buffered saline (TBS) and incubated overnight at 4 °C with primary antibody diluted in block solution. After washing in TBS, membranes were incubated with goat anti-mouse secondary antibody conjugated to horseradish peroxidase (Jackson Immuno Research Labs, Westgrove, PA) diluted in 5% dry milk/TBS for 1 h at room temperature; immunocomplexes were detected using Western Lightning-Plus ECL reagents (PerkinElmer, Waltham, MA) followed by chemiluminescence imaging (PXi, Syngene, Frederick, MD).
Quantitative analysis of immunohistochemical staining
Quantitation of staining positivity for total LRP was performed on cingulate and amygdala sections from 9 LBD cases. A modified form of a quantitation workflow was used, in which for each section three squares of a uniform size (0.25 mm2) were placed within the regions of densest pathology that were at least two box widths apart [75]. Adjacent sections were used, and so boxes were placed at nearly identical locations for sections stained with the different antibodies and retrieval conditions. The positivity within each box for each section was analyzed using the positive pixel count algorithm (Aperio) with intensity threshold values optimized for each antibody to maximize pathology detection and minimize background (Fig. 3). For a given antibody, the same threshold values were used for all amygdala and cingulate cortex sections as same day staining minimized variability in background. For each LBD case, positivity for each antibody with or without formic acid (FA) and for the cingulate versus amygdala were determined. Comparisons were made for positivity with different antibodies and between the cingulate cortex and amygdala using GraphPad Prism software with one-way analysis of variance (ANOVA) and post-hoc analysis using the Sidak multiple comparison test to individually compare each region and antibody.
Results
Pathological A53T αsyn recruits WT αsyn within pathologic inclusions in familial PD/LBD
Unique conformationally or post-translationally modified strain-like forms of pathologic αsyn that aggregate readily, and further seed inclusion pathology may differentiate rapidly progressive forms of synucleinopathy from more innocuous conditions such as AD/ALB and iLBD. In order to test the theory that an aggregation prone form of αsyn could induce endogenous physiologic αsyn to form pathologic inclusions, sections of hippocampus and midbrain were obtained from a Contursi kindred patient in which familial PD/LBD developed due to a heterozygous A53T mutation in the SNCA gene [27]. Antibody 3H11 which does not detect αsyn harboring the A53T mutation [92] was used to label pathologic aggregates comprised of physiologic WT αsyn and compared with LRP seen by antibody 9C10 which binds both WT and A53T αsyn (Fig. 1). Western blot analysis of recombinant WT and A53T αsyn confirmed the specificity of each antibody (Fig. 1a). Additionally, antibody 3H11 was further tested on transgenic mice containing pathologic aggregates comprised of A53T human αsyn (seeded line M83 [94]) or WT human αsyn (seeded line M20 [91]). As 3H11 does not detect mouse αsyn (that contains the A53T substitution) or A53T human αsyn, no pathology was revealed in the M83 mice using 3H11 whereas extensive LRP was labeled in the M20 mice (Fig. 1b). In both the hippocampus and midbrain of the Contursi kindred patient, 9C10 and 3H11 detected similar amounts of LRP indicating that WT αsyn was induced to misfold due to the presence of aggregates initially formed by A53T αsyn and recruited within pathological inclusions (Fig. 1c). These results demonstrate that WT αsyn can be induced to aggregate due to the presence of a more aggregation prone “strain” of αsyn, presumably in a prion-like fashion.

LRP comprised of WT αsyn detected by selective antibody 3H11. a Western blot of 200 ng recombinant WT or A53T human αsyn protein probed with antibody 9C10 (residues 2–21) or antibody 3H11 (residues 43–63); A53T αsyn does not react with antibody 3H11. b Immunohistochemical staining with antibody 3H11 or 9C10 in αsyn transgenic mice. Using antibody 3H11, αsyn aggregates are extensively detected within line M20 mice overexpressing WT human αsyn but not in line M83 mice overexpressing A53T human αsyn demonstrating the histochemical specificity of this antibody. Labeling with antibody 9C10 depicts αsyn pathology in both types of αsyn transgenic mice. Scale bar 50 μm. c Immunohistochemical staining of tissue from the midbrain and hippocampus of a familial case of PD/LBD due to a heterozygous A53T mutation in SNCA. Antibody 9C10 detects both WT and A53T αsyn and detects abundant pathology in both regions; Antibody 3H11 only detects WT αsyn but also labels many pathologic inclusions, indicating that WT αsyn is recruited to aggregate by the presence of the A53T αsyn mutation. Arrowheads indicate LRP. All the sections depicted were treated with FA. Scale bar 50 μm
The LBD and AD/ALB amygdala differ largely in burden of neuropil pathology
In order to compare the immunohistochemical patterns of LRP found within the amygdalas of LBD and AD/ALB cases, 9 pathologically confirmed LBD cases were obtained along with 9 cases of AD/ALB where concurrent LRP is predominantly present within the amygdala upon inspection of 14 different regions ranging from the medulla to the frontal cortex (Tables 1 and 2). Sections from the cingulate cortex, amygdala, and substantia nigra pars compacta (SNpc) were obtained from each case as these 3 regions are heavily affected in LBD and provide confirmation that AD/ALB LRP is mainly localized to the amygdala. The overall burden of LRP within each region of each case was first studied using an antibody specific for pSer129 αsyn (EP1536Y) which is a marker of pathologic inclusion formation [2] (Fig. 2). The presence of pSer129 αsyn-positive inclusions was semi-quantitatively graded at 10x magnification on a four-tiered scale, with “-” representing non-reactivity, “+” mild, “++” moderate, and “+++” representing the strongest level of reactivity (Table 2). Immediately evident is the paucity of LRP in the cingulate cortex and SNpc of AD/ALB cases compared to parallel regions in LBD cases. In AD/ALB, 1/8 cases had rare thread like LNs in the SNpc with no LBs, and no cases of AD/ALB demonstrated any LRP in the cingulate cortex. Comparatively, all LBD cases had extensive LRP in the cingulate cortex and SNpc including LBs and pSer129 positive neurites; semi-quantitative grading for each region is shown (Fig. 2, Table 2). Furthermore, dopaminergic neuronal cell loss is not evident in the examined AD/ALB cases as abundant neuromelanin-laden neurons are present throughout the SNpc (10+ granulated neurons per 10x visual field) whereas overt reduction in dopaminergic neuronal cells was apparent in the LBD cases (1–5 granulated neurons per 10x visual field) (Fig. 2).

Comparison of LRP between LBD and AD/ALB. a Representative sections of the SNpc from 4 AD/ALB and 4 LBD cases stained with pSer129 αsyn antibody EP1536Y. Abundant LNs and LBs (red arrows) are seen within pigmented neurons in all LBD cases, however AD/ALB cases display only rare examples of thread like neurites. Insets display LBs in pigmented neurons. b Representative sections of the amygdala from AD/ALB versus LBD cases labeled with antibody EP1536Y. Cortical-type LBs are apparent in dense patches within amygdalas of either disease type, however abundant thread-like neurites are more apparent in LBD cases compared with AD/ALB. Insets highlight cortical LBs or neurites. c Representative sections of the cingulate cortex from AD/ALB compared with LBD using antibody EP1536Y. LRP is entirely absent in all examined AD/ALB cases, whereas LBD cases display extensive LBs in deeper cortical layers and dot like inclusions in more superficial layers. Case numbers are indicated in lower right corner. Scale bar 100 μm
Within the amygdala, varied LRP was present in the examined AD/ALB cases with 5/8 cases displaying 5–10 cortical LBs per visual field (10x magnification) in regions of densest pathology while 3/8 cases had less inclusions (Table 2). Comparatively, 5/8 LBD amygdala cases had 5–10 cortical LBs in a similar visual field of dense pathology and 3/8 cases had 10–20+ LBs. Although regions of the AD/ALB amygdalas displayed an LB load comparable to that of LBD amygdalas in the regions of densest pathology, abundant neuropil threads positive for pSer129 αsyn were ubiquitous within LBD cases but not to the same extent within AD/ALB cases (Fig. 2). The extensive neuritic pathology within amygdalas in LBD compared with amygdalas in AD/ALB may represent an important difference between these conditions, and aligns with the theory that large LBs serve to sequester misfolded αsyn while smaller neuritic aggregates contain the more active species in disease pathogenesis. Small synaptic aggregates of αsyn have been suggested as containing more toxic species compared with αsyn within compact LBs [14, 59, 89]. The nature of this amygdala neuropil pathology in LBD was further investigated with a panel of monoclonal antibodies raised against αsyn.
The LBD amygdala harbors extensive and unique immunoreactive αsyn pathologies compared with other diseased brain regions and AD/ALB
Sections from the cingulate cortex, midbrain, and amygdala from 9 LBD and 9 AD/ALB cases were utilized for comparison of the immunoreactive profiles of LRP amongst the differing brain regions using a panel of antibodies and different antigen retrieval conditions. This approach has been previously utilized to differentiate between distinct forms of LRP [22, 23, 26, 68, 80]. An N-terminal antibody 9C10 [22], a C-terminal antibody 94-3A10 [22], and a central αsyn antibody 3H11 [23] were chosen for morphologic assessment and quantitative analysis as distinct forms of pathologic αsyn may have differential exposure of various epitopes [8, 80] and/or post-translational modifications such as truncation, ubiquination, or phosphorylation that can result in immunoreactivity differences [2, 76, 93]. Antibody 5G4 that is preferential for oligomeric αsyn [56, 58] and antibody EP1536Y for pser129 αsyn were also utilized. Additionally, the reactivity of select antibodies was compared with and without FA retrieval as unique species of pathologic αsyn are known to be preferentially detected only in the presence of additional antigen retrieval techniques such as formic acid exposure and proteinase K digestion [5, 8, 23, 92, 105]. Staining positivity for each antibody with and without FA was quantitated with a modified Aperio based method [75] where threshold values were determined for each antibody to maximize pathology detection and minimize background (Fig. 3).

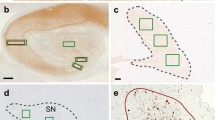
Significant brain regional difference in formic acid antigen retrieval of LRP. a Low magnification sections from the amygdala and cingulate cortex of an LBD case labeled with antibody 9C10 (residues 2–21 αsyn) without or with FA retrieval as indicated. FA retrieval had a major impact in increasing staining of αsyn aggregates in the amygdala compared with the cingulate cortex. Scale bar 1 mm. b A region of dense Lewy pathology within the amygdala was used to optimize threshold values for positive pixel count analysis. All antibodies were tested to ensure similar detection of pathology with minimal background as shown in bottom panel where red colored pixels are positive. Scale bar 50 μm
Quantification and morphologic assessment of staining with each αsyn antibody and no FA retrieval was performed on cingulate cortex and amygdala sections for each LBD case (Figs. 4, 5 and 6, Additional file 1: Figure S1). For all antibodies used, there were no significant differences in positivity between the cingulate cortex and amygdala without FA; averaging the 3 antibodies used the positivity in LBD cingulate cortices was 3.2 ± 3.3% and in LBD amygdalas it was 5.7 ± 4.6%. Semi-quantitative grading using pSer129 antibody EP1536Y without FA retrieval also suggested no difference in the αsyn pathology burden between the cingulate cortex and amygdala in examined LBD cases (Table 2). When FA was used for antigen retrieval, detection of neuropil pathology in the amygdala was greatly increased with all antibodies which is reflected in the highly significant increase in positivity of staining for 9C10, 94-3A10, and 3H11 (Figs. 3, 4 and 6). On average, the largest increase in detection with FA was using the central αsyn antibody 3H11 which went from a positivity in LBD amygdalas of 3.6 ± 3.1% to 20.4 ± 11.4%, a roughly 6-fold increase in detected pathology. Including all antibodies, positivity in LBD amygdalas was 19.5 ± 10.5% with FA compared to 5.7 ± 4.6% without. The LBD cingulate cortex sections did not display this substantial increase in detected pathology when FA was included, and no significant difference in quantification of positivity was present with tested antibodies with or without FA except for antibody 94-3A10 which had a more modest increase in detected neuropil pathology (Figs. 3, 4 and 5). Including all antibodies, positivity in LBD cingulate cortices was 7.3 ± 7.0% with FA compared to 3.2 ± 3.3% without. This pattern of immunoreactivity suggests that this additional αsyn pathology within the LBD amygdalas may be distinct from aggregates elsewhere in the brain, as may be the case with glial αsyn inclusions detected with FA in multiple systems atrophy [23].

Quantitation of LRP in LBD brain regions across a panel of antibodies. a Three areas of dense pathology within the cingulate (C) or amygdala (A) of LBD (N = 9) cases stained with 3 different antibodies without or with (FA) retrieval were subject to positive pixel analysis; average positivity and error bars (std) are displayed for each region and antibody without or with FA. All cases were averaged for each region and antibody for comparison. Without FA retrieval, all antibodies detect similar amounts of LRP within the amygdala versus the cingulate cortex; with FA retrieval a large increase in labeled amygdala pathology is evident with all antibodies whereas a lesser increase in pathology is seen in the cingulate cortex and only with antibody 94-3A10. With FA retrieval, the average amygdala pathology burden is significantly greater than the cingulate cortex for all antibodies. b A statistical summary of positivity comparisons for data presented in A. The average positivity for each antibody between and within regions along with presence or absence of FA retrieval were tested for significant differences using one-way ANOVA and the Sidak post-hoc multiple comparisons test. Mean difference is the absolute difference in positivity values

LRP in LBD SNpc and cingulate cortex is similarly detected across a panel of αsyn antibodies. a Representative sections of the SNpc from an LBD case labeled with 5 different αsyn antibodies as indicted in the top, left corners without or with FA as indicated. Insets display LBs in pigmented neurons. Within the SNpc, abundance of pathology is similar regardless of antibody used or antigen retrieval albeit with minor differences. Scale bar 50 μm. b Representative staining of the cingulate cortex from an LBD case labeled with 5 different αsyn antibodies without or with FA as indicated. Insets display cortical LBs. Within the cingulate, pathology is similar for most antibody and antigen retrieval conditions. αsyn-positive neurites are slightly more apparent with FA retrieval or C-terminal antibodies, particularly 94-3A10 with FA which is reflected in the quantitative positivity analysis. Scale bar 50 μm. c Sections from the cingulate of an AD/ALB case with no LRP were stained with 4 αsyn antibodies without or with FA, as indicated. Little to no positive staining was detected in the AD/ALB cingulate cortex with any of these antibodies. Scale bar 50 μm

Comparison of LRP in the amygdala of LBD versus AD/ALB across a panel of αsyn antibodies. a Representative sections of dense amygdala pathology from a LBD case labeled with 5 different αsyn antibodies without or with FA retrieval, as indicated. Insets display cortical LBs and αsyn-positive neurites. In the LBD amygdala, extensive neuritic pathology is detected for multiple antibodies when FA retrieval was used. The robust increase in apparent Lewy pathology with FA treatment is mainly seen in neuritic and possible glial processes; the amount of cortical LBs per visual field remain the same regardless of antibody or FA treatment. This immunohistochemical staining profile contrasts with the only modest increase in detected pathology seen in the LBD SNpc and cingulate cortex. Scale bar 50 μm. b Stained sections of amygdala pathology from an AD/ALB case labeled with 5 different αsyn antibodies without or with FA retrieval, as indicated. Insets display cortical LBs and αsyn-positive neurites. In the AD/ALB amygdala, neuritic pathology is modestly enhanced for multiple antibodies when FA retrieval is used; however, the density of neuropil αsyn staining pathology in LBD is not apparent in AD/ALB. Scale bar 50 μm
Panels of immunohistochemical sections are shown to compare the distinct morphologies and scale of additional LRP detected within the LBD amygdala using FA treatment compared to without (Figs. 3 and 6). Within multiple LBD amygdalas, a dramatic increase in detected αsyn pathology is appreciated even at low magnification as thousands of neuropil aggregates (per 2x visual field) become apparent with FA compared to without; this increase in not seen in the cingulate cortex within the same case (Fig. 3). Within examined LBD amygdalas at high magnification, without FA all antibodies demonstrate cortical LBs and αsyn-positive neuropil threads similarly to the cingulate cortex (Figs. 5 and 6). With FA retrieval for all antibodies, the number of detected cortical LBs (per 10x visual field) remains largely unchanged in LBD amygdalas; the major increase in stained αsyn pathology is due to abundant αsyn-positive neuropil threads including processes with astrocytic morphologies, and dystrophic neurites within senile plaques (Figs. 6, 7 and 8). For the LBD cingulate cortex sections, at high magnification the addition of FA treatment does not reveal as abundant unique pathologies as is the case with the amygdala (Fig. 5); only a minor increase in detection of αsyn-positive neuropil threads is seen (particularly with C-terminal antibody 94-3A10) and the astrocytic inclusions and neuritic plaque findings are not apparent. In the SNpc, brainstem LBs and engorged LNs were present in all LBD cases detected with all antibodies (Fig. 5). Qualitatively, no additional burden or type of αsyn pathology was detected in the SNpc of LBD cases with any antibodies when FA was used versus without; similarly to the cingulate cortex, only a slight enhancement of neuropil threadlike pathology may be seen particularly with antibody 94-3A10 (Fig. 5). These findings reinforce that abundant and unique forms of pathologic αsyn are present within the amygdala and none of the other examined brain regions in LBD.

αsyn astrocytic pathology is common in LBD amygdala. a Amygdala sections from 4 different LBD cases (1, 2, 7 and 8) were labeled with central αsyn antibodies either 3H11 or 5G4 along with FA retrieval. Typically, more than 5 cells with astrocytic morphology (depicted with arrowheads) stained with these antibodies was readily observed per field; insets show examples for each case and each antibody. Although astrocytic αsyn inclusions may be present in other brain regions, they are only this densely abundant within the MTL, particularly the amygdala. Cases are indicated in lower right corner. Scale bar 50 μm. b Double labeling immunofluorescent analysis of LBD amygdala sections using central αsyn antibody 3H11 in conjunction with astrocytic markers GFAP or vimentin. Astrocytic αsyn often appears as vesicular granules outlining 2–3 astrocytic processes as opposed to the more uniform αsyn staining seen within nearby cortical LBs, suggesting a differing subcellular co-localization or form of aggregates. Scale bar 30 μm

αsyn aggregates within the LBD amygdala often co-localizes with Alzheimer’s disease-type inclusion pathology. a Amygdala sections from 4 different LBD cases (2, 4, 7 and 9) stained with central αsyn antibody 3H11 along with FA retrieval reveal abundant αsyn dystrophic neurites within Aβ senile plaques. These αsyn-positive dystrophic neurites within Aβ plaques were common within the LBD amygdalas but rare in the AD/ALB amygdalas examined. Cases are indicated in lower right corner. Scale bar 50 μm. b Double labeled immunofluorescence microscopy of LBD amygdala sections from 2 different LBD cases using rabbit monoclonal anti-pSer129 αsyn antibody EP1536Y and mouse monoclonal anti-pThr205 tau antibody 7F2 or mouse monoclonal anti-Aβ antibody 33.1.1. Co-localization of tau and αsyn aggregates was common in the LBD amygdala both in neuronal cell bodies and processes. Scale bar 50 μm
As the previously examined AD/ALB cases upon semi-quantitative grading with pSer129 αsyn antibody EP1536Y displayed less baseline neuropil pathology compared with LBD cases in the amygdala (Table 2), FA antigen retrieval was performed on these sections as well. Qualitatively, with FA retrieval there was no dramatic enhancement of neuropil pathology in the AD/ALB amygdalas compared with the LBD amygdalas (Fig. 6). Similarly to LBD, with all antibodies tested the number of cortical LBs per 10x field remained constant with or without FA; upon inspection a slight qualitative increase in neuropil threads may be appreciated in the AD/ALB amygdalas however it is not the multifold quantifiable increase as is seen in LBD. Additionally, αsyn astrocytic inclusions and αsyn-positive dystrophic neurites within senile plaque findings in AD/ALB are fewer compared to LBD. The midbrain and cingulate cortex sections from the AD/ALB cases still had no apparent αsyn pathology using the tested panel of antibodies (9C10, 3H11, 94-3A10, 5G4, EP1536Y) with or without FA. These results suggest that the extensive neuropil pathology present within the LBD amygdala is unique not only regionally within LBD, but also distinguishes between LBD and AD/ALB.
Astrocytic αsyn is distinctively common within the LBD amygdala
The presence of αsyn-positive astrocytes in synucleinopathies has been previously reported, and their presence is thought to be particularly common within MTL structures [92]. Additionally, these astrocytes are reported to contain αsyn predominantly reactive to central αsyn antibodies in conjunction with FA retrieval and not N or C-terminal antibodies [92]. Herein, the presence of these αsyn-positive astrocytes was compared between brain regions within LBD and for LBD versus AD/ALB to determine whether the astrocytic burden of αsyn contributes to the unique enhancement of pathology within the LBD amygdala upon FA antigen retrieval. Astrocytic αsyn pathology identified based on morphology was detected with central αsyn antibody 3H11 following FA antigen retrieval (Fig. 7). Findings were confirmed using a separate central αsyn antibody previously reported to detect αsyn-positive astrocytes in disease, 5G4 [56, 58]. For each LBD and AD/ALB case, astrocytic αsyn pathology was semi-quantitatively graded at 10x magnification on a four-tiered scale, with “-” representing non-reactivity, “+” mild, “++” moderate, and “+++” representing the strongest level of reactivity (Table 3, Fig. 7).
Within the LBD amygdala sections stained with 3H11, in regions of dense pathology there were frequently 5+ cells (per 10x visual field) with an astrocytic morphology positive for αsyn staining (Fig. 7). Overall, 3/9 LBD cases had frequent astrocytic αsyn pathology, 5/9 LBD cases had intermediate, and 1/9 had rare astrocytic αsyn pathology (Table 3). Examples of αsyn-positive inclusions consistent with astrocytic morphology are shown for 4 different LBD cases and stained with 2 central αsyn antibodies (Fig. 7). Consistent with previous findings, αsyn-positive astrocytes were only readily apparent when the combination of a central αsyn antibody and FA retrieval were used [92]; N and C-terminal antibodies 9C10 and 94-3A10 did not obviously label inclusions with an astrocytic morphology in the LBD cases used. Although astrocytic αsyn inclusions have been noted in other studies to be rarely present in the SNpc and other brain regions [92], upon inspection they were not nearly as apparent within these regions compared to the amygdala. Within AD/ALB amygdala sections, 3/8 cases displayed rare examples of αsyn inclusions consistent with astrocytic morphology when using a central αsyn antibody with FA retrieval (Table 3, Fig. 6). These results suggest that astrocytic αsyn pathology is distinctively common within the amygdala in LBD compared with other brain regions and diseases, and additionally the form of αsyn present within these cells may be further unique in the apparent reactivity only with central αsyn antibodies possibly due to N and C-terminal truncation or other structural modifications to the protein.
Using double labeling immunofluorescence for αsyn with the central αsyn 3H11 antibody alongside two different astrocytic markers, GFAP or vimentin, the nature of αsyn within astrocytes in LBD was further investigated (Fig. 7). Within the LBD amygdala, frequent co-localization of αsyn was seen within astrocytic processes in two separate cases. In particular, αsyn labeling was “granular” in astrocytes compared with the more evenly labeled nearby neurites and cortical LBs (Fig. 7). This granular pattern of staining suggests localization of αsyn aggregates to vesicular structures such as lysosomes. These results further suggest that unique αsyn aggregates are present within astrocytes, and these are largely localized to the amygdala in LBD compared with other brain regions and diseases with αsyn pathology.
The amygdala in LBD contains frequent αsyn aggregates co-localized with other AD- associated amyloidogenic proteins
The amygdala has early and heavy involvement in a number of neurodegenerative diseases including AD with pathologic tau neurofibrillary tangles and Aβ senile plaques [73]. MTL regions such as the hippocampus and particularly the amygdala have previously been noted to develop co-pathologies that are not common in other brain regions; for example, tau tangles and cortical LBs have been labeled within the same neuron in these areas [27, 46, 88, 113]. The presence of these co-pathologies in the LBD amygdala was studied herein and qualitatively compared to other LBD brain regions and to AD/ALB. Indeed, within the LBD amygdala there are abundant examples of αsyn immunoreactive dystrophic neurites within senile plaques that were present in all LBD cases examined; examples from four cases are shown (Fig. 8). This finding was confirmed by double labeling analysis using an Aβ antibody 33.1.1 and pSer129 αsyn antibody (Fig. 8). These αsyn-positive senile plaques in LBD are qualitatively far more common within the amygdala compared to the cingulate cortex or SNpc; 2–3 αsyn-positive senile plaques are apparent per 10x visual field within regions of dense pathology in the amygdala but are rarely, if ever seen in the cingulate cortex and the SNpc. Senile plaques positive for αsyn were present more sparsely within AD/ALB. Additionally, as with most other αsyn pathologies in the LBD amygdala these αsyn-positive dystrophic neurites were best appreciated following FA antigen retrieval. When double labeling for both hyper-phosphorylated tau (pThr205) and pSer129 αsyn, aggregates of αsyn and tau were commonly co-localized (Fig. 8). Although this tau and αsyn co-localization has been described for neurofibrillary tangles and cortical LBs, many dystrophic neurites within neuritic plaques contained both αsyn and tau upon inspection of double labeled LBD amygdala sections (Fig. 8). Co-localization of pathologic tau and αsyn was mainly found within the LBD amygdala and not commonly in the LBD cingulate and SNpc. Co-pathologies with tau and Aβ may contribute to unique αsyn alterations present within the amygdala in LBD.
Detergent insoluble αsyn within the medial temporal lobe in LBD is extensively modified
In order to better understand the unique histological immunoreactivity of αsyn within the amygdala in LBD, sequential biochemical fractionation of frozen brain tissue followed by western blot analysis with N-terminal (9C10), C-terminal (94-3A10), and central αsyn (3H11 and 5G4) antibodies was performed. The MTL was retrieved from multiple cases including 5 LBD, 2 AD/ALB, and 2 controls without evidence of synucleinopathy (Table 1). During tissue retrieval, for one case of LBD the amygdala was able to be confidently identified within the MTL and further isolated. For comparison, tissue from the cingulate cortex was also retrieved from 3 cases of LBD and 2 controls without evidence of synucleinopathy (Table 1). Following biochemical fractionation, the HS fractions harboring soluble αsyn and the SDS/urea fractions containing the most insoluble, aggregated αsyn species were compared via western blotting analysis for all aforementioned cases and brain regions (Figs. 9 and 10).

Immunoblotting comparison of αsyn species in LBD and AD/ALB amygdala. High salt (HS) and SDS/urea fractions were obtained from the MTL of 5 LBD cases (lanes 1–5), 2 cases of AD/ALB (lanes 6–7), 2 non-synucleinopathy controls (lanes 8–9), and for one LBD case the amygdala was specifically isolated (lane 10). 20 μg of lysate for each case and fraction were subject to western blot analysis using a panel of 4 antibodies which are indicated. In the HS fraction, all antibodies predominantly revealed full-length (FL) αsyn in all cases. In the SDS/Urea fractions, monomeric FL αsyn is present in high amounts for 4/5 LBD cases (lanes 2, 3, 4, 5, and 10); 1 LBD and 1 AD/ALB case (lanes 1 and 7, respectively) have an intermediate amount of FL αsyn in this fraction and 1 AD/ALB case along with 2 controls (lanes 6, 8, and 9) have very little FL αsyn in this fraction. For the LBD cases, 2 prominent truncation bands are present for all antibodies except for C-terminal antibody 94-3A10 suggesting these are carboxy-truncated forms of αsyn (T1 and T2) in the SDS/urea fractions from the MTL in LBD but not controls or AD/ALB; the truncation bands are strongest in the LBD amygdala. Additional higher molecular mass bands are prominent in LBD cases (M1 and M2); these bands are less visible in AD/ALB or controls. The relative mobilities of molecular mass protein markers are identified on the left of the blots

Immunoblotting comparison of αsyn species in LBD cingulate cortex. High salt (HS) and SDS/urea fractions were obtained from the cingulate cortex of 2 control cases (lanes 1–2) and 3 LBD cases (lanes 3–5). 20 μg of lysate for each case and fraction were subject to western blot analysis using a panel of 4 antibodies, as indicated. In the HS fraction, all antibodies demonstrate similar amounts of monomeric FL αsyn except for one of the LBD cases which had a diminished level. In the SDS/Urea fractions, monomeric FL αsyn is present in high amounts for all LBD cases while the 2 controls have almost no αsyn in this fraction. Although higher molecular mass M1 and M2 bands are present in the cingulate SDS/urea fraction, truncation bands T1 and T2 are not as apparent which represents a biochemical difference between αsyn in the amygdala and MTL compared with cingulate cortex. The relative mobilities of molecular mass protein markers are identified on the left of the blots
Within the HS fractions from the MTL, a single major band is detected for all cases using all 4 antibodies at ~17kda which is monomeric full-length (FL) αsyn (Fig. 9). Although αsyn has a predicted molecular mass of 14.4 kda, it is known to migrate at a larger apparent molecular mass [108]. For LBD lysates, a qualitatively small decrease in amount of HS soluble αsyn is evident compared with the controls and AD/ALB; this is most apparent with the 5G4 antibody. Additionally, minor truncation products are visible particularly with N-terminal antibody 9C10 (Fig. 9). The HS fractions from cingulate cortex lysates for LBD and control cases similarly display a single major band which is monomeric FL αsyn; minor amounts of truncated αsyn are also present in these lysates (Fig. 10). The HS fractions did not reveal any significant differences in the biochemical composition of αsyn between the different brain regions and cases.
Within the SDS/urea fractions from the MTL, 4–5 major bands are present when probed using the various αsyn antibodies. With C-terminal antibody 94-3A10, monomeric FL αsyn is detected in the SDS/urea fraction prominently for 4/5 cases of LBD, with the greatest amount present in the isolated LBD amygdala. For the 2 AD/ALB cases, only 1 had a modest amount of SDS/urea monomeric αsyn which was detected with all 4 antibodies. The 2 control cases contained minimal amounts of SDS/urea monomeric αsyn across all antibodies. In addition to monomeric FL αsyn, N-terminal antibody 9C10 and central αsyn antibodies 3H11 and 5G4 were able to detect 2–3 apparently truncated forms of αsyn within LBD MTL and amygdala fractions (Fig. 9). These truncated species appear to be carboxy-truncated, as they are not detected with the C-terminal antibody 94-3A10. Within the amygdala in particular, the heavier major truncation band (T1) comprises a large portion of all αsyn within the fraction; indeed, antibody 5G4 which is preferential for oligomerized αsyn strongly detects the T1 and FL bands extensively within the amygdala LBD fraction (Fig. 9). Both the T1 band and lighter major truncation band (T2) are present within 4/5 LBD MTL fractions, whereas they are not seen in the AD/ALB or control fractions with any of the 3 antibodies that can detect carboxy-truncated αsyn (9C10, 3H11, and 5G4). Truncated αsyn is not readily apparent within the SDS/urea LBD cingulate cortex fractions, whereas the T1 and T2 truncation bands are abundant in the LBD MTL and especially the amygdala (Figs. 9 and 10). The presence of carboxy-truncated αsyn may play an important role in differing pathologies between the amygdala and other brain regions as carboxy-truncation alters important biochemical aspects of αsyn aggregation [93].
The SDS/urea fractions from the MTL also reveal higher molecular mass αsyn species abundant within the LBD fractions but qualitatively less within control and AD/ALB cases (Fig. 9). Major heavy bands M1 and M2 are approximately ~ 28 kda and ~ 36 kda respectively which could represent ubiquinated forms of αsyn [87]. Alternatively, these bands could be oligomeric αsyn with varying amounts of additional post-translational modifications which is stated to be the preferred target for antibody 5G4 [56]. C-terminal antibody 94-3A10 does not readily detect these high molecular weight bands, whereas N-terminal antibody 9C10 and especially central αsyn antibody 3H11 may detect more αsyn within these bands than in the monomeric αsyn band (Fig. 9). This pattern of band reactivity for M1 and M2 may suggest that the αsyn species contained are modified in multiple ways such as being both N and C-truncated. Furthermore, with antibodies 9C10 and 3H11 there are additional minor amounts of heavy protein bands present in the MTL but not the cingulate cortices of the control cases (Figs. 9 and 10). In particular, αsyn truncation appears most abundant in the LBD amygdala fraction compared with other fractions. These results suggest that post-translational modifications to αsyn within the amygdala may underlie the unique immunohistochemical staining profile for the LBD amygdala.
Discussion
The amygdala may be uniquely prone to pathologic developments in a number of neurodegenerative diseases [73]. Exemplifying this, neuritic senile plaques containing Aβ, tau, and αsyn demonstrate the propensity for pathologic aggregation to occur within the amygdala (Fig. 8). Other studies have noticed unique relationships between the amygdala and αsyn; for example, injections of pre-formed αsyn fibrils into various mouse models at differing locations invariably lead to amygdala pathology [1, 7, 13, 65,66,67, 78, 91, 94], and the amygdala uniquely displays immense upregulation of αsyn in relation to alcohol and opiate abstinence following addiction [114]. The studies herein demonstrate that pathologic αsyn within the amygdala in LBD is unique both in its immunohistochemical properties and immunoblotting profile which may differentiate aggregated forms of αsyn in LBD from the more innocuous AD/ALB. In particular, the presence of aggregation prone carboxy-truncated forms of αsyn within the amygdala may play an initiating role in the disease process as these species are able to misfold and induce endogenous FL αsyn to also aggregate [93]. Our investigations show using diseased human tissue that endogenous WT αsyn is induced to form pathologic inclusions when driven by the more aggregation prone A53T αsyn [15]; we postulate that a more pernicious form of αsyn (due to truncation or other structural modifications) may play a similar role as A53T αsyn in sporadic synucleinopathies. The early involvement of the amygdala in LBD along with sporadic LRP often being entirely restricted to the amygdala indicates that this brain region has the capacity to initiate αsyn misfolding and suggests that aggregation prone forms of αsyn are present and may originate in this region. It is difficult to identify and isolate the more deleterious forms of αsyn from diseased human brain tissue, but comparative immunohistochemical and immunoblotting αsyn profiles amongst various LBD brain regions and between LBD versus AD/ALB amygdalas indicate that there is in fact unique pathological forms of αsyn ubiquitous within the LBD amygdala that differs from other brain regions and disease entities.
The pervasive amount of αsyn neuropil pathology present within the amygdala compared to other brain regions has symptomatic relevance as a number of studies have demonstrated the importance of these smaller aggregates compared with larger LB type inclusions [14, 57, 59, 89]. It has been noted that neuropil pathology, presumably a large portion of which are small pre-synaptic aggregates, correlate more extensively with symptomatic severity compared to LBs [14, 57, 59, 89]. Conversely, it has been observed that the abundance of LBs restricted to the amygdala in AD/ALB have little to no impact on prognosis [74, 86, 102]; our study suggests that the relative sparsity of neuropil pathology in AD/ALB even following FA retrieval compared with LBD might be a key difference between these conditions. Other studies have noted the prevalence of large cortical LBs compared with neuropil pathology in AD/ALB as well, strengthening the findings herein [45, 102]. It is conceivable that structurally distinct forms of αsyn may have increased pathologic potency due to resistance to degradation or sequestration, and/or from more exposed amyloidogenic regions allowing faster prion-like templating of endogenous physiologic αsyn [101]. Differential resistance to proteases is commonly used to distinguish αsyn strains [35, 80], and structurally distinct mutant forms of αsyn are noted to resist lysosomal degradation [17]. Indeed, a pathologic correlate may exist amongst PD and LBD subtypes with more rapid onset and regionally diffuse pathology as it is suggested that rate of aggregate formation is increased likely due to a unique αsyn species [19, 36]. It would be expected that small pre-synaptic aggregates would predominate if a more pathologically potent αsyn was present, as monomeric αsyn is most abundant within the pre-synapse which would be induced to aggregate at that location and subsequent aggregates if resistant to clearance would stay localized to this subcellular location as opposed to sequestration through an aggresome-related process in a cytoplasmic LB which is one theory for authentic LB formation [105]. Some proposed models of αsyn mediated toxicity and prion-like spread center on dysfunction with axonal and dendritic processes where these smaller aggregates are presumably located and not the cell soma where large LBs are typically found [52, 104, 110]. Supporting the importance of neuropil pathology, exposure of neurons to αsyn fibrils is known to induce axonal deficits before any overt cell death [31]. Additionally, we and others have shown that a portion of αsyn-positive neuropil inclusion pathology is within astrocytes which are themselves adversely affected by αsyn aggregates and may have their own role in neurodegeneration and prion-like spread [12, 92]. Indeed, when observing past reports of pathology in Contursi kindred patients who have A53T human αsyn, it has been repeatedly noted that αsyn-positive neuropil threads and dot like inclusions are abundant while true LBs are more sparse [27, 55, 111]. A pathologic presentation of abundant αsyn neuropil aggregates with fewer LBs may be associated with a more aggressive disease course; in light of our findings and prior observations, it is likely that a distinct form of misfolded αsyn may favor the formation of neuropil pathologies which may be the pathologic determinant of disease progression.
Why αsyn misfolds into pathological aggregates remains largely unclear, and what underlies the diversity in the alleged strains of aggregated αsyn is even hazier. However, one major aspect of the amygdala that likely plays a role in influencing strain diversity is the concurrence of tau and Aβ aggregates which may all interact with monomeric or misfolded αsyn to induce structural alterations from which deleterious outcomes may result [33, 50, 55, 73, 77]. Indeed, amongst PD cases with more rapid development of dementia there is greater pathologic burdens of tau or Aβ [36, 52]; similar findings exist for LBD where concurrent AD pathology burden is highly predictive of symptomatic decline [43, 44, 52]. The αsyn-positive senile plaques and co-localized tau and αsyn aggregates shown in this paper are highly prevalent within the MTL and particularly the amygdala [18, 46, 47, 88, 109]. In AD, interactions of Aβ with tau have been postulated to occur within the amygdala and MTL, where induced conformational alterations in tau may refine the misfolded protein’s pathologic properties [73]. A similar phenomenon may occur in LBD, where virtually all cases of LBD display Aβ plaques in the MTL [52]; diversity in the Aβ aggregates may even underlie progression of disease into AD versus LBD as it has been observed that Aβ aggregates in LBD are comprised mainly of the Aβ1–42 peptide and the ratio Aβ1–42 to Aβ1–40 peptides is higher than in AD [61, 63]. AD/ALB may represent off-pathway, attenuated LB pathology induced by an Aβ strain more specific for tau misfolding; conversely LBD is the opposite with its own attenuated form of misfolded tau but more aggressive misfolded αsyn aggregates (Fig. 11). Although concurrent tau pathology is less common than Aβ in diffuse LBD [52], co-fibrils containing both αsyn and tau have been shown to occur which may have unique prion-like properties and could themselves modulate pathologic progression [32]. Interactions between proteins implicated in neurodegeneration are receiving increasing study due to their possible role in promoting strain diversity amongst the diseases, and the findings herein reinforce that such pathologic interactions are primed to occur in the amygdala where ample co-pathologies are present amongst the spectrum of neurodegenerative diseases [51, 73].

Diagram of pathologic determinants associated with further progression of αsyn pathology. The diversity of αsyn pathologies across different brain regions in LBD is shown in orange while the more limited αsyn pathology in AD/ALB is in blue (C = cingulate cortex, A = amygdala, M = midbrain-substantia nigra pars compacta). In LBD, diverse αsyn pathologies are most abundant within the amygdala where common co-localization with tau and Aβ are seen along with dense αsyn-positive neuropil aggregates including in glial processes that may represent a less easily sequestered strain of αsyn. In AD/ALB, αsyn pathology is largely limited to large LB type inclusions that may represented a more sequestered and less pathogenic strain of αsyn
The extensive presence of specific post-translational modifications of αsyn in the amygdala and MTL is another modality by which αsyn in the amygdala could be converted into a more potent form. Biochemical studies revealed that the MTL/amygdala harbored a larger amount of high molecular weight αsyn compared with the cingulate cortex; these bands may represent ubiquinated or oligomerized forms of αsyn. In addition, the presence of 2 major carboxy-truncated forms of αsyn within the MTL/amygdala but not the cingulate cortex may be of importance. Regional specificity in calpain and cathepsin expression and activity may underlie the abundance of these truncation products in the amygdala compared with other regions [28, 82]. In a previous examination of a single case of familial PD/LBD, the localized detection of major truncated αsyn products mainly to the amygdala was also observed which is in line with our own results [55]. Truncated forms of αsyn are known to alter the prion-like and aggregative properties of αsyn [93, 99], and their presence in the amygdala may contribute to unique neuropil pathologies likely as a component of αsyn fibrils also comprised of FL αsyn and other post-translationally modified αsyn proteins. In particular, the very early involvement of the amygdala in LBD and/or cases where only the amygdala has αsyn pathology suggests that the amygdala has the capacity for initial αsyn aggregation and not always necessarily prion-like spread. These differences in prion-like activity could in part be due to the large increase in aggregation propensity truncation of the C-terminus imparts to αsyn.
The amygdala is often considered to be a loosely related collection of separate nuclei with different functions [73], and although regional variation in αsyn pathology was observed within the amygdalas the exact nuclei involved were not distinguished; it has been previously noted that the central and accessory cortical nuclei are the most heavily afflicted in advanced PD [9]. Never the less, the findings of this study agree with an emerging theory that the amygdala is critical in either the initiation of aggregation, or the conversion of pathologic protein aggregates into more potent forms [73]. In addition to cell autonomous factors such as neuro-inflammation and impaired lysosomal autophagy [6, 106], the diversity of pathologic αsyn species occurring within the amygdala due to conformational changes, post-translational modification, or templating from other misfolded proteins such as Aβ should be a key target of study to understand why αsyn pathology may rapidly progress (LBD) or stay localized (AD/ALB). Future studies should utilize appropriate combinations of retrieval techniques and antibodies to maximize detection of neuropil pathology that is seemingly more relevant to disease, and incorporate assays to measure prion-like properties of αsyn within a given brain region to determine which areas of the brain would serve as suitable targets of disease modifying therapeutics designed to halt prion-like spread of pathology.
Conclusions
In summary, the findings of this study first demonstrated that the presence of an aggressive “strain” of αsyn as is the case with A53T αsyn in familial disease is sufficient to start a pathologic cascade incorporating WT naive αsyn. Furthermore, pathologic determinants of progression from isolated LBs within the amygdala to diffuse synucleinopathy were studied which indicated that thread-like neuropil αsyn pathologies including astrocytic αsyn aggregates are more relevant in predicting widespread pathology than the presence of LBs. The amygdala, due to its early involvement in multiple neurodegenerative diseases and unique pathologies discussed herein, is a key location where diverse strains of misfolded αsyn resulting in detrimental neuropil aggregates are likely to occur.
Availability of data and materials
All data generated and analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- AD:
-
Alzheimer’s disease
- AD/ALB:
-
Alzheimer’s disease with amygdala restricted Lewy bodies
- APOE:
-
Apolipoprotein E
- BCA:
-
Bicinchoninic acid assay
- BSA:
-
Bovine serum albumin
- CAA:
-
Cerebral amyloid angiopathy
- FA:
-
Formic acid
- FBS:
-
Fetal bovine serum
- FL:
-
Full-length
- FTLD-TDP:
-
frontotemporal lobar degeneration with TAR DNA-binding protein 43 inclusions
- GFAP:
-
Glial fibrillary acidic protein
- HS:
-
High-salt
- iLBD:
-
incidental Lewy body disease
- LB:
-
Lewy body
- LBD:
-
Lewy body dementia
- LNs:
-
Lewy neurites
- LRP:
-
Lewy related pathology
- MTL:
-
Medial temporal lobe
- PBS:
-
Phosphate buffered saline
- PD:
-
Parkinson’s disease
- PSP:
-
Progressive supranuclear palsy
- RIPA:
-
Radioimmunoprecipitation assay
- SNpc:
-
Substantia nigra pars compacta
- TBS:
-
Tris buffered saline
- WT:
-
Wild type
- αsyn:
-
α-synuclein
References
Abdelmotilib H, Maltbie T, Delic V, Liu Z, Hu X, Fraser KB, Moehle MS, Stoyka L, Anabtawi N, Krendelchtchikova V, Volpicelli-Daley LA, West A (2017) α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic neurodegeneration. Neurobiol Dis 105:84–98. https://doi.org/10.1016/j.nbd.2017.05.014
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-Synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752. https://doi.org/10.1074/jbc.M600933200
Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y (2001) Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res 888:287–296. https://doi.org/10.1016/s0006-8993(00)03082-1
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 152:879–884
Beach TG, White CL, Hamilton RL, Duda JE, Iwatsubo T, Dickson DW, Leverenz JB, Roncaroli F, Buttini M, Hladik CL, Sue LI, Noorigian JV, Adler CH (2008) Evaluation of α-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol 116:277–288. https://doi.org/10.1007/s00401-008-0409-8
Blandini F (2013) Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J NeuroImmune Pharmacol 8:189–201. https://doi.org/10.1007/s11481-013-9435-y
Blumenstock S, Rodrigues EF, Peters F, Blazquez-Llorca L, Schmidt F, Giese A, Herms J (2017) Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol Med 9:716–731. https://doi.org/10.15252/emmm.201607305
Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575. https://doi.org/10.1038/ncomms3575
Braak H, Braak E, Yilmazer D, de Vos RA, Jansen EN, Bohl J, Jellinger K (1994) Amygdala pathology in Parkinson’s disease. Acta Neuropathol 88:493–500
Braak H, Del Tredici K (2017) Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Park Dis 7:S71–S85. https://doi.org/10.3233/JPD-179001
Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Braak H, Sastre M, Del Tredici K (2007) Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol 114:231–241. https://doi.org/10.1007/s00401-007-0244-3
Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamgüney G (2016) Neuroinvasion of α-synuclein prionoids after intraperitoneal and Intraglossal inoculation. J Virol 90:9182–9193. https://doi.org/10.1128/JVI.01399-16
Colom-Cadena M, Pegueroles J, Herrmann AG, Henstridge CM, Muñoz L, Querol-Vilaseca M, Martín-Paniello CS, Luque-Cabecerans J, Clarimon J, Belbin O, Núñez-Llaves R, Blesa R, Smith C, McKenzie C-A, Frosch MP, Roe A, Fortea J, Andilla J, Loza-Alvarez P, Gelpi E, Hyman BT, Spires-Jones TL, Lleó A (2017) Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain 140:3204–3214. https://doi.org/10.1093/brain/awx275
Conway KA, Harper JD, Lansbury PT (1998) Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 4:1318–1320. https://doi.org/10.1038/3311
Crowther RA, Jakes R, Spillantini MG, Goedert M (1998) Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett 436:309–312
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant α-Synuclein by chaperone-mediated autophagy. Science 305:1292–1295. https://doi.org/10.1126/science.1101738
Culvenor JG, McLean CA, Cutt S, Campbell BC, Maher F, Jäkälä P, Hartmann T, Beyreuther K, Masters CL, Li QX (1999) Non-Abeta component of Alzheimer’s disease amyloid (NAC) revisited. NAC and alpha-synuclein are not associated with Abeta amyloid. Am J Pathol 155:1173–1181. https://doi.org/10.1016/s0002-9440(10)65220-0
De Pablo-Fernández E, Lees AJ, Holton JL, Warner TT (2019) Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 76:470. https://doi.org/10.1001/jamaneurol.2018.4377
Deng H, Wang P, Jankovic J (2018) The genetics of Parkinson disease. Ageing Res Rev 42:72–85. https://doi.org/10.1016/j.arr.2017.12.007
Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee S-J (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010–13015. https://doi.org/10.1073/pnas.0903691106
Dhillon J-KS, Riffe C, Moore BD, Ran Y, Chakrabarty P, Golde TE, Giasson BI (2017) A novel panel of α-synuclein antibodies reveal distinctive staining profiles in synucleinopathies. PLoS One 12:e0184731. https://doi.org/10.1371/journal.pone.0184731
Dhillon J-KS, Trejo-Lopez JA, Riffe C, McFarland NR, Hiser WM, Giasson BI, Yachnis AT (2019) Dissecting α-synuclein inclusion pathology diversity in multiple system atrophy: implications for the prion-like transmission hypothesis. Lab Invest 99:982-992. https://doi.org/10.1038/s41374-019-0198-9
Dickson DW (2012) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2:a009258–a009258. https://doi.org/10.1101/cshperspect.a009258
Dickson DW, Fujishiro H, DelleDonne A, Menke J, Ahmed Z, Klos KJ, Josephs KA, Frigerio R, Burnett M, Parisi JE, Ahlskog JE (2008) Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol 115:437–444. https://doi.org/10.1007/s00401-008-0345-7
Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VM, Trojanowski JQ (2000) Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol 59:830–841
Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM-Y, Trojanowski JQ (2002) Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 104:7–11. https://doi.org/10.1007/s00401-002-0563-3
Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT (2007) Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 170:1725–1738. https://doi.org/10.2353/ajpath.2007.061232
Dunn SD (1986) Effects of the modification of transfer buffer composition and the renaturation of proteins in gels on the recognition of proteins on Western blots by monoclonal antibodies. Anal Biochem 157:144–153
Frigerio R, Fujishiro H, Ahn T-B, Josephs KA, Maraganore DM, DelleDonne A, Parisi JE, Klos KJ, Boeve BF, Dickson DW, Ahlskog JE (2011) Incidental Lewy body disease: do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol Aging 32:857–863. https://doi.org/10.1016/j.neurobiolaging.2009.05.019
Froula JM, Henderson BW, Gonzalez JC, Vaden JH, Mclean JW, Wu Y, Banumurthy G, Overstreet-Wadiche L, Herskowitz JH, Volpicelli-Daley LA (2018) α-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol Commun 6:35. https://doi.org/10.1186/s40478-018-0537-x
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM-Y (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640. https://doi.org/10.1126/science.1082324
Giasson BI, Lee VM-Y, Trojanowski JQ (2003) Interactions of Amyloidogenic proteins. NeuroMolecular Med 4:49–58. https://doi.org/10.1385/NMM:4:1-2:49
Giasson BI, Murray IVJ, Trojanowski JQ, Lee VM-Y (2001) A hydrophobic stretch of 12 amino acid residues in the middle of α-Synuclein is essential for filament assembly. J Biol Chem 276:2380–2386. https://doi.org/10.1074/jbc.M008919200
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VMY (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. https://doi.org/10.1016/j.cell.2013.05.057
Halliday GM, Holton JL, Revesz T, Dickson DW (2011) Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 122:187–204. https://doi.org/10.1007/s00401-011-0852-9
Hamilton RL (2000) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10:378–384
Han H, Weinreb PH, Lansbury PT (1995) The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem Biol 2:163–169
Hawkes CH, Del Tredici K, Braak H (2010) A timeline for Parkinson’s disease. Parkinsonism Relat Disord 16:79–84. https://doi.org/10.1016/j.parkreldis.2009.08.007
Hoyer W, Cherny D, Subramaniam V, Jovin TM (2004) Impact of the acidic C-terminal region comprising amino acids 109−140 on α-Synuclein aggregation in vitro. Biochemistry 43:16233–16242. https://doi.org/10.1021/bi048453u
Hoyer W, Cherny D, Subramaniam V, Jovin TM (2004) Rapid self-assembly of α-synuclein observed by in situ atomic force microscopy. J Mol Biol 340:127–139. https://doi.org/10.1016/j.jmb.2004.04.051
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging–Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8:1–13. https://doi.org/10.1016/j.jalz.2011.10.007
Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, Keene CD, Montine TJ, Zabetian CP, Trojanowski JQ (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16:55–65. https://doi.org/10.1016/S1474-4422(16)30291-5
Irwin DJ, Hurtig HI (2018) The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. J Alzheimer’s Dis Parkinson 8. https://doi.org/10.4172/2161-0460.1000444
Iseki E (2004) Dementia with Lewy bodies: reclassification of pathological subtypes and boundary with Parkinson’s disease or Alzheimer’s disease. Neuropathology 24:72–78
Iseki E, Marui W, Kosaka K, Uéda K (1999) Frequent coexistence of Lewy bodies and neurofibrillary tangles in the same neurons of patients with diffuse Lewy body disease. Neurosci Lett 265:9–12. https://doi.org/10.1016/S0304-3940(99)00178-0
Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003) Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397
Jellinger KA (2008) A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol 116:1–16. https://doi.org/10.1007/s00401-008-0406-y
Jellinger KA (2009) Formation and development of Lewy pathology: a critical update. J Neurol 256:270–279. https://doi.org/10.1007/s00415-009-5243-y
Jellinger KA (2011) Interaction between α-synuclein and other proteins in neurodegenerative disorders. Sci World J 11:1893–1907. https://doi.org/10.1100/2011/371893
Jellinger KA (2012) Interaction between pathogenic proteins in neurodegenerative disorders. J Cell Mol Med 16:1166–1183. https://doi.org/10.1111/j.1582-4934.2011.01507.x
Jellinger KA (2018) Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J Neural Transm 125:615–650. https://doi.org/10.1007/s00702-017-1821-9
Kellie JF, Higgs RE, Ryder JW, Major A, Beach TG, Adler CH, Merchant K, Knierman MD (2015) Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep 4:5797. https://doi.org/10.1038/srep05797
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506. https://doi.org/10.1038/nm1747
Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ, Lee VM-Y (2004) Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Exp Neurol 187:279–288. https://doi.org/10.1016/j.expneurol.2004.01.007
Kovacs GG, Breydo L, Green R, Kis V, Puska G, Lőrincz P, Perju-Dumbrava L, Giera R, Pirker W, Lutz M, Lachmann I, Budka H, Uversky VN, Molnár K, László L (2014) Intracellular processing of disease-associated α-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol Dis 69:76–92. https://doi.org/10.1016/j.nbd.2014.05.020
Kovacs GG, Milenkovic IJ, Preusser M, Budka H (2008) Nigral burden of α-synuclein correlates with striatal dopamine deficit. Mov Disord 23:1608–1612. https://doi.org/10.1002/mds.22207
Kovacs GG, Wagner U, Dumont B, Pikkarainen M, Osman AA, Streichenberger N, Leisser I, Verchère J, Baron T, Alafuzoff I, Budka H, Perret-Liaudet A, Lachmann I (2012) An antibody with high reactivity for disease-associated α-synuclein reveals extensive brain pathology. Acta Neuropathol 124:37–50. https://doi.org/10.1007/s00401-012-0964-x
Kramer ML, Schulz-Schaeffer WJ (2007) Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410. https://doi.org/10.1523/JNEUROSCI.4564-06.2007
Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL, Lee MK (2005) Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A 102:2162–2167. https://doi.org/10.1073/pnas.0406976102
Lippa CF, Ozawa K, Mann DM, Ishii K, Smith TW, Arawaka S, Mori H (1999) Deposition of beta-amyloid subtypes 40 and 42 differentiates dementia with Lewy bodies from Alzheimer disease. Arch Neurol 56:1111–1118
Liu C-W, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ (2005) A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem 280:22670–22678. https://doi.org/10.1074/jbc.M501508200
Mann DM, Brown SM, Owen F, Baba M, Iwatsubo T (1998) Amyloid beta protein (a beta) deposition in dementia with Lewy bodies: predominance of a beta 42(43) and paucity of a beta 40 compared with sporadic Alzheimer’s disease. Neuropathol Appl Neurobiol 24:187–194
Markesbery WR, Jicha GA, Liu H, Schmitt FA (2009) Lewy body pathology in Normal elderly subjects. J Neuropathol Exp Neurol 68:816–822. https://doi.org/10.1097/NEN.0b013e3181ac10a7
Mason DM, Nouraei N, Pant DB, Miner KM, Hutchison DF, Luk KC, Stolz JF, Leak RK (2016) Transmission of α-synucleinopathy from olfactory structures deep into the temporal lobe. Mol Neurodegener 11:49. https://doi.org/10.1186/s13024-016-0113-4
Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M (2014) Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun 2:88. https://doi.org/10.1186/s40478-014-0088-8
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DMA, Hasegawa M (2013) Prion-like spreading of pathological α-synuclein in brain. Brain 136:1128–1138. https://doi.org/10.1093/brain/awt037
Mavroeidi P, Arvanitaki F, Karakitsou A-K, Vetsi M, Kloukina I, Zweckstetter M, Giller K, Becker S, Sorrentino ZA, Giasson BI, Jensen PH, Stefanis L, Xilouri M (2019) Endogenous oligodendroglial alpha-synuclein and TPPP/p25α orchestrate alpha-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol 138:415-441. https://doi.org/10.1007/s00401-019-02014-y
McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, DeLucia M, Lin W-L, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T (2005) Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47:191–199. https://doi.org/10.1016/j.neuron.2005.06.030
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, VMY L, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O’Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K (2017) Diagnosis and management of dementia with Lewy bodies. Neurology 89:88–100. https://doi.org/10.1212/WNL.0000000000004058
Mishizen-Eberz AJ, Norris EH, Giasson BI, Hodara R, Ischiropoulos H, Lee VM-Y, Trojanowski JQ, Lynch DR (2005) Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry 44:7818–7829. https://doi.org/10.1021/bi047846q
Murray IVJ, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VM-Y (2003) Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42:8530–8540. https://doi.org/10.1021/bi027363r
Nelson PT, Abner EL, Patel E, Anderson S, Wilcock DM, Kryscio RJ, Van Eldik LJ, Jicha GA, Gal Z, Nelson RS, Nelson BG, Gal J, Azam MT, Fardo DW, Cykowski MD (2018) The amygdala as a locus of pathologic misfolding in neurodegenerative diseases. J Neuropathol Exp Neurol 77:2–20. https://doi.org/10.1093/jnen/nlx099
Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR (2010) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20:66–79. https://doi.org/10.1111/j.1750-3639.2008.00244.x
Neltner JH, Abner EL, Schmitt FA, Denison SK, Anderson S, Patel E, Nelson PT (2012) Digital pathology and image analysis for robust high-throughput quantitative assessment of Alzheimer disease neuropathologic changes. J Neuropathol Exp Neurol 71:1075–1085. https://doi.org/10.1097/NEN.0b013e3182768de4
Nonaka T, Iwatsubo T, Hasegawa M (2005) Ubiquitination of α-synuclein. Biochemistry 44:361–368. https://doi.org/10.1021/bi0485528
Ono K, Takahashi R, Ikeda T, Yamada M (2012) Cross-seeding effects of amyloid β-protein and α-synuclein. J Neurochem 122:883–890. https://doi.org/10.1111/j.1471-4159.2012.07847.x
Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE (2015) Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199. https://doi.org/10.1016/j.nbd.2015.06.003
Peelaerts W, Bousset L, Baekelandt V, Melki R (2018) ɑ-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences. Cell Tissue Res 373:195–212. https://doi.org/10.1007/s00441-018-2839-5
Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, Trojanowski JQ, Lee VM-Y (2018) Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 557:558–563. https://doi.org/10.1038/s41586-018-0104-4
Peng C, Gathagan RJ, Lee VM-Y (2018) Distinct α-Synuclein strains and implications for heterogeneity among α-Synucleinopathies. Neurobiol Dis 109:209–218. https://doi.org/10.1016/j.nbd.2017.07.018
Pieri L, Chafey P, Le Gall M, Clary G, Melki R, Redeker V (2016) Cellular response of human neuroblastoma cells to α-synuclein fibrils, the main constituent of Lewy bodies. Biochim Biophys Acta 1860:8–19. https://doi.org/10.1016/j.bbagen.2015.10.007
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Popescu A, Lippa CF, Lee VM-Y, Trojanowski JQ (2004) Lewy bodies in the amygdala: increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol 61:1915–1919. https://doi.org/10.1001/archneur.61.12.1915
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJA, Kraneveld AD (2017) Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 8:37. https://doi.org/10.3389/fneur.2017.00037
Roudil J, Deramecourt V, Dufournet B, Dubois B, Ceccaldi M, Duyckaerts C, Pasquier F, Lebouvier T, Brainbank Neuro-CEB Neuropathology Network (2018) Influence of Lewy pathology on Alzheimer’s disease phenotype: a retrospective Clinico-pathological study. J Alzheimers Dis 63:1317–1323. https://doi.org/10.3233/JAD-170914
Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM-Y (2003) Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol 163:91–100. https://doi.org/10.1016/s0002-9440(10)63633-4
Schmidt ML, Martin JA, Lee VM-Y, Trojanowski JQ (1996) Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer’s disease and Lewy body disorders. Acta Neuropathol 91:475–481. https://doi.org/10.1007/s004010050454
Schulz-Schaeffer WJ (2010) The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143. https://doi.org/10.1007/s00401-010-0711-0
Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000) Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A 97:4897–4902
Sorrentino ZA, Brooks MMT, Hudson V, Rutherford NJ, Golde TE, Giasson BI, Chakrabarty P (2017) Intrastriatal injection of α-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol Neurodegener 12:40. https://doi.org/10.1186/s13024-017-0182-z
Sorrentino ZA, Giasson BI, Chakrabarty P (2019) α-Synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol 138:1-21. https://doi.org/10.1007/s00401-019-01977-2
Sorrentino ZA, Vijayaraghavan N, Gorion K-M, Riffe CJ, Strang KH, Caldwell J, Giasson BI (2018) Physiological carboxy-truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J Biol Chem 293:18914-18932. https://doi.org/10.1074/jbc.RA118.005603
Sorrentino ZA, Xia Y, Funk C, Riffe CJ, Rutherford NJ, Ceballos Diaz C, Sacino AN, Price ND, Golde TE, Giasson BI, Chakrabarty P (2018) Motor neuron loss and neuroinflammation in a model of α-synuclein-induced neurodegeneration. Neurobiol Dis 120:98–106. https://doi.org/10.1016/j.nbd.2018.09.005
Soto C, Pritzkow S (2018) Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci 21:1332–1340. https://doi.org/10.1038/s41593-018-0235-9
Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388:839–840. https://doi.org/10.1038/42166
Spires-Jones TL, Attems J, Thal DR (2017) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 134:187–205. https://doi.org/10.1007/s00401-017-1709-7
Strang KH, Goodwin MS, Riffe C, Moore BD, Chakrabarty P, Levites Y, Golde TE, Giasson BI (2017) Generation and characterization of new monoclonal antibodies targeting the PHF1 and AT8 epitopes on human tau. Acta Neuropathol Commun 5:58. https://doi.org/10.1186/s40478-017-0458-0
Terada M, Suzuki G, Nonaka T, Kametani F, Tamaoka A, Hasegawa M (2018) The effect of truncation on prion-like properties of α-synuclein. J Biol Chem 293:13910–13920. https://doi.org/10.1074/jbc.RA118.001862
Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, Lee EB, Arnold SE, Duda JE, Hurtig H, Lee VM-Y, Adler CH, Beach TG, Trojanowski JQ (2016) Pathological α-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol 131:393–409. https://doi.org/10.1007/s00401-015-1526-9
Uchihara T, Giasson BI (2016) Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131:49–73. https://doi.org/10.1007/s00401-015-1485-1
Uchikado H, Lin W-L, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697. https://doi.org/10.1097/01.jnen.0000225908.90052.07
Valdinocci D, Radford RAW, Siow SM, Chung RS, Pountney DL (2017) Potential modes of intercellular alpha-synuclein transmission. Int J Mol Sci 18. https://doi.org/10.3390/ijms18020469
Volpicelli-Daley LA (2017) Effects of α-synuclein on axonal transport. Neurobiol Dis 105:321–327. https://doi.org/10.1016/j.nbd.2016.12.008
Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H (2013) The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol 47:495–508. https://doi.org/10.1007/s12035-012-8280-y
Wang B, Abraham N, Gao G, Yang Q (2016) Dysregulation of autophagy and mitochondrial function in Parkinsons disease. Transl Neurodegener 5:19. https://doi.org/10.1186/s40035-016-0065-1
Waxman EA, Giasson BI (2011) Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci 31:7604–7618. https://doi.org/10.1523/JNEUROSCI.0297-11.2011
Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT (1996) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35:13709–13715. https://doi.org/10.1021/bi961799n
Wirths O, Weickert S, Majtenyi K, Havas L, Kahle PJ, Okochi M, Haass C, Multhaup G, Beyreuther K, Bayer TA (2000) Lewy body variant of Alzheimer’s disease: alpha-synuclein in dystrophic neurites of a beta plaques. Neuroreport 11:3737–3741
Wong YC, Krainc D (2017) α-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13. https://doi.org/10.1038/nm.4269
Yamaguchi K, Cochran EJ, Murrell JR, Polymeropoulos MH, Shannon KM, Crowther RA, Goedert M, Ghetti B (2005) Abundant neuritic inclusions and microvacuolar changes in a case of diffuse Lewy body disease with the A53T mutation in the α-synuclein gene. Acta Neuropathol 110:298–305. https://doi.org/10.1007/s00401-005-1042-4
Yamamoto R, Iseki E, Marui W, Togo T, Katsuse O, Kato M, Isojima D, Akatsu H, Kosaka K, Arai H (2005) Non-uniformity in the regional pattern of Lewy pathology in brains of dementia with Lewy bodies. Neuropathology 25:188–194
Yan X, Uronen R-L, Huttunen HJ (2018) The interaction of α-synuclein and tau: a molecular conspiracy in neurodegeneration? Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2018.05.005
Ziolkowska B, Gieryk A, Wawrzczak-Bargiela A, Krowka T, Kaminska D, Korkosz A, Bienkowski P, Przewlocki R (2008) α-Synuclein expression in the brain and blood during abstinence from chronic alcohol drinking in mice. Neuropharmacology 54:1239–1246. https://doi.org/10.1016/J.NEUROPHARM.2008.04.001
Acknowledgements
Brittany Ulm provided guidance in usage of Olympus IX81-DSU spinning disk confocal microscope. Tissue samples were supplied by the University of Florida Neuromedicine Human Brain Tissue Bank.
Funding
These studies were supported by grants P50AG047266, R01NS089622 and R01NS100876 from the National Institutes of Health and the University of Florida Moonshot initiative. JSD was supported by grant T32NS082168. ZAS was supported by grant F30AG063446.
Author information
Authors and Affiliations
Contributions
ZAS conducted experiments, analyzed the results, contributed to the experimental designs and writing of the manuscript. MSG and JSD designed and performed tissue fractionation procedures and analyzed results. CJR, NV, KG, YX performed immunohistochemistry and assisted with analysis. KNM performed the genotyping analysis. LIG and ATY contributed tissue from patients. BIG contributed to writing the manuscript and the experimental design. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Quantitation of LRP in LBD brain regions across a panel of antibodies individual cases. Three areas of dense pathology within the cingulate (C) or amygdala (A) of 9 LBD cases stained with 3 different antibodies without or with (FA) retrieval were subject to positive pixel analysis; average positivity and error bars (std) are displayed for each case, region, and antibody without or with FA. Without FA retrieval, all antibodies detect similar amounts of LRP within the amygdala versus the cingulate cortex; with FA retrieval a large increase in labeled amygdala pathology is evident with all antibodies whereas a lesser increase in pathology is seen in the cingulate cortex and only with antibody 94-3A10. With FA retrieval, the average amygdala pathology burden is significantly greater than the cingulate cortex for all antibodies. (TIF 4434 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sorrentino, Z.A., Goodwin, M.S., Riffe, C.J. et al. Unique α-synuclein pathology within the amygdala in Lewy body dementia: implications for disease initiation and progression. acta neuropathol commun 7, 142 (2019). https://doi.org/10.1186/s40478-019-0787-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0787-2