
Abstract
RNA-binding proteins (RBPs) are associated with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), but the underlying disease mechanisms remain unclear. In an unbiased screen in Drosophila for RBPs that genetically interact with TDP-43, we found that downregulation of the mRNA export factor Ref1 (fly orthologue to human ALYREF) mitigated TDP-43 induced toxicity. Further, Ref1 depletion also reduced toxicity caused by expression of the C9orf72 GGGGCC repeat expansion. Ref1 knockdown lowered the mRNA levels for these related disease genes and reduced the encoded proteins with no effect on a wild-type Tau disease transgene or a control transgene. Interestingly, expression of TDP-43 or the GGGGCC repeat expansion increased endogenous Ref1 mRNA levels in the fly brain. Further, the human orthologue ALYREF was upregulated by immunohistochemistry in ALS motor neurons, with the strongest upregulation occurring in ALS cases harboring the GGGGCC expansion in C9orf72. These data support ALYREF as a contributor to ALS/FTD and highlight its downregulation as a potential therapeutic target that may affect co-existing disease etiologies.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are neurodegenerative diseases that share clinical, genetic, and pathologic hallmarks [50]. In recent years, mutations in a number of RNA binding proteins (RBPs) have been discovered in ALS/FTD, highlighting RNA-centric mechanisms in disease [62]. Of note, TDP-43 (encoded by the TARDBP gene) was identified as the primary component of ubiquitinated inclusions in ALS cases and a subset of FTD cases [40]. As an RBP, TDP-43 has been found to mediate a number of pathways related to RNA metabolism [28].
Recent investigations have revealed factors that may mediate TDP-43 associated ALS/FTD. An intermediate length polyglutamine repeat expansion (PolyQ) within another RBP, Ataxin-2, was defined as a risk factor for ALS that enhances the toxicity of TDP-43 [15]. Further, in genetic cases of familial ALS and FTD with TDP-43 pathology (FTD-TDP), a GGGGCC hexanucleotide repeat expansion (termed G4C2) in C9orf72 was identified as the most common mutation [13, 42]. Interestingly, mice expressing expanded G4C2 have TDP-43 pathology [7]. Links between TDP-43 and G4C2 may converge on RNA metabolism as G4C2 expansions contribute to disruptions in various aspects of RNA processes [2, 27, 59, 61]. Altogether, accumulating studies strongly point to altered RNA biology as a critical component of disease etiology in ALS and FTD [9, 21].
Here, using a directed screen aimed at RNA-interacting proteins in Drosophila, we identified that RNA and export factor binding protein 1 (Ref1), an orthologue of human ALYREF (also known as THOC4), modulates toxicity associated with TDP-43, TDP-43 with Ataxin-2, and G4C2. ALYREF is a component of the TRanscription and EXport (TREX) complex, a conserved complex that links transcription and processing of mRNAs to their export from the nucleus into the cytoplasm [20, 23, 48, 63]. Impaired nucleocytoplasmic shuttling has been identified as a major pathway impacted in C9+ ALS/FTD [8, 16, 43, 60]. Evidence also suggests that ALYREF may play a more direct role in transcription beyond mRNA export [5, 41, 49, 52, 58].
Using Drosophila, we show that knockdown of Ref1 suppresses the toxicity of multiple related ALS/FTD genes (TDP-43, TDP-43 with Ataxin-2, and G4C2), providing the first evidence that it can mediate TDP-43-associated toxicity independent of G4C2 [16]. Further, depletion of Ref1 using RNAi caused reductions in mRNA level and concomitant reduction in disease protein levels produced from TDP-43 and G4C2 disease genes, but not from a Tau disease gene. Interestingly, endogenous Ref1 mRNA became upregulated in the TDP-43 and G4C2 fly models and ALYREF protein (the human Ref1 orthologue) is upregulated by immunohistochemistry in ALS motor neurons. Upregulation is strongest in C9+ ALS cases, which harbor the G4C2 expansion and, presumably, have TDP-43 pathology [1, 13, 38, 42], compared to C9- ALS cases, which are expected to have only TDP-43 pathology [14, 32]. These data argue that a feed-forward loop may exist between the expression of ALYREF and disease genes, while highlighting ALYREF as an important disease modifier that may represent a therapeutic target of multiple co-existing disease etiologies.
Results
RNAi screen identifies Ref1 as a strong suppressor of TDP-43 associated toxicity
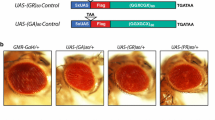
We established an in vivo RNAi screen in Drosophila melanogaster to identify RNA interacting proteins that can modify the toxicity of ALS-associated genes (Fig. 1a). The screen was performed against two Drosophila models of TDP-43 proteinopathy. Human TDP-43 was expressed with or without added ATXN2 bearing an intermediate polyglutamine repeat expansion of 32 glutamines (Q), an established risk factor for ALS [15, 39]. The disease-associated genes, as well as the RNAi transgenes, were expressed in the fly eye using the GAL4/UAS system [4]. 107 Drosophila genes that encode proteins with an RNA recognition motif (RRM) and have a direct human orthologue were targeted by RNAi. We focused on RRM containing genes because many of the genes associated with ALS contain such domains [21].

An in vivo RNAi screen identified Ref1 among RNA-binding protein genes as a modifier of TDP-43 and ATXN2-32Q toxicity. a Screen overview. b Heatmap of modifiers identified in the screen. Enhancers in shades of red and suppressors in green. c Ref1 knockdown does not affect the mRNA level of a reporter LacZ transgene. n.s. not statistically significant, two-tailed Student’s t-test. d Ref1 knockdown does not affect the β-galactosidase protein levels produced from a reporter LacZ transgene. n.s. not statistically significant, two-tailed Student’s t-test. Genotypes for (c) and (d). Control RNAi: GMR-GAL4, UAS-LacZ/+; UAS-control RNAi (JF01355)/+. Ref1 RNAi: GMR-GAL4, UAS-LacZ/+; UAS-Ref1 RNAi (HMS01301)/+. e External image (top) and cross section (bottom) of the fly eye. Genotype: YH3-GAL4/UAS-control RNAi (JF01355). f Ref1 RNAi suppresses TDP-43-mediated neurodegeneration. Genotypes. Control: UAS-TDP-43/+; YH3-GAL4/UAS-control RNAi (JF01355). Ref1: UAS-TDP-43/+; YH3-GAL4/UAS-Ref1 RNAi (HMS01301). Quantification of retinal area from Drosophila head sections shown on the right. ***p < 0.001, two-tailed Student’s t-test. g Ref1 RNAi suppresses TDP-43+ ATXN2(32Q)-mediated neurodegeneration. Genotypes. Control: UAS-TDP-43, UAS-ATXN2-32Q/+; YH3-GAL4/UAS-control RNAi (JF01355). Ref1: UAS-TDP-43, UAS-ATXN2-32Q /+; YH3-GAL4/UAS-Ref1 RNAi (HMS01301). Quantification of retinal area shown on the right. ***p < 0.001 two-tailed Student’s t-test. For all graphs, individual data points are shown with mean +/− standard deviation
The screen identified a total of 22 modifiers (Fig. 1b, full screen results described in Additional file 1: Table S1). Of these, 3 showed strong and consistent effects between the TDP-43 and TDP-43/ATXN2-32Q models: Half pint (hfp, also known as pUF68) RNAi enhanced TDP-43 and TDP-43/ATXN2-32Q toxicity, while Ref1 and second mitotic wave missing (swm) RNAi suppressed the degenerative eye effect. We used a reporter gene, LacZ, to rule out whether these modifiers affected the GAL4/UAS expression system of the transgenes. This showed that RNAi to hfp increased and swm decreased the levels of β-galactosidase protein (Additional file 2: Figure S1A); importantly, Ref1 had no effect on the RNA or protein expression from the control LacZ gene (Fig. 1c, d). We confirmed efficient knockdown of Ref1 by the RNAi line (Additional file 2: Figure S1B). Ref1 was the strongest hit from the screen as it suppressed neurodegeneration caused by TDP-43 (Fig. 1c, d) and TDP-43/ATXN2-32Q (Fig. 1e), without an effect on expression of an unrelated control protein.
Ref1 knockdown suppresses TDP-43 toxicity by reducing its expression
As ALYREF is a known mediator of RNA metabolism, we hypothesized that loss of Ref1 could alter the level of expression of the TDP-43 mRNA. Total RNA was extracted from fly heads co-expressing TDP-43. TDP-43 mRNA levels were determined using quantitative realtime polymerase chain reaction (qRT-PCR). Knockdown of Ref1 significantly reduced TDP-43 mRNA levels (Fig. 2a). Western immunoblot analysis was then used to determine the effects of Ref1 knockdown on TDP-43 protein levels. Consistent with its effects on TDP-43 mRNA, Ref1 RNAi reduced TDP-43 protein levels (Fig. 2b).

Ref1 reduction lowers TDP-43 RNA and protein abundance. a Ref1 Knockdown reduces TDP-43 mRNA levels in Drosophila heads. ***p < 0.001 two-tailed Student’s t-test. b Ref1 knockdown reduces TDP-43 protein levels in Drosophila heads. Western immunoblot analysis of human TDP-43 and β-Tubulin as loading control. *p < 0.05 two-tailed Student’s t-test. Genotypes for (a) and (b). Control RNAi: UAS-TDP-43/+; YH3-GAL4/UAS-control RNAi (JF01355). Ref1: UAS-TDP-43/+; YH3-GAL4/UAS-Ref1 RNAi (HMS01301). c Loss of Ref1 does not alter Tau mRNA levels. n.s. not statistically significant, two-tailed Student’s t-test. d Loss of Ref1 does not alter Tau protein levels. n.s not statistically significant, two-tailed Student’s t-test. Genotypes for (c) and (d). Control RNAi: YH3, UAS-Tau/ UAS-control RNAi (JF01355). Ref1 RNAi: YH3, UAS-Tau/ UAS-Ref1 RNAi (HMS01301). For all graphs, individual data points are shown with mean +/− standard deviation
To gain an understanding if the effect of Ref1 knockdown was universal to all disease genes, we tested for effects of Ref1 depletion on expression of the disease gene Tau. Tau (encoded by the MAPT gene) is hyperphosphorylated and aggregates in a class of neurodegenerative diseases termed Tauopathies, which includes a subset of FTD cases lacking abnormal TDP-43 inclusions [47]. Thus, Tau represents an unrelated disease etiology as it does not co-occur with TDP-43 pathology. The reduced expression from the TDP-43 transgene in response to Ref1 RNAi was specific to this disease gene as Ref1 RNAi had no effect on mRNA expression (Fig. 2c) or protein expression (Fig. 2d) from a wild-type Tau transgene. These data indicated that Ref1 mitigated toxicity by selectively reducing levels of the disease TDP-43 mRNA.
Ref1 knockdown reduced expression from a G4C2 transgene, resulting in reduced G4C2-toxicity
ALYREF (human Ref1 protein homolog) was previously reported to bind G4C2 RNA [11, 19] and the presence of G4C2 expansions is found in TDP-43-associated ALS/FTD. Thus, we hypothesized that Ref1 may be able to also modify toxicity caused by expression of > 30 G4C2 repeats. Ref1 had previously been proposed as a modifier of G4C2-induced toxicity, causing accumulation of total mRNA within the nucleus when depleted [16]. However, Ref1 downregulation was not able to significantly modify toxicity associated with a shorter (G4C2)36 gene [19]. To further examine Ref1 as a potential modifier of expanded G4C2, we used a fly model expressing (G4C2)49 that induces neurodegeneration [6, 18, 26, 37]. Co-expression of Ref1 RNAi and expanded G4C2 resulted in suppressed degeneration in the fly eye (Fig. 3a). Importantly, this effect was consistent in both the external eye – seen by reduced pigment loss and recovered ommatidial organization – and in the internal retina tissue – seen by increased tissue integrity.

Ref1 reduction lowers G4C2 RNA and dipeptide abundance. a Ref1 RNAi suppresses G4C2 repeat-mediated neurodegeneration. Genotypes. Control: UAS-(G4C2)49, YH3-GAL4/UAS-control RNAi (JF01355). Ref1: UAS-(G4C2)49, YH3-GAL4/UAS-Ref1 RNAi (HMS01301). Quantification of retinal area shown on the right. ***p < 0.001, two-tailed Student’s t-test. b Ref1 knockdown reduces G4C2 mRNA levels in Drosophila heads. d Ref1 knockdown reduces GR-GFP protein levels in the Drosophila eye. e Quantification of GR-GFP signal. Genotypes for (b) and (c). Control RNAi: (G4C2)44 [GR-GFP], YH3/ UAS-control RNAi (JF01355). Ref1 RNAi: (G4C2)44 [GR-GFP], YH3/ UAS-Ref1 RNAi (HMS01301). ***p < 0.001 two-tailed Student’s t-test. For all graphs, individual data points are shown with mean +/− standard deviation
Given the effect of Ref1 RNAi on expression of the TDP-43 mRNA (see Fig. 2) and that recent evidence supports that ALYREF may play a more global role in transcription [5, 41, 49, 52, 58], we hypothesized that Ref1 may also alter expression of expanded G4C2. To assess the effects of Ref1 on expression, total RNA was extracted from fly heads co-expressing the G4C2 repeat transgene together with RNAi to Ref1 or a control. G4C2 RNA levels were determined using qRT-PCR [18]. Knockdown of Ref1 caused a significant reduction in the mRNA levels of G4C2 repeats (Fig. 3b).
G4C2 RNA can produce three dipeptide repeats (DPR) by non-AUG (RAN-) translation: glycine-arginine (GR), glycine-alanine (GA), glycine-proline (GP) [1, 34]. These DPR can form aggregates in ALS/FTD tissue. Of these, GR dipeptides have been consistently shown to be toxic in Drosophila with GA showing mild effects, and GP no toxicity [16, 36]. To determine if Ref1 knockdown effected GR protein levels, we utilized a Drosophila transgenic line that expresses a G4C2 repeat with a green fluorescent protein (GFP) tag in frame with the GR dipeptide [18]. Fluorescent imaging revealed that knockdown of Ref1 dramatically reduced GR-GFP accumulation (Fig. 3c). Blinded quantification of the GFP signal revealed a significant and consistent downregulation of the GR-GFP signal.
Overall, modification of G4C2-toxicity was consistent with a previous report [16], while our data further show that Ref1 loss alters the mRNA level of G4C2. Importantly, as Ref1 also altered expression of TDP-43 mRNA, it may serve as a unique target that can modify these two co-existing pathologies simultaneously.
Ref1 is upregulated in response to TDP-43 or G4C2 expression in the adult fly brain
As our data supported that knockdown of Ref1 modulates toxicity of TDP-43 and G4C2, we considered whether expression of endogenous Ref1 might be impacted by expression of these disease genes. To examine this, we expressed TDP-43 or a G4C2 repeat expansion in neurons of adult flies for 16d using the drug-inducible neuronal driver ElavGS, and then measured endogenous Ref1 mRNA levels in fly heads by qRT-PCR. Interestingly, endogenous Ref1 mRNA levels were significantly increased in animals expressing TDP-43 (Fig. 4a). Further, Ref1 mRNA levels were significantly increased upon expression of long and toxic (49 units), but not short and inert (8 units), G4C2 repeats (Fig. 4b). Taken together, these data suggest that Ref1 activity is upregulated by TDP-43 and toxic expanded G4C2, and that reduction of that activity by RNAi in Drosophila is beneficial.

Ref1 mRNA level is increased in Drosophila ALS/FTD models. a Ref1 mRNA levels in Drosophila heads are increased by TDP-43 expression. *p < 0.05 two tailed Student’s t-test. Genotypes. Control: elavGS/+. TDP-43: elavGS/UAS-TDP-43. b Ref1 mRNA levels in Drosophila heads are increased by long (49), but not short (8) G4C2 repeats expression. One-way ANOVA with Dunnett’s multiple comparisons test, F(2,15) = 52.09, p < 0.001. ***p < 0.001, n.s not statistically significant. Genotypes. Control: elavGS/+. (G4C2)8: elavGS/UAS-(G4C2)8. (G4C2)49: elavGS/UAS-(G4C2)49. For all graphs, individual data points are shown with mean +/− standard deviation
ALYREF protein levels are increased in human ALS motor neurons
Thus far, we found that Ref1 depletion in flies could suppress toxicity caused by expression of ALS/FTD disease genes TDP-43 and G4C2. Suppression was the result of reduced mRNA levels of these disease genes. Moreover, expression of TDP-43 and expanded G4C2 in the adult fly nervous system caused upregulation of endogenous Ref1. Given these findings, we were curious as to whether there may be an alteration in the expression of ALYREF, the human Ref1 orthologue, in human disease.
Immunofluorescence imaging for ALYREF protein was performed on lumbar spinal cord tissue sections to define any changes in localization and/or expression of ALYREF in disease (Fig. 5a). ALYREF is known to predominantly localize to the nucleus, although it can translocate to the cytoplasm as it accompanies exported nuclear mRNAs into the cytoplasm [3, 54, 57, 63]. Using two independent antibodies to ALYREF, we found that ALYREF localized to both the nucleus and cytoplasm in ALS motor neurons (Fig. 5b; patient information can be found in Additional file 3: Table S2). Importantly, ALYREF protein levels were significantly upregulated in ALS motor neurons compared to controls. Blinded quantification of ALYREF intensity revealed that upregulation of ALYREF was stronger in C9+ ALS cases, which have both TDP-43 pathology and the G4C2 repeat expansion present in C9orf72 [1, 13, 38, 42], compared to C9- ALS cases, which have only TDP-43 pathology [14, 32] (Fig. 5c). These data are consistent with the fly data showing that Ref1 (the orthologue of ALYREF in Drosophila) is upregulated upon expression of TDP-43 and G4C2. Moreover, these data support ALYREF dysregulation in human ALS/FTD.

ALYREF protein levels are increased in human ALS patient motor neurons. a Schematic depicting the location of spinal cord motor neurons imaged in (b). b Fluorescent immunostaining of ALYREF in lumbar spinal cord sections from control and ALS cases. 2 independent ALYREF antibodies were used. Scale bar 50 μm. c Image intensity quantification using the rabbit polyclonal antibody targeting ALYREF protein. Control n = 83 cells from n = 9 cases. C9- ALS n = 27 cells from n = 3 cases. C9+ ALS n = 38 cells from n = 3 cases. ****p < 0.0001, **p < 0.01, *p < 0.05 One-way ANOVA, Tukey’s multiple comparisons test, F(2,145) = 23.87. For all graphs, individual data points are shown with mean +/− standard deviation
Discussion
Herein, we identified that knockdown of Ref1 is a suppressor of toxicity of TDP-43 and TDP-43 co-expressed with Ataxin-2 in a fly-based, modifier screen of 107 RNA binding proteins containing RNA recognition motifs (RRMs). Suppression of TDP-43 toxicity was associated with downregulation of TDP-43 on both the RNA and protein levels in Ref1-depleted animals. Neither RNA nor protein expression from a control LacZ gene or an unrelated disease gene, Tau, were altered upon Ref1 RNAi. Additional investigation revealed that Ref1 depletion also suppressed toxicity of a related ALS/FTD mutation: the expanded G4C2 repeat (also [16]). Significant reductions in G4C2 RNA and concomitant reductions in toxic GR-dipeptide were seen upon Ref1 downregulation in G4C2 expressing animals. Intriguingly, expression of TDP-43 or expanded (G4C2)49 disease transgenes within the adult fly nervous system were associated with an upregulation of endogenous Ref1 RNA, suggesting a feed-forward mechanism may be occurring. Consistent with these results, previous work on TDP-43 expressing human cells also reported ALYREF upregulation in the cytoplasmic fraction [35]. Importantly, our data indicate that ALYREF is upregulated at the protein level in motor neurons of ALS patients, with patients bearing the expanded G4C2 repeat mutation showing significantly higher ALYREF protein levels (see Fig. 5).
ALYREF is involved in several related pathways that may lead to suppression of TDP-43 and G4C2 toxicity when depleted. As a member of the TREX complex, ALYREF is best known for its function in mediating nucleocytoplasmic transport of mRNAs [20, 23, 48, 63]. Within this complex, ALYREF serves as an adaptor protein between mRNA and export factors NXF1/p15. Importantly, disruptions in nucleocytoplasmic transport may be a mechanism of both TDP-43- and G4C2-associated disease [8, 16, 43]. In contrast to its role in transport, ALYREF has also been reported to mediate RNAPII-driven transcription because it interacts with a number of transcription factors and depletion of ALYREF can reduce RNAPII occupancy for a subset of genes [20, 23, 49]. Our findings further highlight transcription as an additional mechanism underlying the role of Ref1/ALYREF in disease, as we see downregulation of the TDP-43 and G4C2 repeat transgene mRNA levels (see Figs. 2, 3).
Interestingly, ALYREF can bind both sense G4C2 and antisense G2C4 RNA [10, 11, 19, 30], suggesting that an interaction between ALYREF/Ref1 and G4C2 can be direct. For TDP-43, we present the first evidence that ALYREF also modulates TDP-43-associated toxicity. Further investigation into whether TDP-43 RNA interacts with ALYREF protein are needed to determine whether this may also be by direct binding. Overall, our data suggest that there are commonalities in the ability of ALYREF to modify expression of these two disease genes over a non-disease transcript. While it is clear that ALYREF is selective to specific transcripts [17, 24, 33, 49], what defines an ALYREF interacting gene is currently unknown. Only recently have there been studies that shed light on underlying mechanisms, defining ALYREF as an m5C reader [57] and potential ALYREF binding motifs [46].
ALYREF may serve as a unique therapeutic target in ALS as its depletion was able to suppress both TDP-43- and G4C2-induced toxicity. Further investigations into the role of ALYREF in global transcription, global mRNA export, and effects on disease-associated pathways are needed to define it as a potential therapeutic target [24, 53, 55]. Previous work showed that ALYREF is not essential for bulk mRNA export from the nucleus in Drosophila and C. elegans [17, 33] and only a subset of mRNAs are affected when ALYREF is depleted in human cells [41, 49]. Importantly, our data indicate that ALYREF is upregulated in motor neurons of ALS patients, with patients bearing the expanded G4C2 repeat mutation showing significantly higher ALYREF levels (see Fig. 5). Overall, these data support previous findings that there may be overlapping mechanisms underlying these related disease etiologies [7, 12, 29, 44]. Interesting to C9+ disease, ALYREF has been reported to interact with Iws1 – a transcription factor that binds SPT4/5 RNAPII-elongation factors [31, 58]. SPT4/5 has recently been identified as unique transcriptional regulators of expanded G4C2 [26], suggesting that ALYREF is positioned to be a protein that may couple G4C2 transcription to nuclear export machinery. TREX proteins (including ALYREF) were also found to interact with Matrin 3 [3], another RBP that is mutated in ALS [22], suggesting that ALYREF may play a role in multiple types of ALS/FTD.
Despite recent advances in our understanding of the molecular mechanisms underlying ALS/FTD, there is an urgent and unmet need to develop effective therapeutics. Our results identify ALYREF as a potential novel target that is increased in ALS motor neurons, and whose downregulation may suppress the toxicity of multiple ALS and FTD associated genes.
Material and methods
Drosophila stocks and crosses
Flies were grown on standard cornmeal molasses agar with dry yeast. Stock lines were maintained at 18 °C. Transgenic lines used in this study were: UAS-TDP-43/CyO; GMR-GAL4 (YH3)/TM6B. UAS-TDP-43(37 M), UAS-hATXN2-32Q (F26)/CyO; GMR-GAL4 (YH3)/TM6B and GMR-GAL4 (YH3)/TM3, Sb [25]. UAS-(G4C2)49, GMR-GAL4 (YH3)/TM6, Sb [18, 26, 37]. UAS-LDS-(G4C2)4,42,44[GR-GFP] [18]. UAS-TauWT [56]. RNAi lines from the Transgenic RNAi Project (TRiP) and mutant lines were obtained from the Bloomington Drosophila Stock Center. Additional RNAi lines were obtained from the Vienna Drosophila Resource Center. Complete list of RNAi lines used in this study is found in Additional file 4: Table S3.
For the genetic screen, virgin female flies were selected from each disease model line or from driver-only lines and were crossed to males harboring RNAi transgenes at 25 °C, under normal light/dark cycles. Male progeny of the appropriate genotypes were selected, aged to 3-5d at 25 °C. For external eye imaging, flies were anesthetized with ether for 10 min, placed on a glass slide and imaged with Leica Z16 APO. For internal eye morphology, male flies from the same crosses were fixed in Bouin’s solution (Sigma-Aldrich) for 120 h, embedded in paraffin, sectioned on a Leica RM2255 microtome and mounted on SuperFrost Plus slides (Fischer Scientific). Slides were dried overnight at room temperature, baked for 1 h at 56 °C, and paraffin was removed with Histoclear (National Diagnostics). Slides were mounted with coverslips using Cytoseal XYL (Thermo Scientific) and imaged using an upright Leica fluorescent microscope.
Western immunoblots
20 fly heads were homogenized in 50 μl LDS sample buffer (Invitrogen) including 5% beta-mercaptoethanol (Sigma-Aldrich). Samples were boiled at 95 °C for 5 min and centrifuged at 15,000 g for 5 min at 4 °C. The supernatant was collected and stored at − 20 °C until loaded on 4–12% Bis-Tris NuPAGE gels (Invitrogen) using 5 μl of sample per well. Gel electrophoresis was performed at 140 V for 70 min and the gels were blotted on a PVDF membrane using XCell II (Invitrogen) at 30 V for 1 h. Membranes were blocked in 3% bovine serum albumin in tris buffered saline with 0.1% Tween20 (TBST) for 30 min and incubated with primary antibodies in blocking buffer over-night at 4 °C. Following washes in TBST, membranes were incubated with HRP-conjugated secondary antibodies (Jackson Immunoresearch) at 1:10,000 for 2 h, washed and the luminescent signal was developed using ECL prime (Amersham) and detected with Amersham Imager 600. Primary antibodies: anti β-tubulin (CAT#E7, DSHB, 1:500), anti β-Galactosidase (CAT#Z378A, Promega, 1:2000), anti-TDP-43 (CAT#10782, Proteintech, 1:1000), anti Tau (CAT#A0024, Dako, 1:1000).
Real-time PCR
RNA was extracted using Trizol Reagent (ThermoFisher Scientific), according to the manufacturer’s instructions. RNA concentration was determined using Nanodrop (Nanodrop) and RNA quality was assessed using 1% agarose gel-electrophoresis. 400 ng RNA was used per reverse-transcription reaction using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) in a 20 μl total reaction volume using random primers. cDNA was then used as template for real-time qPCR using Fast SYBR Green Master Mix (Applied Biosystems). Real-time PCR was performed on the Applied Biosystems ViiA7 machine using 384-well format in technical duplicates. For each primer set, a serial dilution curve validated primer efficiency. Melting curve analysis confirmed the existence of one amplicon. Primers were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/).
Primer sequences are listed in Additional file 5: Table S4.
Human tissue – immunofluorescence
For immuno-fluorescence, Superfrost slides (Fisher Scientific) with paraffin sections were deparaffinized in xylene, 100, 90 and 70% ethanol. Following a brief rinse in water, antigen retrieval was performed by boiling the samples for 10 min in citric buffer pH 6 (10 mM citric acid, pH 6). Slides were cooled to room temperature, rinsed with water and incubated in blocking buffer for 20 min (Tris buffered saline (TBS) with 2% bovine serum). Sections were circled with a liquid blocker PAP pen (Daido Sangyo) and primary antibodies in blocking buffer were incubated overnight at 4 °C. Following 3 washes in TBS, slides were blocked for 5 min and incubated with secondary antibodies for 2 h at room-temperature. Slides were washed 3 times in TBS. To quench autofluorescence, slides were washed for 1 min in 70% ethanol, incubated for 1 min in Sudan Black (0.3% in 70% ethanol), and washed 3 times in 70% ethanol. Final TBS wash was followed by 1 μg/ml DAPI to stain DNA, slides were rinsed in water, mounted with anti-fade mounting media (20 mM Tris pH 8.0, 0.5% N-propyl gallate, 80% glycerol), and sealed with clear nail polish. Quantification of fluorescent signal was performed with Fiji [45]. Imaging and quantification were performed blinded to disease status. Primary antibodies were used at 1:200 dilution: anti-ALYREF (mouse monoclonal, CAT# ab6141, abcam), anti-ALYREF (rabbit polyclonal, CAT# ab202894, abcam). Secondary antibodies were used at 1:200 dilution: anti-Rabbit IgG Alexa Fluor 594 (#A-11012, 1:200, Invitrogen). Details of human samples are described in Additional file 3: Table S2. Donor spinal cord samples following neuropathological evaluation were selected from the brain bank at the Center for Neurodegenerative Disease Research at the University of Pennsylvania [51]. Phosphorylated TDP-43 deposits were evaluated using the pS409/410 antibody (mAb, 1:500) [40]. Controls were defined as subjects who were cognitively normal and did not meet the threshold for a neurodegenerative or vascular dementia diagnosis during the neuropathological examination. Informed consent for autopsy was obtained for all patients from their next of kin.
Statistical analysis
Statistical analysis was performed using Prism (Version 6, GraphPad). One-way ANOVA with Dunnett’s or Tukey’s multiple comparisons test or two tailed Student’s t-test were used as appropriate, with significance level set at 0.05.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- DPR:
-
Dipeptide repeats
- FTD:
-
Frontotemporal Dementia
- G4C2:
-
Expanded (GGGGCC)30+ mutation found within C9orf72
- GFP:
-
Green fluorescent protein
- qRT-PCR:
-
Quantitative realtime polymerase chain reaction
- RAN-translation:
-
Repeat Associated Non-AUG translation
- RBP:
-
RNA binding protein
- Ref1:
-
RNA and export factor binding protein 1; fly orthologue to human ALYREF
- RNP:
-
Ribonucleoprotein
- RRM:
-
RNA recognition motif
- TDP-43:
-
TAR DNA-binding protein 43
- TREX:
-
TRanscription and EXport
References
Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin W-L, DeJesus-Hernandez M et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 77:639–646
Barker HV, Niblock M, Lee Y-B, Shaw CE, Gallo J-M (2017) RNA misprocessing in C9orf72-linked neurodegeneration. Front Cell Neurosci 11:195
Boehringer A, Garcia-Mansfield K, Singh G, Bakkar N, Pirrotte P, Bowser R (2017) ALS associated mutations in Matrin 3 Alter protein-protein interactions and impede mRNA nuclear export. Sci Rep 7:14529
Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415
Bruhn L, Munnerlyn A, Grosschedl R (1997) ALY, a context-dependent coactivator of LEF-1 and AML-1, is required for TCRalpha enhancer function. Genes Dev 11:640–653
Burguete AS, Almeida S, Gao F-B, Kalb R, Akins MR, Bonini NM (2015) GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife 4:e08881
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang Y-J, Castanedes-Casey M et al (2015) C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 348:1151–1154
Chou C-C, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F et al (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21:228
Conlon EG, Manley JL (2017) RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev 31:1509–1528
Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB et al (2015) Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol 130:63–75
Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D et al (2014) Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 137:2040–2051
Davidson YS, Barker H, Robinson AC, Thompson JC, Harris J, Troakes C et al (2014) Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 2:70
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked frontotemporal dementia and amyotrophic lateral sclerosis. Neuron. 72:245–256
Edbauer D, Haass C (2016) An amyloid-like cascade hypothesis for C9orf72 ALS/FTD. Curr Opin Neurobiol 36:99–106
Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 466:1069–1075
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133
Gatfield D, Izaurralde E (2002) REF1/Aly and the additional exon junction complex proteins are dispensable for nuclear mRNA export. J Cell Biol 159:579–588
Goodman LD, Prudencio M, Kramer NJ, Martinez-Ramirez LF, Srinivasan AR, Lan M, et al. (2019) Expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nature Neuroscience, in press.
Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez-Martinez A, Cooper-Knock J, Higginbottom A et al (2017) SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8:16063
Heath CG, Viphakone N, Wilson SA (2016) The role of TREX in gene expression and disease. Biochem J 473:2911–2935
Ito D, Hatano M, Suzuki N (2017) RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci Transl Med 9:eaah5436
Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nature Neuroscience 17:664
Katahira J (2012) mRNA export and the TREX complex. Biochim Biophys Acta 1819:507–513
Katahira J, Inoue H, Hurt E, Yoneda Y (2009) Adaptor Aly and co-adaptor Thoc5 function in the tap-p15-mediated nuclear export of HSP70 mRNA. EMBO J 28:556–567
Kim H-J, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ et al (2014) Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nature Genetics 46:152–160
Kramer NJ, Carlomagno Y, Zhang Y-J, Almeida S, Cook CN, Gendron TF et al (2016) Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 353:708–712
Kumar V, Hasan GM, Hassan MI (2017) Unraveling the role of RNA mediated toxicity of C9orf72 repeats in C9-FTD/ALS. Front Neurosci 11:711
Lee EB, Lee VM-Y, Trojanowski JQ (2011) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13:38–50
Lee Y-B, Baskaran P, Gomez-Deza J, Chen H-J, Nishimura AL, Smith BN et al (2017) C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet 26:4765–4777
Lee Y-B, Chen H-J, Peres JN, Gomez-Deza J, Attig J, Štalekar M et al (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5:1178–1186
Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA et al (2003) Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol 23:1368–1378
Liscic RM (2017) ALS and FTD: insights into the disease mechanisms and therapeutic targets. Eur J Pharmacol 817:2–6
Longman D, IL J, JF C (2003) The ref/Aly proteins are dispensable for mRNA export and development in Caenorhabditis elegans. RNA. 9:881–891
Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS et al (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 1:68
Mihevc SP, Baralle M, Buratti E, Rogelj B (2016) TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci Rep 6:33996
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 345:1192–1194
Mordes DA, Prudencio M, Goodman LD, Klim JR, Moccia R, Limone F et al (2018) Dipeptide repeat proteins activate a heat shock response found in C9ORF72-ALS/FTLD patients. Acta Neuropathol Commun 6:55
Mori K, Weng S-M, Arzberger T, May S, Rentzsch K, Kremmer E et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 339:1335–1338
Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM (2014) Amyotrophic lateral sclerosis risk for spinocerebellar Ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol 71:1529–1534
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Osinalde N, Olea M, Mitxelena J, Aloria K, Rodriguez JA, Fullaondo A et al (2013) The nuclear protein ALY binds to and modulates the activity of transcription factor E2F2. Mol Cell Proteomics 12:1087–1098
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 72:257–268
Rossi S, Serrano A, Gerbino V, Giorgi A, Francesco LD, Nencini M et al (2015) Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci 128:1787–1799
Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D et al (2018) Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol 135:459–474
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al (2012) Fiji: an open-source platform for biological-image analysis. Nature Methods 9:676–682
Shi M, Zhang H, Wu X, He Z, Wang L, Yin S et al (2017) ALYREF mainly binds to the 5′ and the 3′ regions of the mRNA in vivo. Nucleic Acids Res 45:9640–9653
Strang KH, Golde TE, Giasson BI (2019) MAPT mutations, tauopathy, and mechanisms of neurodegeneration. https://doi.org/10.1038/s41374-019-0197-x
Sträßer K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, et al (2002) TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417:304
Stubbs SH, Conrad NK (2015) Depletion of REF/Aly alters gene expression and reduces RNA polymerase II occupancy. Nucleic Acids Res 43:504–519
Taylor JP, Brown RH, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature. 539:197–206
Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL et al (2014) A platform for discovery: the University of Pennsylvania integrated neurodegenerative disease biobank. Alzheimer’s & Dementia 10:477–484.e1
Virbasius C-MA, Wagner S, Green MR (1999) A human nuclear-localized chaperone that regulates dimerization, DNA binding, and transcriptional activity of bZIP proteins. Mol Cell 4:219–228
Walker C, El-Khamisy SF (2018) Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain. 141:1247–1262
Wickramasinghe VO, Laskey RA (2015) Control of mammalian gene expression by selective mRNA export. Nat Rev Mol Cell Biol 16:431–442
Wickramasinghe VO, Savill JM, Chavali S, Jonsdottir AB, Rajendra E, Grüner T et al (2013) Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell 51:737–750
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M et al (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714
Yang X, Yang Y, Sun B-F, Chen Y-S, Xu J-W, Lai W-Y et al (2017) 5-methylcytosine promotes mRNA export — NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res 27:606
Yoh SM, Cho H, Pickle L, Evans RM, Jones KA (2007) The Spt6 SH2 domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicing and export. Genes Dev 21:160–174
Zhang K, Coyne AN, Lloyd TE (2018) Drosophila models of amyotrophic lateral sclerosis with defects in RNA metabolism. Brain Res 1693:109–120
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61
Zhang Y-J, Gendron TF, MTW E, O’Raw AD, Yue M, Jansen-West K et al (2018) Poly (GR) impairs protein translation and stress granule dynamics in C9orf72 -associated frontotemporal dementia and amyotrophic lateral sclerosis. Nature Medicine 24:1136–1142
Zhao M, Kim JR, van Bruggen R, Park J (2018) RNA-binding proteins in amyotrophic lateral sclerosis. Mol Cell 41:818–829
Zhou Z, Luo M, Straesser K, Katahira J, Hurt E, Reed R (2000) The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature 407:401–405
Acknowledgements
We thank John Robinson from the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania for help selecting and analyzing human tissue samples, and members of the Bonini laboratory for ideas and comments. This work was funded by a NIH Ruth L. Kirschstein NRSA postdoctoral fellowship (F32-NS084667-03) (to AB), an NIH training grant on Systems and Integrative Biology (T32-GM07517) (to LDG) and a NIH Maximizing Investigators’ Research Award (R35-NS097275) (to NMB).
Author information
Authors and Affiliations
Contributions
AB and NMB initiated the project. AB, LDG and NMB designed the project. NMB supervised and advised on the project. AB, LDG, AS, CGO, and JA performed studies and analyzed the data. VM-YL and JQT contributed reagents and materials. AB, LDG and NMB wrote and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. RBP screen details. (PDF 162 kb)
Additional file 2:
Figure S1. Extended data characterizing Ref1 RNAi. Figure S2. Uncropped western blot images. (DOCX 977 kb)
Additional file 3:
Table S2. Patient information. (PDF 56 kb)
Additional file 4:
Table S3. Drosophila RNAi lines. (PDF 98 kb)
Additional file 5:
Table S4. Primer sequences. (PDF 57 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Berson, A., Goodman, L.D., Sartoris, A.N. et al. Drosophila Ref1/ALYREF regulates transcription and toxicity associated with ALS/FTD disease etiologies. acta neuropathol commun 7, 65 (2019). https://doi.org/10.1186/s40478-019-0710-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0710-x