
Abstract
Mitochondria play a key role in common neurodegenerative diseases and contain their own genome: mtDNA. Common inherited polymorphic variants of mtDNA have been associated with several neurodegenerative diseases, and somatic deletions of mtDNA have been found in affected brain regions. However, there are conflicting reports describing the role of rare inherited variants and somatic point mutations in neurodegenerative disorders, and recent evidence also implicates mtDNA levels. To address these issues we studied 1363 post mortem human brains with a histopathological diagnosis of Parkinson’s disease (PD), Alzheimer’s disease (AD), Frontotemporal dementia – Amyotrophic Lateral Sclerosis (FTD-ALS), Creutzfeldt Jacob disease (CJD), and healthy controls. We obtained high-depth whole mitochondrial genome sequences using off target reads from whole exome sequencing to determine the association of mtDNA variation with the development and progression of disease, and to better understand the development of mtDNA mutations and copy number in the aging brain. With this approach, we found a surprisingly high frequency of heteroplasmic mtDNA variants in 32.3% of subjects. However, we found no evidence of an association between rare inherited variants of mtDNA or mtDNA heteroplasmy and disease. In contrast, we observed a reduction in the amount of mtDNA copy in both AD and CJD. Based on these findings, single nucleotide variants of mtDNA are unlikely to play a major role in the pathogenesis of these neurodegenerative diseases, but mtDNA levels merit further investigation.
Similar content being viewed by others


Introduction
Mitochondria are critical intracellular organelles involved in calcium signaling, lipid biosynthesis, apoptosis [31], and the generation of adenosine triphosphate (ATP) via the mitochondrial respiratory chain [28]. Over the last decade it has become clear that mitochondria play a key role in the pathogenesis of common neurodegenerative disorders. Mitochondria contain their own 16.5 kb circular mitochondria genome (mtDNA), which codes for key components of the mitochondrial proteome. MtDNA is present in 10s-1000s of copies in each cell and undergoes lifelong replication in post-mitotic cells including neurons [10]. With rudimentary mechanisms for repair, mtDNA is vulnerable to mutations, which accumulate within cells and also within the germ line.
There is emerging evidence that genetic variation of mtDNA contributes to the pathogenesis of neurodegenerative disorders [14]. Common population genetic variants divide the human population into a geographically-defined ‘haplogroups’ [30], which have been associated with several neurodegenerative disorders including Alzheimer’s disease (AD) and Parkinson’s disease (PD), conferring a small increase in disease risk [13, 22]. Furthermore, high-density genotyping arrays provide preliminary evidence that rare (minor allele frequency, MAF <5%) mtDNA polymorphisms are also associated with neurodegenerative disease, supporting previous work on AD [12].
In addition to maternally inherited polymorphisms, acquired somatic mutations of mtDNA have also been associated with neurodegenerative disorders. Unlike the maternally inherited germ line variants (which are ‘homoplasmic’), the somatic mutations are usually present alongside the original wild-type molecules (heteroplasmy), and the proportion of mutated alleles determines whether a biochemical defect is manifest at the cellular level. MtDNA deletions accumulate in the ageing brain, reaching higher levels in regions vulnerable to neurodegeneration [3, 26], but evidence describing the accumulation of somatic point mutations or small insertion-deletion mutations (indels) is less compelling, with conflicting reports in the literature [6, 20]. Finally, several recent studies have described abnormal amounts of mtDNA both in cerebrospinal fluid or the brains of patients with neurodegenerative diseases [23, 24], but only in a limited number of individuals.
To provide definitive evidence in all three areas, we studied the entire mtDNA sequence and quantity in1363 post mortem brains with Alzheimer’s disease (AD), Amyotrophic-frontotemporal dementia (ALS-FTD), Creutzfeldt-Jakob Disease (CJD), Parkinson’s disease (PD) and Dementia with Lewy Bodies (DLB), and compared them to healthy age-matched control brains. We found no evidence that rare inherited polymorphisms or mtDNA heteroplasmy contributes to the pathogenesis of neurodegenerative diseases, although differences in mtDNA content provide a clue to disease mechanism in AD and CJD.
Materials and methods
Study samples
Full mitochondrial genome sequence data was extracted from Exome Sequencing data of 1363 case or control brain tissue samples from the Medical Research Council Brain Tissue Resource [15]. Cases fulfilling both ante-mortem and post-mortem diagnostic criteria for major neurodegenerative disease were included (Table 1, Additional file 1: Methods, Additional file 1: Table S1). DNA was extracted from the cerebellum in 87.3% of cases (n = 1190), cerebral cortex in 6.5% of cases (n = 89) with other brain regions in 6.16% (n = 84).
Mitochondrial sequences extraction and variant calling
Exome sequencing was performed using Nextera 62 Mb Rapid Capture Expended Exome kits on the Illumina HiSeq 2000 with 100 bp paired-end reads. Sequence data were aligned to the human reference sequence (UCSC hg19) (http://genome.ucsc.edu) using Burrows-Wheeler Aligner (BWA) v0.7.6 [17], formatted using Samtools v0.1.19 [18], and duplicates removed using Picard v1.707 (http://picard.sourceforge.net).
MToolBox [4] recovered mtDNA from off-target sequences, remapped onto the revised Cambridge Reference Sequence [2], and discarded those mapping to regions of (GRCh37/hg19), and considered Nuclear mitochondrial Sequences [29].
Known insertions or deletions (ins/dels) were defined, and all variants scored in HmtDB [27] and MITOMAP [28]. Remaining reads reconstructed the mitochondrial genome. Nucleotide mismatches and ins/del with quality scores (QS ≥25) and read depth (rd ≥5) were included.
Determining heteroplasmy and homoplasmy
We determined the proportion of variant alleles at each site of the mitochondrial genome. We then calculated the heteroplasmic fraction (HF, %) by dividing the number of variant reads by the total number of reads (for SNVs and deletions) or of the total number of 5′ flanking reads (for insertions). If the HF was <10% or >90%, we conservatively considered the variant site to be homoplasmic. If the HF was between 10 and 90%, the site was considered to be heteroplasmic, and the HF was studied further.
Defining mtDNA haplogroups
Haplogroup assignment was performed using the mt-classifier tool [32], and a maximum likelihood (ML) phylogenetic tree was created using the PHYLIP software (v3.696) from sequence alignments built by Clustal Omega (v1.2.0) (http://www.clustal.org/omega). Defined haplogroups were compared to the haplogroups genotyped in 2360 individuals from the 1958 Birth Cohort (WTCCC58C) as previously determined by Hudson et al. [11].
Defining rare variants
Minor allele frequencies for each base of the mitochondrial genome were calculated from 30,506 full-length mitochondrial sequences in NCBI-GenBank using custom Python scripts. Rare homoplasmic variations were defined as those alleles present in less than 5% of individuals within their haplogroup using MITOMASTER [21], and novel variants those not present in the NCBI-GenBank dataset, 1000 genomes [9], MITOMAP [28] or HmDB [27].
Functional prediction
Pathogenicity scores for all possible non-haplogroup defining SNVs were calculated using MutPred [19] and Polyphen-2 [1].
MtDNA copy number estimation
The relative amount of mtDNA in each brain (referred to as mtDNA copy number) was calculated as the ratio between the mean mtDNA read depth and the mean exome read depth as previously described [5].
Statistical analysis
Mean variant counts or fractions both within and between groups were performed using Mann-Whitney, Fisher’s exact, or Spearman’s rank test tests as appropriate and as defined at the uncorrected threshold. A Poisson loglinear model was used to test the association between the number of variants and age among individuals within each main group. All statistical analyses were performed using R (v3.0.2) (http://CRAN.R-project.org/doc/FAQ/R-FAQ.html), and plots made using Python.
Results
Coverage and quality of assembled mitochondrial genomes
Complete assembly of the mitochondrial genome was obtained in all cases, with a mean read depth across the genome of 289 (SD = 169.8) (range 66–1328) and a mean base quality score of 36.8 (sd = 0.25) (range 36.0–37.5) (Additional file 1: Figures S1 and S2). There was no difference in mean read depth or mean base quality score for any group vs controls (Additional file 1: Figure S3).
Haplogroup associations
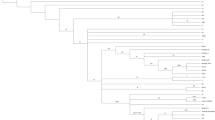
Haplogroups and phylogenetic relationships were determined for all 1363 samples (Fig. 1). There was no difference in major overall haplogroup frequency when compared to 2360 UK population controls (Additional file 1: Table S2), confirming the accuracy of haplogroup calling and that the cohort as representative of the UK population. We saw no association between any disease cohort and specific haplogroups in our study (Additional file 1: Table S3).

Phylogenetic tree of the 1363 mtDNA sequenced derived from the MRC Brain Tissue Resource. Major haplogroups are shown
Homoplasmic variants
One thousand, nine hundred twenty-three homoplasmic variants were detected within the cohort, with a mean of 22.9 (sd = 10.8) variants per sample. Four hundred sixty-seven variants were defined as ‘common’ (minor population Allele Frequency, MpAF >0.05 within their haplogroup), and included known haplogroup defining variants. One thousand, four hundred fifty-six homoplasmic variants were defined as rare (MpAF <0.05 within their haplogroup). Twenty-five of the rare variants were novel, not seen in the NCBI database (n = 30,506), MITOMAP (n = 30,589) or the 1000 genomes database. Four of the novel variants were in non-coding regions, two were in rRNA genes, and 19 were synonymous (Additional file 1: Table S4).
Association with disease – common homoplasmic variants
Here we considered all homoplasmic variants, and a subgroup analysis of all non-synonymous homoplasmic variants. We saw no evidence of a disease association with any single variant, the burden of homoplasmic variants in any gene, nor the burden of homoplasmic variants in groups of genes forming a respiratory chain complex (Additional file 1: Figures S4–S8, Additional file 1: Table S5). However, when stratifying by age, there was a trend towards young onset AD cases (age of death <60) having a greater number of total variants in MT-TR compared to controls (6/13 vs 9/139) (p = 0.002) (Additional file 1: Figure S5).
Association with disease - rare homoplasmic variants
No single rare homoplasmic variant was present at greater frequency in any disease compared to controls (Additional file 1: Figure S8). There was a trend towards a greater number of rare homoplasmic point mutations in two genes in AD compared to controls; MT-RNR1 (AD; 30/282 (10.6%), Controls; 16/344 (4.7%)) (p = 0.005)) and again MT-TR (AD; 6/282 (2.1%), Controls; 0/344) (p = 0.008), although both failed to reach significance at the corrected threshold of p = 0.0014. (Additional file 1: Figure S8, Additional file 1: Table S5). When stratified by age, this suggested that the trend towards an excess burden of rare homoplasmic variants in MT-RNR1 was likely driven by variants in young onset AD cases vs controls (AD: 9/53 (16.9%), Controls (2/65), p = 0.012 (3%) (Additional file 1: Figure S9). We also saw that young onset PD-DLB cases (death aged <70) had a significantly greater number of rare homoplastic mutations in MT-CO2 (PD-DLB: 5/23 (21.7%), Controls: 5/213 (2.3%), p = 0.0010 (Additional file 1: Figure S9). The majority of the variants in MT-CO2 in both cohorts were in non-coding D-loop, but when combined this did not reach the corrected threshold for significance. There was no association between any rare non-synonymous variant, nor the burden of rare non-synonymous variants in any gene or respiratory chain complex in any disease group vs controls.
Heteroplasmic variants
Three hundred eleven heteroplasmic variants (>10% MAF) were detected (mean HF = 7%, sd = 1.0), in 440 cases, with 10 of these variants entirely novel. 55.7% of all heteroplasmic variants occurred within the D-loop, 33.3% in coding regions, 4.8% in rRNA genes and 6.2% in tRNA genes (Fig. 2).

Circos plot summarizing all of the genetic data from 1363 sequences derived form the MRC Brain Tissue Resource. From outside the circle to inside: (1) mtDNA position, (2) mtDNA genes, (3) mtDNA Complex, (4) frequency of variants in 40,440 mitochondrial sequences in NCBI-GenBank, (5) mean read depth of 1363 samples per base, (6) Total variants in all 1363 samples [circles], (7) Total Rare variants in 1363 samples [triangles], (8) Total novel variants in 1363 samples [squares]. Colour code for circles (6) – (8): Red - AD, green - ALS-FTD, blue - CJD, yellow - DLB-PD, grey - others; from inner to outer, HF increasing. Key – AD –Alzheimer’s disease, CJD – Creutzfeldt Jacob Disease, DLB-PD – Dementia with Lewy Bodies or Parkinson’s disease, FTD-ALS – Frontotemporal Dementia or Amyotrophic Lateral Sclerosis
There was no association between any disease group and controls for any single heteroplasmic variant, the total number of heteroplasmic variants, or the mean variant pathogenicity score. There was also no association between the number of non-synonymous heterozygous variants in any gene or complex and any disease group compared to controls (Additional file 1: Tables S6–S8, Additional file 1: Figure S12).
Heteroplasmy and age
We subsequently used a Poison loglinear model to determine the relationship between heteroplasmy and age within each group. There was no age correlation with the total number of heteroplasmic variants, mean level of heteroplasmy (HF), nor the mean variant pathogenicity score in any disease group (Fig. 3).

The distribution and nature of major heteroplasmic mtDNA variation within 1363 cases from the MRC Brain Bank Tissue Resource. a Top left – The mean number of heteroplasmic variants per sample in each cohort. b Top right - The distribution of heteroplasmic fraction (ratio of mutant to wild-type allele) for heteroplasmic variants for each disease cohort. c Bottom left – The mean number of heteroplasmic point mutations by age for each disease cohort. d Bottom right – The mean heteroplasmic fraction by age for each disease cohort. Key – AD –Alzheimer’s disease, CJD – Creutzfeldt Jacob Disease, DLB-PD – Dementia with Lewy Bodies or Parkinson’s disease, FTD-ALS – Frontotemporal Dementia or Amyotrophic Lateral Sclerosis
mtDNA number
mtDNA copy number was significant lower in AD and CJD compared to controls (p = 2.85 × 10−7 (AD), p = 3.34 × 10−7 (CJD)), and we observed a strongly positive correlation between age and mtDNA copy number in CJD (p = 2.7 × 10−11). No association with age was seen in other groups (Fig. 4). The frequency of cerebellar samples in the AD cohort was no different to controls (p = 0.64) or the FTD-ALS cohort (p = 0.87). However, the CJD cohort did show a greater proportion of cases from the cerebellum compared to all other cohorts (100%, p < 0.001 vs all other groups).

The relative mtDNA copy number in each disease cohort. a Top – Relative copy number of each cohort calculated as the ratio between the mean mtDNA read depth and the mean exome read depth as previously described [5]. ** (P < 0.01). b Bottom - The association between relative mtDNA copy number and age for all CJD cases (n = 182) with the Spearman Rank ρ and p-value shown. Key – AD –Alzheimer’s disease, CJD – Creutzfeldt Jacob Disease, DLB-PD – Dementia with Lewy Bodies or Parkinson’s disease, FTD-ALS – Frontotemporal Dementia or Amyotrophic Lateral Sclerosis
Discussion
Here we report a comprehensive study of mtDNA sequence variation and abundance in brain tissue from 1363 neuropathologically characterized post mortem samples from MRC Brain Tissue Resource. After correction for multiple significance testing, we saw no difference in the frequency of mtDNA haplogroups, no difference in the frequency of common or rare homoplasmic variants, and no difference in the presence or degree of mtDNA heteroplasmy between different disease groups and control subjects. Overall, we conclude that neither rare homoplasmic variants nor heteroplasmic variants play a substantial role in the pathogenesis of these disorders. However, further work is required to clarify whether mtDNA copy number is important for the pathogenesis of AD and CJD.
Our most interesting finding was the frequent occurrence of mtDNA heteroplasmy in human brain tissue (HF >10% found in 32.3% of cases and controls), but contrary to previous reports, this did not change with age, nor was it associated with disease. This suggests that, although ongoing replication errors in mtDNA occur with age [16, 25], moderate level heteroplasmic variants (>10%) are likely to have either occurred de novo in early development, or have been inherited within the germ-line, perhaps clonally expanding to these levels in early life. Resolving this issue will be difficult in human subjects, but will require the analysis of serial samples from the same tissue in the same subject. We cannot exclude the possibility that low level heteroplasmy (MAF <10%) is associated with these different disorders, or that different regions of the brain contain a significant burden of mtDNA variants, as recently described [8]. However, if low level heteroplasmic variants are important (perhaps because the low mean level masks high levels in individual neurons or glia), it is surprising that the higher levels of heteroplasmy we detected were not associated with disease, and that patient brains did not contain more pathogenic mutations than control subjects.
Finally we observed a significant reduction in mtDNA copy number in both Alzheimer’s disease and CJD brains. Our data support previous work in AD [7], but to our knowledge is the first study of mtDNA copy number in CJD brain tissue. It is possible that the CJD finding reflects the higher proportion of cerebellar samples studied when compared to controls. In addition, the mtDNA copy number in the variant CJD (vCJD) cases (n = 40, mean mtDNA copy number = 4.14, sd = 2.96) was lower than all other CJD types (mean = 10.03, sd = 7.67; p = 2.8 × 10−6). Therefore, it is therefore also possible that the trend observed for CJD actually reflects unusually low levels of mtDNA in the brains from patients with vCJD, who died at a younger age than other forms of CJD.
Conclusion
When taken together, these findings support the role of mtDNA content in the pathogenesis of neurodegeneretive diseases, but with caveats. First, the abnormalities may be restricted to specific brain regions. Previous studies in PD showed a reduction in mtDNA copy number within the substantia nigra, but not frontal cortex, raising the possibility that the altered mtDNA levels are a consequence of the underlying pathology, and not the cause. This could be an indirect effect reflecting a change in the proportion of different cell types within a tissue homogenate that is linked to the neurodegeneration. Nonetheless, the observed association between mtDNA copy number and age in CJD brains supports a causal role which requires further investigation. Although this could reflect compensatory mitochondrial biogenesis in older subjects, an alternative explanation is that younger-onset cases have more aggressive pathology, with associated neuronal loss.
References
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–9
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23(2):147
Bender A, Krishnan KJ, Morris CM et al (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38(5):515–7
Calabrese C, Simone D, Diroma MA et al (2014) MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 30(21):3115–7
Castle JC, Biery M, Bouzek H et al (2010) DNA copy number, including telomeres and mitochondria, assayed using next-generation sequencing. BMC Genomics 11:244
Chinnery PF, Taylor GA, Howell N, Brown DT, Parsons TJ, Turnbull DM (2001) Point mutations of the mtDNA control region in normal and neurodegenerative human brains. Am J Hum Genet 68(2):529–32
Coskun PE, Beal MF, Wallace DC (2004) Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A 101(29):10726–31
Coxhead J, Kurzawa-Akanbi M, Hussain R, Pyle A, Chinnery P, Hudson G (2016) Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol Aging 38:217, e1-6
Diroma MA, Calabrese C, Simone D et al (2014) Extraction and annotation of human mitochondrial genomes from 1000 Genomes Whole Exome Sequencing data. BMC Genomics 15(Suppl 3):S2
Elson JL, Samuels DC, Turnbull DM, Chinnery PF (2001) Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet 68(3):802–6
Gleeson JG, Minnerath S, Kuzniecky RI et al (2000) Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am J Hum Genet 67(3):574–81
Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF (2014) Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet 10(5):e1004369
Hudson G, Nalls M, Evans JR et al (2013) Two-stage association study and meta-analysis of mitochondrial DNA variants in Parkinson disease. Neurology 80(22):2042–8
Keogh MJ, Chinnery PF (2015) Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta 1847(11):1401–11
Keogh MJ, Wei W, Wilson I, et al. Genetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource. Genome Res. 2016. In press.
Larsson NG (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79:683–706
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–60
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25(16):2078–9
Li B, Krishnan VG, Mort ME et al (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25(21):2744–50
Lin MT, Cantuti-Castelvetri I, Zheng K et al (2012) Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol 71(6):850–4
Lodato MA, Woodworth MB, Lee S et al (2015) Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350(6256):94–8
Maruszak A, Canter JA, Styczynska M, Zekanowski C, Barcikowska M (2009) Mitochondrial haplogroup H and Alzheimer’s disease--is there a connection? Neurobiol Aging 30(11):1749–55
Pyle A, Anugrha H, Kurzawa-Akanbi M, Yarnall A, Burn D, Hudson G (2016) Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging 38:216, e7-10
Pyle A, Brennan R, Kurzawa-Akanbi M et al (2015) Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann Neurol 78(6):1000–4
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414(6859):105–11
Ross JM, Stewart JB, Hagstrom E et al (2013) Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501(7467):412–5
Rubino F, Piredda R, Calabrese FM et al (2012) HmtDB, a genomic resource for mitochondrion-based human variability studies. Nucleic Acids Res 40(Database issue):D1150–9
Ruiz-Pesini E, Lott MT, Procaccio V et al (2007) An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res 35(Database issue):D823–8
Simone D, Calabrese FM, Lang M, Gasparre G, Attimonelli M (2011) The reference human nuclear mitochondrial sequences compilation validated and implemented on the UCSC genome browser. BMC Genomics 12:517
Torroni A, Richards M, Macaulay V et al (2000) mtDNA haplogroups and frequency patterns in Europe. Am J Hum Genet 66(3):1173–7
van der Giezen M, Tovar J (2005) Degenerate mitochondria. EMBO Rep 6(6):525–30
van Oven M, Kayser M (2009) Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30(2):E386–94
Acknowledgements
This work was funded by the UK Medical Research Council (13044). MJK is a Wellcome Trust Clinical Research Training Fellow. PFC is a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z), and a UK NIHR Senior Investigator, who receives support from the Medical Research Council Mitochondrial Biology Unit (MC_UP_1501/2), the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease (G0601943), EU FP7 TIRCON, and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. We thank Thahira Rahman and Mikyung Jang for assistance in initial the database management. We are grateful to Dr Mark Head, Ms Helen Yull and Ms Suzanne Lowrie for tissue sampling, DNA extraction and management of the CJD samples used in this study.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WW performed the primary bioinformatic analysis with MJK and HG, supervised by MSK. IW performed statistical analysis. MJK co-ordinated the study with JC, S Ryan, S Rollinson, M K-A, C-A McK, & CT. KT, MRT, JA, CS, SAS, CMM, and OA supervised the neuropathological and collection of the clinical data. SPB supervised the C9ORF72 repeat analysis. PFC and JI supervised the entire study study and obtained funding. MJK and PFC wrote the first draft manuscript which was modified following contributions from the other authors. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
The original version of this article was revised: Marzena Kurzawa-Akinibi has been updated to Marzena Kurzawa-Akanbi.
An erratum to this article is available at http://dx.doi.org/10.1186/s40478-017-0419-7.
Additional file
Additional file 1:
Supplementary Material. (DOCX 6597 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wei, W., Keogh, M.J., Wilson, I. et al. Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. acta neuropathol commun 5, 13 (2017). https://doi.org/10.1186/s40478-016-0404-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-016-0404-6