
Abstract
Primary central nervous system lymphoma (PCNSL) is an aggressive variant of diffuse large B-cell lymphoma (DLBCL) confined to the CNS. TP53 mutations (MUT-TP53) were investigated in the context of MIR34A/B/C- and DAPK promoter methylation status, and associated with clinical outcomes in PCNSL patients. In a total of 107 PCNSL patients clinical data were recorded, histopathology reassessed, and genetic and epigenetic aberrations of the p53-miR34-DAPK network studied. TP53 mutational status (exon 5–8), with structural classification of single nucleotide variations according to the IARC-TP53-Database, methylation status of MIR34A/B/C and DAPK, and p53-protein expression were assessed. The 57/107 (53.2 %) patients that were treated with combination chemotherapy +/− rituximab (CCT-treated) had a significantly better median overall survival (OS) (31.3 months) than patients treated with other regimens (high-dose methotrexate/whole brain radiation therapy, 6.0 months, or no therapy, 0.83 months), P < 0.0001. TP53 mutations were identified in 32/86 (37.2 %), among which 12 patients had hotspot/direct DNA contact mutations. CCT-treated patients with PCNSL harboring a hotspot/direct DNA contact MUT-TP53 (n = 9) had a significantly worse OS and progression free survival (PFS) compared to patients with non-hotspot/non-direct DNA contact MUT-TP53 or wild-type TP53 (median PFS 4.6 versus 18.2 or 45.7 months), P = 0.041 and P = 0.00076, respectively. Multivariate Cox regression analysis confirmed that hotspot/direct DNA contact MUT-TP53 was predictive of poor outcome in CCT-treated PCNSL patients, P = 0.012 and P = 0.008; HR: 1.86 and 1.95, for OS and PFS, respectively. MIR34A, MIR34B/C, and DAPK promoter methylation were detected in 53/93 (57.0 %), 80/84 (95.2 %), and 70/75 (93.3 %) of the PCNSL patients with no influence on survival. Combined MUT-TP53 and MIR34A methylation was associated with poor PFS (median 6.4 versus 38.0 months), P = 0.0070. This study suggests that disruption of the p53-pathway by MUT-TP53in hotspot/direct DNA contact codons is predictive of outcome in CCT-treated PCNSL patients, and concomitant MUT-TP53 and MIR34A methylation are associated with poor PFS.
Similar content being viewed by others


Introduction
Primary central nervous system lymphoma (PCNSL) of the diffuse large B-cell lymphoma (DLBCL) subtype is an aggressive variant of DLBCL confined to the CNS [44]. Patients with extra CNS DLBCL display diverse clinical courses, which may be explained by considerable heterogeneity of the clinical, morphological and cytogenetic phenotypes [29, 37, 40, 52]. PCNSL or CNS DLBCL has specific features that further distinguish this subtype from DLBCL [11, 44]. Despite extensive research, the clinical outcome of patients diagnosed with PCNSL is still dismal, a fact that cannot only be explained by the specific anatomical site or the difficulty of drug-delivery across the blood-brain-barrier [14, 15, 21, 30, 32, 36, 39, 54]. The pathogenic mechanisms driving this disease are still not clear, and only few genetic or epigenetic aberrations have been suggested as predictive or prognostic markers in PCNSL [1, 29, 31].
The TP53 tumor suppressor gene plays important roles in the regulation of cell proliferation, apoptosis and genomic integrity. Under stressful conditions, i.e. DNA damage, hypoxia, or oncogenic activation, the wild-type TP53 (WT-TP53) gene product p53 mediates cell-cycle arrest and preserves genomic stability via transcription-dependent activity (TA) and transcription-independent activity (TIA) in the cell. Reversely, the mutant p53 protein may be non-functional or have gain-of-function properties both leading to genomic instability and uncontrolled proliferation of damaged cells [27, 33, 34, 43, 51].
In DLBCL outside the CNS, TP53 mutations (MUT-TP53) have mostly been studied with emphasis on the disruption of the coding sequences of the DNA binding domain (DBD) (exons 5–8) with a reported mutation frequency of ~20–25 % [35, 52]. Structural classification and functional characteristics of MUT-TP53 assessed by yeast-based functional assays, available in the International Agency for Research on Cancer (IARC)-TP53-Database (http://www.iarc.fr), have been reported in DLBCL, with worse prognosis associated with mutations in the DBD of TP53 [35, 51, 52]. Indeed, the majority of cancer-related TP53 mutations occur in the region of the DBD, which contains the Loop (L1)-sheet-helix (LSH) motif, two large loops L2 and L3, the direct DNA contact areas including the zinc-binding site, and the hotspots [5, 9, 51, 52]. Few previous studies that report TP53 mutation status in PCNSL reveal incidences <10 % [10, 31], while, paradoxically, other investigations show higher incidences of p53 protein expression by immunhistochemistry (IHC) (29–60 %) as a surrogate marker of MUT-TP53 [3, 6].
The members of the miR34-family (A/B/C) have been recognized as tumor suppressors, which are implicated in a variety of cellular processes that control carcinogenesis in concert with p53 [13, 19]. The miR34s have been placed at the center of the cell-cycle and apoptosis regulation, and loss of miR34A has been associated with poor response to therapy [4, 20, 25]. We recently investigated the MIR34-family in a cohort of 150 primary DLBCL patients, and identified promoter methylation, and hence inactivation, of MIR34A in 28 % of the patients, and thereby showed that concomitant (”double hit”) TP53 mutation and MIR34A methylation were related to an exceedingly poor median survival of only 9.4 months [4, 20]. We also investigated the methylation status of DAPK (death-associated protein kinase), another player in the p53 signaling network in DLBCL, and showed that both overall- and allele specific DNA methylation correlate with poor outcome. Here, patients with concomitant TP53 mutation also had a tendency of poorer survival [22].
Since these studies suggest that genetic- and epigenetic disruption of the p53-miR34A-DAPK network may be associated with worse outcome in DLBCL patients, we speculated whether this pathway was also disrupted in CNS DLBCL. Thus, the purpose of the current study was to investigate and characterize the structural subsets of TP53 mutations, and to evaluate these data in the context of the MIR34 family- and DAPK promoter methylation status to the clinical outcome in a large cohort of patients with PCNSL of the DLBCL subtype.
Methods and materials
Study population and patient samples
All patients, consecutively diagnosed with PCNSL of the DLBCL subtype at our institution (University Hospital of Copenhagen, Rigshospitalet) during a period of 13 years (2001–2014), were included. A total of 107 histological specimens, which were eligible for further histopathological examination and DNA extraction for molecular analyses, were reclassified by the authors HDMP, HB, and ER (one training- and two specialist neuro- and hematopathologists) according to the WHO-classification [44]. In addition, data were extracted and critically reviewed from the patient files, the national Danish Pathology Database (Patobank), and the national Danish Lymphoma Database (LYFO).
The clinical data included date of diagnosis, age, sex, WHO performance status, serum lactate dehydrogenase (LDH), International Prognostic Index (IPI), 1st line treatment by chemo- and immunotherapy, whole brain radiotherapy (WBRT) including number of fractions and dose, adjuvant WBRT, response to treatment, relapse, and death or last follow-up date.
Immunohistochemistry
Assessment of protein expression by immunohistochemistry (IHC) was done by re-evaluating a panel of antibodies against: CD3, CD5, CD20, CD79A, CD10, BCL2, BCL6, MUM1, and MIB-1. A general cut-off level of 30 % was applied for all, except BCL2, where the cut-off was set at 70 % [16]. p53 protein expression was assessed by immunohistochemistry (IHC), see Additional file 1: Methods 1, and evaluated semiquantitatively. On-slide controls were used. No expression was defined as 0–9 %, and presence of expression was evaluated in intervals of 10–25 %, 25–50 %, 50–75 %, 75–100 %. In the survival analysis cut-off thresholds above 10 % were applied, as described by others [52, 53]. For the proliferation factor MIB-1 the percentage of stained nuclei was evaluated.
DNA extraction from FFPE tissue
From all samples, DNA was isolated from 4 to 8 10 μm FFPE (formalin-fixed paraffin-embedded) tissue sections using the RecoverAllTM Total Nucleic Acid Isolation Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturers’ guidelines. Paraffin was removed from the samples by Xylene incubation at 50 °C followed by ethanol wash. Proteins were degraded by digestion buffer and Proteinase-K digestion at 50°/80 °C. Subsequent isolation of DNA was performed by addition of isolation buffer and ethanol wash, followed by binding to a spin-column system, and RNA degradation by RNAse treatment. Total DNA was eluted in a 60 μL elution volume. The quantity (260 nm) and quality (260/280 ratio) of total DNA was measured by spectrophotometry on a NanoDrop-1000 spectrophotometer (Thermo Scientific, Delaware, USA).
Detection and structural classification of TP53 mutations
The coding sequences and splice sites of exons 5–8 of the TP53 gene were screened for mutations by PCR and denaturing gradient gel electrophoresis (DGGE) as described previously [4, 17, 52]. By covering exon 5–8 it is expected that ~95 % of TP53 mutations will be detected [51, 52]. Positive samples were subjected to Sanger sequencing [51]. All mutations were confirmed twice from DNA of the original sample.
Structural classification and nomenclature were applied using the IARC-TP53-Database (www.p53.iarc.fr) version R17 [35]. The types of TP53 mutation (missense, nonsense, deletion, insertion) and the sites of mutation (exon, codon, functional domain, conserved area, CpG-site relation, functional transactivation activity, and SIFT-class) were determined. For tumors with more than one mutation, the data for each mutation were recorded as separate entries in the absolute number of mutations, but analyzed as single events for survival. The most deleterious, i.e. functionally inactivating, mutations were selected as representative for survival analysis in these double mutated cases as in previously published studies [52]. Single nucleotide variations (SNVs) were scanned in the IARC-TP53-Database for detection of validated single nucleotide polymorphisms (SNPs) [35, 47], and excluded. As described thoroughly by Young et al. [52], we compared the survival of patients with MUT-TP53 within mutational subgroups or with the survival of WT-TP53 patients. Structural subsets, which were applied as factors in the survival analysis, included TP53 mutations located at hotspots in the DBD including codons involved in direct DNA contact, at the zinc-binding site, as well as frequent destabilizing mutations in β-sheets. In the analysis, TP53 mutations located in Loop-L2, Loop-L3, and LSH-motifs, conserved areas 2–5, and CpG-sites were also incorporated, as well as functional properties including p53 activity and SIFT-class, for details, see Additional file 1: Methods 2.
Bisulfite conversion
For initial bisulfite conversion, 250 ng (MIR34A/B/C) and 125 ng (DAPK) of DNA/sample were applied using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturers’ recommendations.
Methylation-specific melting curve analysis of MIR34A/B/C
The methylation status of the MIR34A and MIR34 B/C promoters was examined using methylation-specific melting curve analysis (MS-MCA); The amplification was carried out on a LightCycler® 480 instrument II (Roche Diagnostics) [4, 18, 24, 49]. The DNA sequences analyzed for promoter hypermethylation are located near the transcription start sites and in the promoter CpG-islands as previously described [4, 7, 24, 46]. The melting peaks were calculated using the LightCycler® 480 Software, Release 1.5.0SP3. Methylated DNA (Chemicon, Millipore, Billerica, MA, USA), unmethylated DNA (Qiagen), and a no template control (NTC) were included in all experiments.
DAPK methylation analysis using allelic MSP-pyrosequencing
The DAPK methylation specific PCR (MSP) primers target the antisense strand and amplify the region surrounding the rs13300553 SNP (A/G), as described previously [23, 24]. For primer sequences and detailed methods see [22, 23], and Additional file 1: Methods 3.
Statistics
The statistical software used for the analyses was SPSS version 22 for Windows (SPSS Inc. IBM, Armonk, NY, USA) and R version 3.2.1 for Mac OS X, Leopard (Apple, Cupertino, CA, USA). Analysis of categorical data and their in between associations was done by Fisher’s exact test, whereas t-test was used for continuous data. Survival was calculated based on survival status 5 years (60 months) after date of diagnosis. Survival analyses were carried out using the Kaplan-Meier method, and the estimates were evaluated by the log-rank test. The main outcomes were median overall survival (OS) and progression free survival (PFS). Cox proportional hazard method was applied to evaluate which variables were independent prognostic factors. The co-factors of the fitted models (sex, age, IPI) were tested for proportionality using the Cox-Aalen test. The significance level was defined as P-values < 0.05. All P-values were two-sided.
Ethics, consent and permissions
The project was conducted according to the Declaration of Helsinki, and was approved by the regional ethics committee (H-3-2012-127) and by the Danish data protection committee (2007-58-0015) according to the Danish ethical regulations.
Results
Clinical and survival parameters in 107 PCNSL patients
TP53 mutation analysis was successful in 86/107 patients, and promoter methylation studies of the MIR34A, MIR34B/C, and DAPK were successful in 93/107, 84/107 and 75/107 patients, respectively. The clinicopathological data of the whole cohort (N = 107) and the TP53, MIR34A, and DAPK subgroups are listed in Additional file 2: Table S1a-d. These tables also include description of the different treatment regimens and relevant doses that patients received as 1st line therapy.
The median OS for the whole cohort was 9.2 months [CI95%: 4.0–14.4], and the PFS was 6.7 months [CI95%: 3.7–9.6]. The mean and median follow-up time were 22.4 and 9.1 months (range: 0.1–157.5 months), respectively.
Survival differed significantly according to 1st line treatment modality. The 70 patients that received combination chemotherapy with or without rituximab (CCT-treated +/− rituximab) had a significantly longer median OS of 31.3 months [CI 95 %: 12.6–44.3], as compared to patients receiving monotherapy with high-dose methotrexate (HD-MTX), 6.0 months [CI 95 %: 1.6-NA], whole brain radiation therapy (WBRT), 6.0 [CI 95 %: 2.1-NA], or no therapy, 0.83 months [CI 95 %: 0.57–2.2], P < 0.0001, Fig. 1.

Overall survival associated with 1st line treatment in 107 PCNSL patients. Kaplan-Meier plot of 107 PCNSL patients, who were treated with combination chemotherapy +/− rituximab, whole brain radiation therapy (WBRT), high-dose (HD) methotrexate, and no therapy, P < 0.0001
Disease status according to 1st line treatment is recorded in Additional file 2: Table S2a. At the end of follow-up, 23 (21.5 %) patients were alive, and 20/23 in partial- or complete remission; 84 (78.5 %) patients were deceased. Twenty-two patients suffered a relapse after 1st line treatment (19 CCT +/− rituximab, and 3 HD-MTX only), and received a second treatment, Additional file 2: Table S2a-b. The median time to relapse after 1st treatment in this group was 8.4 months (range: 1–60.8). Among these patients, 2 (9.1 %) patients were in complete remission and alive 51.5 and 52.0 months after diagnosis, respectively, the remaining 20 (90.9 %) had deceased. The two surviving patients were originally treated with CCT + rituximab.
Histopathology and immunohistochemistry
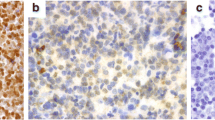
HE-staining showed classical patterns of CNS DLBCL with malignant angiocentric lymphocytic infiltration or diffuse sheets of B-type lymphoma (CD20+, CD79+, CD3-, CD5-) with immuno- or centroblast morphology, Fig. 2a-d.

Histology and immunohistochemistry of PCNSL of the DLBCL-subtype. PCNSL composed of large lymphocytic cells with irregular immunoblastic and centroblastic nuclei, increased number of mitoses, and (a) perivascular-, or (b) diffuse lymphoma infiltration. Immunohistochemical (IHC) marker expression includes (c) CD20+ (B-cell staining) and (d) CD3- (B-cells not staining, reactive T-cells+). IHC staining pattern of p53 protein in PCNSL tissue samples: (e) p53++ (black arrow) in patient ID18 with a hotspot/direct DNA contact MUT-TP53, p.R175H, (f) p53++ in patient ID14 with a hotspot/direct DNA contact MUT-TP53, p.R248Q. (g) p53-, in a patient with WT-TP53, and (h) p53+, in a patient with WT-TP53. Perivascular staining of p53 and neoplastic B-cells (black arrow)
IHC marker expression of the whole cohort and the three subgroups are shown in Additional file 2: Table S3 a-d. Briefly, the PCNSLs had an IHC-profile with the majority expressing BCL2+ (62.3 %), BCL6+ (74.6 %), CD10- (81.7 %), MUM1+ (96.7 %). Staining intensity of p53-IHC varied within specimens, and the number of nuclei expressing p53 protein was highly variable, suggesting tumor heterogeneity (Fig. 2e-h). In 86 % of the specimens p53-IHC positivity was present.
Structural mutation profile of the TP53 gene in PCNSL patients
Thirty-two of 86 (37.2 %) patients had one or more TP53 mutations, resulting in 37 missense mutations, one deletion, and one nonsense mutation. Thus, 7 (8 %) patients carried two mutations. Two SNVs occurred three times each; p.R175H (hotspot) and p.M169I, and p.T140I twice, leaving 34 different SNVs detected. The classification of the absolute number of mutations is shown in detail in Table 1, which is divided in two; the upper part (patient ID’s 1–24) representing patients that received CCT-treatment, and the lower part (patient ID’s 25–32) representing patients that received other treatment, but no CCT-therapy.
The structural analysis was restricted to the 24 CCT-treated patients in concordance with the survival analyses described below. The distribution pattern of these 28 SNVs (4 patients had two TP53 mutations) was analyzed according to their position in the 3-dimensional crystal model; 22/28 (78.6 %) were localized in codons involved in DNA-binding motifs of the central core domain. These included 5 (17.9 %) in the Loop-L3 (codons 237–250) that interacts with the minor groove of the DNA, and 5 (17.9 %) in the LSH-motif (codons 119–135 and 272–287) that interacts with the major groove of the DNA, and 12 (42.9 %) in the Loop-L2 (codons 164–194), which enhances the binding affinity between the DNA-helix and TP53 under physiological conditions. In 9 (32.1 %) of the absolute numbers of mutations, the mutated amino acid residues were involved in areas with hotspot properties, direct DNA contact or the central Zn-binding site, Table 1. All hotspot mutations were integrated in the direct DNA contact group. Location of MUT-TP53 according to exons, highly conserved areas and the distribution of basepair shifts are illustrated in Additional file 3: Figure S1a-c. The missense mutations could be classified according to their functionality, revealing the capacity of transactivating the promoter regions of several p53 target genes. Among the missense mutations, 14 (50.0 %) were predicted to inactivate p53, 7 (25.0 %) were associated with partially active p53, and 5 (17.9 %) with active p53. Among the remaining 2 (7.1 %), a single base deletion was allocated to the Zn-binding site. The SIFT-class prediction model designated 22 (78.6 %) of the TP53 mutations as being deleterious, Table 1.
Association between TP53 mutations, clinical characteristics and survival
In the 86 patients with a successful TP53 mutational analysis, complete or partial clinical data was available, Additional file 2: Table S2b. There were no significant differences in the clinical presentation patterns between the 32 patients that carried one or more TP53 mutations compared to the 54 patients with WT-TP53.
In these 86 patients, survival analysis did not reveal any differences in OS between PCNSL cases with MUT-TP53 versus WT-TP53 (Median OS: 13.1 vs. 9.1 months respectively, P = 0.625). At the end of follow up, 27/32 (84.4 %) of the MUT-TP53 cases had deceased as opposed to 41/54 (75.9 %) of the WT-TP53 cases, and no differences in OS were seen between MUT-TP53 cases with 1 or 2 mutations, respectively (P = 0.46).
When the analyses were confined to the 57/86 (65.5 %) CCT-treated patients, a potential biological difference between PCNSL harboring MUT-TP53 versus WT-TP53 was observed, however, this was non-significant, Fig. 3a, b. This observation prompted us to perform survival analyses based on the predicted structural subsets in the group of CCT-treated PCNSL patients.

Overall- and progression free survival of 57 PCNSL CCT-treated patients subjected to TP53 mutation analysis. Patients with MUT-TP53 (red) (n = 24) showed a non-significant trend towards worse (a) OS, and (b) PFS compared to patients with WT-TP53 (blue), P = 0.16 and P = 0.10, respectively. Presence of a TP53 hotspot/or direct DNA contact mutations (n = 9) was indicative of poorer (c) OS, and (d) PFS compared to patients with non-hotspot/non-direct DNA contact TP53 mutations and WT-TP53, P = 0.041 and P = 0.00076, respectively
Prognostic impact of TP53 hotspot mutations/direct DNA contact in CCT-treated patients
Because all hotspot mutations (n = 8) were an integrated part of the direct DNA contact group (n = 9), we chose to merge these groups into one. When the survival analyses were confined to the cohort of the 57 CCT-treated PCNSL, the OS and PFS differed significantly for patients with hotspot/direct DNA contact- (n = 9) vs. non-hotspot/non-direct DNA contact MUT-TP53 (n = 15) vs. WT-TP53 (n = 32); in the OS analysis the median was 6.9 vs 33.7 vs 53.4 months, respectively, P = 0.041 whereas for the PFS, the median was 4.6 months vs. 18.2 vs 45.7 months, P = 0.00076, Fig. 3c, d. In an univariate- and multivariate Cox regression model, presence of a hotspot/direct DNA contact MUT-TP53 was highly predictive for poor outcome in both univariate- and multivariate analyses for both PFS P = 0.0046 (HR: 1.97, [CI95%: 1.97–3.01]) and P = 0.0083 (HR: 1.95, [CI95%: 1.19–3.21]), respectively, and OS P = 0.028 (HR: 1.67, [CI95%: 1.06–2.67]) and P = 0.012 (HR: 1.86, [CI95%: 1.14–3.01]), respectively, Table 2.
MUT-TP53 located to Loop-L2, Loop-L3, and LSH-motifs, as well as mutations in the conserved regions 2–5, or according to functional properties did not have any association with survival outcomes (data not shown).
MIR34A and MIR34B/C methylation status and survival outcome
Promoter methylation status of MIR34A and MIR34B/C was successfully investigated in 93 and 84 patients. The clinical data of patients with or without methylation of MIR34A are shown in Additional file 2: Table S2c. Fifty-three of 93 (57.0 %) patients had promoter methylation of MIR34A, and 80/84 (95.2 %) patients had promoter methylation of MIR34B/C. No survival analysis was carried out for MIR34B/C, since only 2 events occurred among the 4 patients designated as un-methylated.
No association between PFS or OS and promoter methylation of MIR34A alone was detected, neither in the entire cohort, P = 0.63 and P = 0.84, respectively, nor in the CCT-treated subcohort, P = 0.58 and P = 0.19, respectively, Additional file 3: Figure S2a, b.
Impact of concomitant TP53 mutation and MIR34A methylation on survival
Seventy-eight PCNSL cases, which had been analyzed for both MUT-TP53 and MIR34A promoter methylation, were matched pairwise to detect presence of “double hit”. Eleven of 78 (14.1 %) had “double hit”. The remaining 67/78 patients (85.9 %) that either had a TP53 mutation or a MIR34A promoter methylation, i.e. “single hit”, or neither of them, were grouped together as “non-double hit”. In the CCT-treated subcohort, 52/54 (96.3 %) of the patients entered the analysis, of whom 8/52 (15.4 %) had “double hit”. Presence of a “double hit” was associated with a poorer PFS in the CCT-treated subgroup, as compared to “non-double hit”. Eight of 8 (100 %) CCT-patients with “double hit” had all experienced an event at follow-up (median PFS 6.4 months), in comparison to 24/44 (54.5 %) in the “non-double hit” subgroup, (median PFS 38.0 months), P = 0.0070, Fig. 4.

Concomitant TP53 mutation and MIR34A methylation “double-hit” associated with poorer progression free survival in CCT-treated PCNSL patients. Kaplan-Meier plot of 52/57 PCNSL patients, of which the 8 patients with “double-hit” TP53 mutation and MIR34A promoter methylation had a significantly poorer PFS. P = 0.0070
Noteworthy, among the CCT-treated patients 5/8 of the “double hit” cases were hotspot/direct DNA contact mutations (ID’s 5, 11, 20, 21, 23), and 3 non-hotspot/non-direct DNA contact mutations (ID’s: 2, 7, 8). In the Cox regression model, presence of a “double-hit” was significant only in the univariate-, but not in the multivariate model, P = 0.010 (HR: 2.96, [CI95%: 1.29–6.76] and P = 0.066 (HR: 2.30, [CI95%: 0.95–5.57]), Table 2.
DAPK methylation status and survival
Seventy-five of 107 (70.1 %) PCNSL FFPE samples were successfully tested for DAPK promoter methylation and 70/75 (93.3 %) were methylated. The clinical data of patients with or without methylation of DAPK are presented in Additional file 2: Table S2d. There was no significant difference between OS/PFS of patients with or without methylation of DAPK in the entire cohort, when analyzed in CCT-treated patients only, or when combined with MUT-TP53 data (data not shown). Data on the rs13300553 SNP genotypes and allelic DAPK methylation patterns are presented in Additional file 4: Data.
Discussion
Here, we present data on a large cohort of 107 consecutively diagnosed PCNSL cases, with comprehensive analyses of clinical data, histopathology, TP53 mutation status, and MIR34A/B/C and DAPK promoter methylation. At the time of diagnosis 80 of 107 (75 %) cases had disruption of one or more of the investigated genes, indicating that disrupted p53-signaling may be an essential part of PCNSL pathogenesis, which may contribute to the aggressive phenotype.
We did not observe a general negative effect on outcome by TP53 mutations in our entire cohort. However, when analyzing the subcohort of CCT-treated PCNSL patients, Kaplan-Meier analysis showed an inverse relationship with both PFS and OS in cases with MUT-TP53 in hotspots/direct DNA contact residues, compared to cases with WT-TP53 or other types of MUT-TP53. This observation was emphasized in multivariate Cox regression analyses, which showed that presence of a hotspot/direct DNA contact mutation was an independent negative prognostic factor for survival in CCT-treated PCNSL patients when adjusting for sex, age and IPI-status.
Previous studies have shown that TP53 mutation status plays a pivotal role in predicting outcome in patients with peripheral DLBCL treated with R-CHOP/CHOP-like chemotherapy: Patients with MUT-TP53 have a 2-fold higher relative risk of death compared to patients with WT-TP53, and patients with TP53 mutations that involved hotspot residues, and overlapping direct DNA contact residues of the DBD had a high mortality rate [51–53]. Our study of PCNSL consolidates the importance of not only reporting the mere MUT-TP53 number, but combines the structural classification in the survival analyses, stressing the inverse association to outcome when PCNSL patients harbor hotspot/direct DNA contact MUT-TP53, as formerly shown in DLBCL and other cancer subtypes [12, 26, 35, 42].
The reported frequency of somatic TP53 mutations in lymphomas varies significantly [50], even within individual lymphoma subtypes across different studies, which is likely due to differences in the quality of study material and the applied methods [26]. TP53 mutation status in PCNSL has only sporadically been reported in two other cohorts revealing much lower incidences compared to our study [10, 31]. In Cobbers’ pioneering work, 20 PCNSL patient samples (11 freshly frozen and 9 FFPE) were subjected to single-strand conformation polymorphism analysis followed by gel electrophoresis and DNA sequencing of TP53 exons 5–8, discovering only one missense mutation at codon 248 in one of twenty (5 %) patients [10]. In a recent work, 71 PCNSL patient samples (48 freshly frozen and 23 FFPE) were screened for mutations in 21 genes using the Ion Torrent NGS-platform, revealing a TP53 mutation rate of 7 %. However, validation by alternative methods was not performed for TP53, and no detailed structural classification of MUT-TP53 was provided in this study [31]. The primers of the current commercial Ion Torrent NGS-platform would a priori only cover 70.6 % of the detected TP53 mutations in our study; some genomic areas may be difficult to assess (e.g. homopolymeric tracts), and intra- and intertumor heterogeneity may require additional depth and coverage [28, 38].
When considering the reported low frequency of MUT-TP53 in PCNSL, we speculated whether the observed high frequency could be influenced by sequencing artifacts resulting from the use of FFPE DNA [2], or if it represented a genuine high frequency of driving mutations with clinical impact accompanied by random passenger mutations of unknown relevance [8, 12, 41, 45]. The fact that up to one mutation artifacts per 500 bases have been reported in methodological analyses of FFPE tissue in DNA sequencing with the majority of transitions being of C > T or G > A type [48] could be an indication of the former situation [12]. However, the previous studies mentioned above also included a significant proportion of FFPE samples in their cohorts, and we have not observed notable higher frequencies of TP53 mutations in FFPE compared to fresh frozen samples in peripheral DLBCL using the same methodology [22]. Finally, it could be argued that we would not observe the clear inverse relationship between hotspot/direct DNA contact TP53 mutations and clinical survival parameters, confirmed by multivariate Cox regression analysis, if a significant proportion of the detected mutations were randomly occurring artifacts. Thus, taken together, our data suggest that the frequency of TP53 mutations in PCNSL is higher than what has previously been appreciated and consolidate the prognostic impact of hotspot/direct DNA contact MUT-TP53 in PCNSL.
Interestingly, we also observed higher frequencies of MIR34A/B/C and DAPK promoter methylation than we previously detected in DLBCL outside the CNS [4, 22]. As in extra-CNS DLBCL we identified cases with concomitant MUT-TP53 and MIR34A methylation to be associated with a poor PFS. When combined with MIR34A methylation, also MUT-TP53 outside the hotspot/direct DNA contact loci seem to influence outcome emphasizing the synergistic function of these aberrations in lymphomagenesis. However, although we provide one of the largest molecular studies of PCNSL to date, the numbers of cases in the individual groups are still small. Ideally, the prognostic impact of hotspot/direct DNA contact TP53 mutations and concomitant MUT-TP53 and MIR34A methylation observed in this study, should be confirmed in other independent cohorts. NGS is a preferred methodology in future MUT-TP53 investigations, however, at least initially; the observed mutational spectrum should be compared to that obtained by other standard methods.
Conclusion
In conclusion, our data indicate that PCNSL have comprehensive molecular alterations in the p53-pathway, and that the high disruption frequency of the p53-pathway contributes to the poor outcome in CCT-treated PCNSL patients. In a clinical perspective, TP53 mutational and MIR34A methylation analyses of PCNSL could serve as additional tools in the prognostic setting when choosing treatment strategy. With referral to the accumulating evidence that p53 hotspot mutants have multiple gain of function properties [43], they may potentially serve as therapeutic targets. Thus it is tempting to speculate whether disruption of the p53-pathway may identify patients that are candidates for emerging, alternative treatment options in PCNSL.
Abbreviations
- CCT:
-
combination chemotherapy (Additional Table 1a-d, footnote)
- CI 95 %:
-
confidence interval 95 %
- CNS:
-
central nervous system
- CNS DLBCL:
-
central nervous system diffuse large b-cell lymphoma (used synonymously with PCNSL)
- DAPK:
-
death-associated protein kinase
- DBD:
-
DNA binding domain
- DGGE:
-
denaturing gradient gel electrophoresis
- DLBCL:
-
diffuse large b-cell lymphoma
- FFPE:
-
formalin fixed paraffin embedded
- HD-MTX:
-
high-dose methotrexate
- HR:
-
hazard ratio
- IARC:
-
international agency for research on cancer
- IHC:
-
immunohistochemistry
- IPI:
-
international prognostic index
- L2:
-
loop-L2
- L3:
-
loop-L3
- LDH:
-
lactate dehydrogenase
- LSH:
-
Loop (L1)-sheet-helix
- miR/MIR:
-
miRNA/MIRNA
- MSP:
-
methylation specific PCR
- MTX:
-
methotrexate
- MUT-TP53:
-
TP53 mutations
- NGS:
-
next generation sequencing
- OS:
-
overall survival
- PCNSL:
-
primary central nervous system lymphoma (used synonymously with CNS DLBCL)
- PCR:
-
polymerase chain reaction
- PFS:
-
progression free survival
- SNP:
-
single nucleotide polymorphy
- SNV:
-
single nucleotide variation
- WBRT:
-
whole brain radiation therapy
- WHO:
-
world health organization
- WT-TP53:
-
wildtype TP53
References
Adachi J-I, Mishima K, Wakiya K, Suzuki T, Fukuoka K, Yanagisawa T, Matsutani M, Sasaki A, Nishikawa R. O6-methylguanine-DNA methyltransferase promoter methylation in 45 primary central nervous system lymphomas: quantitative assessment of methylation and response to temozolomide treatment. J Neurooncol. 2012;107:147–53. doi:10.1007/s11060-011-0721-3.
Akbari M, Hansen MD, Halgunset J, Skorpen F, Krokan HE. Low copy number DNA template can render polymerase chain reaction error prone in a sequence-dependent manner. J Mol Diagn. 2005;7:36–9.
Arai M, Sasaki A, Saito N, Nakazato Y. Immunohistochemical analysis of cleaved caspase-3 detects high level of apoptosis frequently in diffuse large B-cell lymphomas of the central nervous system. Pathol Int. 2005;55:122–9. doi:10.1111/j.1440-1827.2005.01808.x.
Asmar F, Hother C, Kulosman G, Treppendahl MB, Nielsen HM, Ralfkiaer U, Pedersen A, Møller MB, Ralfkiaer E, de Nully Brown P, Grønbæk K. Diffuse large B-cell lymphoma with combined TP53 mutation and MIR34A methylation: Another “double hit” lymphoma with very poor outcome? Oncotarget. 2014;5:1912–25.
Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19:1245–56. doi:10.1038/sj.onc.1203434.
Chang C-C, Kampalath B, Schultz C, Bunyi-Teopengco E, Logan B, Eshoa C, Dincer AP, Perkins SL. Expression of p53, c-Myc, or Bcl-6 suggests a poor prognosis in primary central nervous system diffuse large B-cell lymphoma among immunocompetent individuals. Arch Pathol Lab Med. 2003;127:208–12. doi:10.1043/0003-9985(2003)127<208:EOPMOB>2.0.CO;2.
Chang T-C, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi:10.1016/j.molcel.2007.05.010.
Chanock SJ, Thomas G. The devil is in the DNA. Nat Genet. 2007;39:283–4. doi:10.1038/ng0307-283.
Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55.
Cobbers JM, Wolter M, Reifenberger J, Ring GU, Jessen F, An HX, Niederacher D, Schmidt EE, Ichimura K, Floeth F, Kirsch L, Borchard F, Louis DN, Collins VP, Reifenberger G. Frequent inactivation of CDKN2A and rare mutation of TP53 in PCNSL. Brain Pathol. 1998;8:263–76.
Deckert M, Montesinos-Rongen M, Brunn A, Siebert R. Systems biology of primary CNS lymphoma: from genetic aberrations to modeling in mice. Acta Neuropathol. 2013. doi:10.1007/s00401-013-1202-x.
Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, Sundström M, Micke P, Botling J, Soussi T. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci U S A. 2012;109:9551–6. doi:10.1073/pnas.1200019109.
Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE, Valeri N, Calore F, Sampath D, Fanini F, Vannini I, Musuraca G, Dell’Aquila M, Alder H, Davuluri R V, Rassenti LZ, Negrini M, Nakamura T, Amadori D, Kay NE, Rai KR, Keating MJ, Kipps TJ, Calin GA, Croce CM. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi:10.1001/jama.2010.1919.
Ferreri AJM, Zucca E, Armitage J, Cavalli F, Batchelor TT. Ten years of international primary CNS lymphoma collaborative group studies. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:3444–5. doi:10.1200/JCO.2013.51.7557.
Feugier P, Van Hoof A, Sebban C, Solal-Celigny P, Bouabdallah R, Fermé C, Christian B, Lepage E, Tilly H, Morschhauser F, Gaulard P, Salles G, Bosly A, Gisselbrecht C, Reyes F, Coiffier B. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 2005;23:4117–26. doi:10.1200/JCO.2005.09.131.
Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, Nielsen O, Gadeberg O V, Mourits-Andersen T, Frederiksen M, Pedersen LM, Møller MB. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:3460–7. doi:10.1200/JCO.2011.41.4342.
Guldberg P, Nedergaard T, Nielsen HJ, Olsen AC, Ahrenkiel V, Zeuthen J. Single-step DGGE-based mutation scanning of the p53 gene: application to genetic diagnosis of colorectal cancer. Hum Mutat. 1997;9:348–55. doi:10.1002/(SICI)1098-1004(1997)9:4<348::AID-HUMU8>3.0.CO;2-1.
Guldberg P, Worm J, Grønbaek K. Profiling DNA methylation by melting analysis. Methods. 2002;27:121–7.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi:10.1038/nature05939.
Hother C, Asmar F, Kulosman G, Marianne B Treppendahl, Fuhrman G, Ralfkiaer U, Pedersen A, Møller MB, Brown P, Grønbæk K miR34s in Normal and Malignant B-Cells: miR34A Plays a Dominant Role in Normal B-Cells, and aggressive Diffuse Large B-Cell Lymphoma Carry Combined Lesions of TP53, MIR34A, and MIR34B/C. American Society of Hematology (ASH) Annual Meeting. Abstract. ISBN 4525211598. 2012a.
Kasenda B, Ferreri AJM, Marturano E, Forst D, Bromberg J, Ghesquieres H, Ferlay C, Blay JY, Hoang-Xuan K, Pulczynski EJ, Fosså A, Okoshi Y, Chiba S, Fritsch K, Omuro A, O’Neill BP, Bairey O, Schandelmaier S, Gloy V, Bhatnagar N, Haug S, Rahner S, Batchelor TT, Illerhaus G, Briel M. First-line treatment and outcome of elderly patients with primary central nervous system lymphoma (PCNSL) - A systematic review and individual patient data meta-analysis. Ann Oncol. 2015. doi:10.1093/annonc/mdv076.
Kristensen LS, Asmar F, Dimopoulos K, Nygaard MK, Aslan D, Hansen JW, Ralfkiaer E, Grønbæk K. Hypermethylation of DAPK1 is an independent prognostic factor predicting survival in diffuse large B-cell lymphoma. Oncotarget. 2014;5:9798–810.
Kristensen LS, Johansen JV, Grønbæk K. Allele-specific DNA methylation detection by pyrosequencing®. Methods Mol Biol. 2015;1315:271–89. doi:10.1007/978-1-4939-2715-9_20.
Kristensen LS, Treppendahl MB, Asmar F, Girkov MS, Nielsen HM, Kjeldsen TE, Ralfkiaer E, Hansen LL, Grønbæk K. Investigation of MGMT and DAPK1 methylation patterns in diffuse large B-cell lymphoma using allelic MSP-pyrosequencing. Sci Rep. 2013;3:2789. doi:10.1038/srep02789.
Kumar M, Lu Z, Takwi AAL, Chen W, Callander NS, Ramos KS, Young KH, Li Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene. 2011;30:843–53. doi:10.1038/onc.2010.457.
Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat. 2014;35:672–88. doi:10.1002/humu.22552.
Lu T-X, Young KH, Xu W, Li J-Y. TP53 dysfunction in diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2015. doi:10.1016/j.critrevonc.2015.08.006.
Mafficini A, Amato E, Fassan M, Simbolo M, Antonello D, Vicentini C, Scardoni M, Bersani S, Gottardi M, Rusev B, Malpeli G, Corbo V, Barbi S, Sikora KO, Lawlor RT, Tortora G, Scarpa A. Reporting tumor molecular heterogeneity in histopathological diagnosis. PLoS One. 2014;9:e104979. doi:10.1371/journal.pone.0104979.
Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, Corbett RD, Chan FC, Mendez-Lago M, Trinh DL, Bolger-Munro M, Taylor G, Hadj Khodabakhshi A, Ben-Neriah S, Pon J, Meissner B, Woolcock B, Farnoud N, Rogic S, Lim EL, Johnson NA, Shah S, Jones S, Steidl C, Holt R, Birol I, Moore R, Connors JM, Gascoyne RD, Marra MA. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122:1256–65. doi:10.1182/blood-2013-02-483727.
Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, Engelhardt B, Hallenbeck JM, Lonser RR, Ohlfest JR, Prat A, Scarpa M, Smeyne RJ, Drewes LR, Neuwelt EA. Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab. 2013;33:13–21. doi:10.1038/jcbfm.2012.153.
Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, Tanaka K, Fukushima S, Takami H, Arita H, Kubo A, Shuto T, Ohno M, Miyakita Y, Kocialkowski S, Sasayama T, Hashimoto N, Maehara T, Shibui S, Ushijima T, Kawahara N, Narita Y, Ichimura K. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2015. doi:10.1111/nan.12259.
Olszewski AJ, Winer ES, Castillo JJ. Improved survival with rituximab-based chemoimmunotherapy in older patients with extranodal diffuse large B-cell lymphoma. Leuk Res. 2014;38:866–73. doi:10.1016/j.leukres.2014.04.009.
Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi:10.1101/cshperspect.a001107.
Petitjean A, Achatz MIW, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi:10.1038/sj.onc.1210302.
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian S V, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi:10.1002/humu.20495.
Pfreundschuh M, Kuhnt E, Trümper L, Osterborg A, Trneny M, Shepherd L, Gill DS, Walewski J, Pettengell R, Jaeger U, Zinzani P-L, Shpilberg O, Kvaloy S, de Nully Brown P, Stahel R, Milpied N, López-Guillermo A, Poeschel V, Grass S, Loeffler M, Murawski N. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. 2011;12:1013–22. doi:10.1016/S1470-2045(11)70235-2.
Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, Yang L, Wilson WH, Jaffe ES, Simon R, Klausner RD, Powell J, Duffey PL, Longo DL, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Montserrat E, López-Guillermo A, Grogan TM, Miller TP, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt LM, Lymphoma/Leukemia Molecular Profiling Project. Lymphoma/Leukemia Molecular Profiling Project (2002) The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. doi:10.1056/NEJMoa012914.
Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14:R51. doi:10.1186/gb-2013-14-5-r51.
Rubenstein JL, Hsi ED, Johnson JL, Jung S-H, Nakashima MO, Grant B, Cheson BD, Kaplan LD. Intensive chemotherapy and immunotherapy in patients with newly diagnosed primary CNS lymphoma: CALGB 50202 (Alliance 50202). J Clin Oncol. 2013;31:3061–8. doi:10.1200/JCO.2012.46.9957.
Scott DW, Wright GW, Williams PM, Lih C-J, Walsh W, Jaffe ES, Rosenwald A, Campo E, Chan WC, Connors JM, Smeland EB, Mottok A, Braziel RM, Ott G, Delabie J, Tubbs RR, Cook JR, Weisenburger DD, Greiner TC, Glinsmann-Gibson BJ, Fu K, Staudt LM, Gascoyne RD, Rimsza LM. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123:1214–7. doi:10.1182/blood-2013-11-536433.
Soussi T. Locus-specific databases in cancer: what future in a post-genomic era? The TP53 LSDB paradigm. Hum Mutat. 2014;35:643–53. doi:10.1002/humu.22518.
Soussi T, Asselain B, Hamroun D, Kato S, Ishioka C, Claustres M, Béroud C. Meta-analysis of the p53 mutation database for mutant p53 biological activity reveals a methodologic bias in mutation detection. Clin Cancer Res. 2006;12:62–9. doi:10.1158/1078-0432.CCR-05-0413.
Soussi T, Wiman KG. TP53: an oncogene in disguise. Cell Death Differ. 2015;22:1239–49. doi:10.1038/cdd.2015.53.
Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4. th edit. Lyon: IARC Press; 2008.
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Macconnaill LE, Lee JC, Nicoletti R, Hatton C, Goyette M, Girard L, Majmudar K, Ziaugra L, Wong K-K, Gabriel S, Beroukhim R, Peyton M, Barretina J, Dutt A, Emery C, Greulich H, Shah K, Sasaki H, Gazdar A, Minna J, Armstrong SA, Mellinghoff IK, Hodi FS, Dranoff G, Mischel PS, Cloughesy TF, Nelson SF, Liau LM, Mertz K, Rubin MA, Moch H, Loda M, Catalona W, Fletcher J, Signoretti S, Kaye F, Anderson KC, Demetri GD, Dummer R, Wagner S, Herlyn M, Sellers WR, Meyerson M, Garraway LA. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. doi:10.1038/ng1975.
Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008;68:4123–32. doi:10.1158/0008-5472.CAN-08-0325.
Whibley C, Pharoah PDP, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107. doi:10.1038/nrc2584.
Williams C, Pontén F, Moberg C, Söderkvist P, Uhlén M, Pontén J, Sitbon G, Lundeberg J. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol. 1999;155:1467–71. doi:10.1016/S0002-9440(10)65461-2.
Worm J, Aggerholm A, Guldberg P. In-tube DNA methylation profiling by fluorescence melting curve analysis. Clin Chem. 2001;47:1183–9.
Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, Greiner TC, McDonnell TJ, Young KH. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119:3668–83. doi:10.1182/blood-2011-11-366062.
Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu W, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Zhao XF, Choi WWL, Zhao X, van Krieken JH, Huang Q, Huh J, Ai W, Ponzoni M, Ferreri AJM, Zhou F, Kahl BS, Winter JN, Xu W, Li J, Go RS, Li Y, Piris MA, Møller MB, Miranda RN, Abruzzo L V, Medeiros LJ, Young KH. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120:3986–96. doi:10.1182/blood-2012-05-433334.
Young KH, Leroy K, Møller MB, Colleoni GWB, Sánchez-Beato M, Kerbauy FR, Haioun C, Eickhoff JC, Young AH, Gaulard P, Piris MA, Oberley TD, Rehrauer WM, Kahl BS, Malter JS, Campo E, Delabie J, Gascoyne RD, Rosenwald A, Rimsza L, Huang J, Braziel RM, Jaffe ES, Wilson WH, Staudt LM, Vose JM, Chan WC, Weisenburger DD, Greiner TC. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008;112:3088–98. doi:10.1182/blood-2008-01-129783.
Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, Campo E, Delabie J, Gascoyne RD, Ott G, Rimsza L, Müller-Hermelink HK, Jaffe ES, Rosenwald A, Staudt LM, Chan WC, Greiner TC. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood. 2007;110:4396–405. doi:10.1182/blood-2007-02-072082.
Zacher J, Kasenda B, Engert A, Skoetz N. The role of additional radiotherapy for primary central nervous system lymphoma. Cochrane Database Syst Rev. 2014. doi:10.1002/14651858.CD009211.pub2.
Acknowledgments
HDMP received study grants from the following sources: The Research Foundation of the University Hospital of Copenhagen, Rigshospitalet (R85-A3122), the Danish Cancer Research Fund, the Dagmar Marshall Foundation, the Fabrikant Einar Willumsen Foundation, the Agnes and Poul Friis Foundation, and the Christian Andersen grant. KG received grants from the Novo Nordisk foundation (NNF130C0003435), the Dan Stem collaboration (10–092798), and the van Andel Research Institute, KG and LSK from the Danish Cancer Society, and FA from the Danish Council of Independent Research (DFF – 4092–00443).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HDMP contributed to the design of the study, wrote the manuscript, and collected the clinical data, the FFPE samples, purified the DNA, and scored the IHC. HDMP carried out the molecular genetic and epigenetic studies of TP53 mutations, MIR34 A/B/C and DAPK, made the principal data analysis of all assembled data and the statistics. FA contributed to the design of the study, supervised the TP53 analyses, and participated in the TP53 data analysis. KD contributed to the data interpretation and statistical analyses. AA participated with practical lab work and preparation of FFPE tissue. PB contributed to the design of the study and interpretation of data. MSG carried out the the molecular genetic studies in the DAPK subpart. AP supervised the TP53 study. LDS & SH participated in the design of the study. HB & ER participated in the design of the study and reviewed the histological samples (HE & FFPE). LSK contributed to the design of the study supervised the DAPK substudy, and participated in the writing of the draft of the manuscript. KG contributed to the design of the study, was the main supervisor and coordinator of the whole project, and wrote the manuscript. All authors have critically read and commented the draft during the process of writing, and accepted the final version of the manuscript.
Additional files
Additional file 1:
Methods 1. Immunohistochemistry. Methods 2. Detection and structural classification of TP53 mutations. Methods 3. DAPK methylation analysis using allellic MSP-pyrosequencing. (PDF 379 kb)
Additional file 2: Tables S1a-d.
Clinical data in 107 PCNSL patients and TP53, MIR34A, and DAPK subgroups. Tables S2a-b. Disease status (a), and treatment at relapse/progression (b). Tables S3a-d. Immunohistochemical marker expression in 107 PCNSL patients and TP53, MIR34A, and DAPK subgroups. (PDF 534 kb)
Additional file 3: Figure S1a-c.
Supplemental figures 1a-c. Figure S2a-b. Supplemental figures. (PDF 474 kb)
Additional file 4:
Data methylation status and survival. (PDF 262 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Munch-Petersen, H.D., Asmar, F., Dimopoulos, K. et al. TP53 hotspot mutations are predictive of survival in primary central nervous system lymphoma patients treated with combination chemotherapy. acta neuropathol commun 4, 40 (2016). https://doi.org/10.1186/s40478-016-0307-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-016-0307-6