
Abstract
A 28-month female with a clinical diagnosis of neurocutaneous melanosis and numerous intracranial abnormalities (including a right choroid plexus tumor and left hemimegalencephaly) presented with a rapidly expanding tumor in the left occipital cerebrum. Microscopic examination of the resected specimen revealed a myxoid mesenchymal neoplasm consisting of fusiform cells that were immunoreactive for vimentin, CD34, and P53 but no melanocyte markers. Focused amplicon deep sequencing on DNA extracted from the brain tumor and a cutaneous nevus revealed a heterozygous (c.37G > C; p.G13R) substitution in the NRAS gene. DNA sequencing of “normal” skin and buccal swab showed the identical NRAS change albeit at lower allelic frequency. Her parents did not harbor the NRAS mutation. The skin lesion, but not the brain tumor, had a BRAF mutation (c.1397G > T; p.G466V). A germline single nucleotide polymorphism in MET was found in the child and her father (c.3209C > T; p.T1010I). The findings suggest NRAS mosaicism that occurred sometime after conception and imply an oncogenic role of the activating NRAS mutation in both the brain and skin lesions in this child.
Similar content being viewed by others


Background
Neurocutaneous melanosis (NCMS; Mendelian Inheritance in Man MIM# 249400) is a rare congenital phakomatosis consisting of numerous giant cutaneous nevi along with extensive leptomeningeal melanosis. Approximately 100 cases have been described in the literature [1]. The pathogenesis of NCMS likely involves a morphogenetic error in which melanocyte precursors derived from the neural crest migrate abnormally and proliferate locally [2],[3]. Most cases with an identifiable cause have a somatic gain-of-function mutation in codon 61 of the NRAS gene (MIM# 164790) located on chromosome 1p13 [4]-[7]. Approximately 30% of affected children have melanin deposits detectable in the leptomeninges or brain [8] and half have epilepsy [9].
We present a case of a female infant with NCMS who developed an unusual myxoid mesenchymal brain tumor. DNA sequencing showed shared mutations in codon 13 of NRAS in the melanocytic nevi and the brain tumor.
Case report
Clinical details
This female child was born to a healthy non-consanguineous couple after an uneventful full-term pregnancy. At birth she had numerous slightly raised, hairy melanocytic lesions on the scalp, neck, upper trunk, upper extremities, and hands; the largest was 4–5 cm in greatest dimension (Figure 1). Skin lesions from the neck, scalp, and arm were previously excised and diagnosed as intradermal and compound nevi with congenital features. She had normal height but was macrocephalic (97th percentile head circumference). She had been admitted to hospital numerous times for uncontrolled seizures starting at age 2 months. Magnetic resonance (MR) imaging of the brain and spine was performed at 3.5 months age. T1 weighted images showed a solitary 2 mm focus of increased signal intensity in the right cerebellopontine cistern; this was thought to represent melanin deposition. Cystic lesions, the largest of which was 2.7 cm diameter, with signal characteristics the same as cerebrospinal fluid (CSF) were present around the atria of the lateral ventricles in the right cerebellopontine cistern along with small cysts around the bodies of the lateral ventricles. The left cerebral hemisphere was larger than the right, and parietal white matter volume was abnormally small. The combination of giant congenital nevi along with cerebral melanin deposition led to a clinical diagnosis of NCMS [10]. A CT scan of the head performed at 4 months age during an episode of status epilepticus showed no additional abnormalities. An abdominal sonogram performed the following day showed no abnormalities. Repeat MR imaging showed the presumed melanocytic nodule had increased to 3 mm; it was hyperintense on T1, hypointense on T2, and hyperintense on FLAIR. The periventricular cysts had increased in size and quantity. A new lesion in continuity with the choroid plexus was expanding the temporal horn of the right lateral ventricle. MR imaging of the brain at 21 months age showed a new left occipital brain tumor that was T2 hyperintense and enhanced strongly following administration of gadolinium-DTPA. It grew from 1.6 × 1.5 × 1.2 cm to 4.0 × 3.6 × 3.2 cm 4 months later (Figure 2). A left occipital craniotomy was performed at 24.5 months age. The tumor was not in contact with the meninges. It had a single vascular pedicle. There was an easily identified plane between it and the adjacent brain. The tumor was removed in one piece without complication. MR imaging of the brain 26 months after surgery (51 months age) showed no recurrence of the tumor; a vague region of non-enhancing T2 signal abnormality in the right medial occipital lobe had expanded slightly.

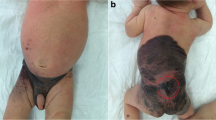
Photograph of the patient at 14 months age showing giant congenital melanocytic nevi on each shoulder and numerous smaller satellite nevi on the neck, scalp, arms, and upper trunk.

Magnetic resonance image of the brain. At 14 months age (upper image, coronal, T2 weighted) an enlarged left temporal lobe and a tumor of the right choroid plexus (arrow) were apparent. At 23 months age (lower image, horizontal, T1-weighted) a 4 cm tumor is present in the left occipital lobe. Periventricular cysts are located adjacent to the right occipital horn (arrow).
Histopathological characterization
The tumor was a 4.5 × 3.5 × 3 cm firm tan nodule with a smooth external surface and uniform cut surface (Figure 3A). The brain tumor and skin biopsies were fixed in 10% neutral buffered formalin, dehydrated and embedded in paraffin wax for sectioning (5 uM in thickness). All samples were stained with hematoxylin and eosin for histologic evaluation. Immunohistochemistry was performed using primary antibodies against vimentin (mouse monoclonal, V9, Dako), CD34 (mouse monoclonal, QBEND-10, Dako), alpha B crystallin (mouse monoclonal, G2JF, Novocastra), CD56 (mouse monoclonal, CD564, Dako), D240 (mouse monoclonal, D2-40, Dako), CD99 (mouse monoclonal, 12E71, Dako), Bcl2 (mouse monoclonal, 124, Dako), p53 (mouse monoclonal, D0-7, Dako), Ki67 (mouse monoclonal, MIB-1, Dako), HLA-DR (mouse monoclonal, CR3/43, Dako), Factor 13a (mouse monoclonal, EP3372, Cellmarque), GFAP (rabbit polyclonal, Dako), S100 (rabbit polyclonal, Dako), Collagen IV (mouse monoclonal, C1V22, Dako), pan-cytokeratin (mouse monoclonal, AE1/AE3, Dako), EMA (mouse monoclonal, E29, Dako), CD57 (mouse monoclonal, TB01, Dako), HMB-45 (mouse monoclonal, HMB45, Dako), MART-1/Melan-A (mouse monoclonal, A103, Dako), calretinin (rabbit polyclonal, Cellmarque), neurofilament (mouse monoclonal, 2 F11, Dako), synaptophysin (mouse monoclonal, 27G12, Novocastra), NeuN (mouse monoclonal, A60, Lifespan Biosciences). Antigen retrieval was performed in a Bull’s Eye Decloaking chamber (Biocare Medical, Concord, CA) for 1 minute at 125°C utilizing a Dako pH9 retrieval solution. All antibodies were detected using the Dako Envision system (Dako) and diaminobenzidine precipitation solution. A sample of the tumor was fixed in 2.5% buffered glutaraldehyde, post-fixed in osmium tetroxide, dehydrated in graded ethanol, and embedded in epoxy resin. Semithin sections (0.5 µm) were stained with toluidine blue, and ultrathin sections were contrasted with uranyl acetate and lead citrate then viewed with electron microscopy using a JEM 1010 transmission electron microscope (JEOL Ltd., Tokyo, Japan).

Macroscopic and microscopic appearance of the tumor. (A) The firm tumor was easily dissected from the surrounding brain tissue and had a smooth tan-yellow glistening cut surface. (B) Microscopic examination revealed a paucicellular tumor consisting of predominantly fusiform cells in a myxoid background. There is moderate nuclear pleomorphism with a multinucleated cell in the center (hematoxylin & eosin). (C) The loose myxoid background stained positive for Alcian blue. (D) Immunohistochemical staining for CD34 is diffusely positive. (E) Electron microscopic examination showed stellate cells with long delicate processes and occasional multinucleation. (F) The extracellular material is abundant with clusters of collagen fibers. (Original magnifications: B-C, ×400; D, ×200; E, ×2000; F ×30000).
Microscopic examination showed an indistinct interface with the brain parenchyma. The tumor had a diffuse pattern of stellate and elongated cells with delicate processes in a loose myxoid background. There were no pigmented cells. Numerous small, multinucleated cells were evenly distributed within the lesion (Figure 3B). Rarely the cells clustered around blood vessels. Very rare mitotic figures were present and there was no necrosis or endothelial hyperplasia. Focal areas of the extracellular material stained with Alcian blue (Figure 3C). Reticulin staining was negligible. By immunohistochemistry, the cells were positive for vimentin, CD34 (Figure 3D), CD56, D240, Bcl2, CD99 (weak), and P53. A subpopulation of cells was Factor 13a positive. An estimated 5-10% of nuclei were Ki67 positive. Only perivascular cells (likely astrocytes) were positive for glial fibrillary acidic protein (GFAP), S100, and alpha B crystallin. Blood vessel walls were positive for E cadherin and collagen IV. There was no immunoreactivity for cytokeratin (AE1/3), epithelial membrane antigen (EMA), CD57, HMB-45, MART-1/Melan-A, calretinin, neurofilament, synaptophysin, or NeuN. Scattered HLA-DR positive cells were likely infiltrating microglia. Electron microscopic examination showed fusiform cells with abundant rough endoplasmic reticulum and prominent intermediate filaments but no specific secretory organelles or obvious intercellular junctions. The cells lacked a well-defined basement membrane and were surrounded by a flocculent extracellular material with rare clusters of striated collagen bundles (Figure 3E, 3F). The diagnostic categorization arrived at locally and supported by external consultation was myxoid mesenchymal brain tumor of uncertain growth potential.
DNA extraction and sequencing
Genomic DNA was extracted from formalin-fixed paraffin-embedded samples of the intracranial tumor, an excised cutaneous nevus lesion, and grossly unaffected skin. DNA was also extracted from swabbed buccal cells of the patient and both of her parents. DNA was quantitated using the Qubit 2.0 fluorometer (Life Technology). Focused deep sequencing of 10 ng of genomic DNA extracted from the above samples was performed using the Ion AmpliSeq Cancer panel (Life Technology). This includes PCR primers covering 739 potential cancer-related hotspot mutations in 46 genes including KRAS, NRAS, BRAF, PIK3CA, and IDH1[11]. This technology permits the interrogation of genetic alterations including mutations and insertions/deletions, even minor alleles in complex samples, in suboptimal specimens including formalin-fixed paraffin-embedded tissues. Processing of all samples was performed according to the manufacturer’s protocol. Construction and enrichment of the emulsion PCR library was performed using the Ion OneTouch instrument. Sequencing was done on Ion 314 and Ion 316 sequencing chips using the Ion Torrent Personal Genome Machine (Life Technology) following the manufacturer’s protocol. Data analysis including alignment to hg19 human reference genome, base calling, and identification of variants was done using the Ion Variant Caller (version 2.2). Somatic variants from the brain tumor and the melanocytic nevus were identified after filtering out germline changes identified from the patient’s buccal swab. Allele frequency of a variant is calculated by dividing the number of variant reads by total reads in the same nucleotide position. All variants are covered by a minimum of 500 reads.
The panel identified a missense mutation (chr1:115258745; c.37G > C) in NRAS that results in a p. G13R amino acid substitution. The allelic frequency of the nucleotide change in the brain tumor and the cutaneous nevus (58% and 33% respectively) is consistent with a heterozygous mutation (Table 1). The same change was observed at an allelic frequency of 4.8% in the normal skin and 2.7% in the buccal swab of the patient. Deep sequencing of DNA extracted from the buccal swabs from both parents did not reveal NRAS mutations. A BRAF somatic nucleotide change (chr7: 140481441; c.1397G > T) resulting in amino acid substitution p. G466V was detected only in the nevus at an allelic frequency of 32%. A single nucleotide polymorphism (SNP) resulting in a missense mutation (chr7: 116411990; c. 3209C > T; p.T1010I;) was found in the MET gene of the child’s tumor and buccal swab as well as in the buccal swab of the father.
Discussion and conclusions
Neurocutaneous melanosis (NCMS), first described by Rokitansky in 1861 [12], is a rare congenital disorder consisting of multiple large melanocytic cutaneous nevi and melanocytic proliferations in the leptomeninges [13]. Malformative lesions of the posterior fossa have rarely been described [14],[15]. Recent genetic findings have clarified the pathogenesis of NCMS and large/giant congenital melanocytic nevi [7],[16]. Single postzygotic mutations of NRAS codon 61 and associated mosaicism are responsible for the majority of NCMS cases [5] and large/giant congenital melanocytic nevi [17]. The same mutation is common in congenital melanocytic nevi [18]. The NRAS p.G13R somatic mosaicism in this patient is unusual and, to our knowledge, is the first instance reported in association with NCMS. The presence mosaicism in combination with the absence of mutation in the parents suggests this mutation likely occurred after conception. This is consistent with the mosaicism hypotheses for NCMS and other phakomatoses [19]. This child does not show any dysmorphic features associated with germline mutations in NRAS, which are usually similar to Noonan syndrome [20]. NRAS mutations are considered important in the genesis of melanoma. NRAS activates four major signaling pathways including RAF-MEK-ERK, RalGDS, PI3K-AKT/PDK1, and PLC/PKC [21]. Mutations affecting codons 12, 13 and 61, lead to constitutive activation of RAS GTPase in the absence of growth factor signaling and ultimately neoplastic growth. The specific NRAS G13R mutation identified in this case has been rarely found in melanomas of the skin [22]-[24] and esophagus [25] as well as in 1/27 patients with large congenital melanocytic nevi [26],[27]. In melanoma the most common NRAS (Q61R, Q61L, Q61K) and BRAF (V600E, V600K) mutation sites and substitutions differ from those found in this patient [28].
In addition to NCMS, this child had a low-grade mesenchymal brain tumor, which itself is very rare. Typically children with NCMS do not develop sarcomas, although one rhabdomyosarcoma was described in a congenital giant nevus [29]. Children with NCMS are reported to develop choroid plexus papilloma and meningioma [5]. Primary sarcomas of the brain represented only 0.36% of brain tumors in one very large series [30] and 0.7% of all sarcomas in another series [31]. Most likely arise from the meninges or blood vessels; among them are rhabdomyosarcoma, fibrosarcoma, leiomyosarcoma, and angiosarcoma. Primary non-meningeal myxoid mesenchymal intracranial tumors are especially rare. Reported cases include two low-grade fibromyxoid sarcomas [32],[33] and one myxofibrosarcoma, the diagnosis of which was based on fluorescent in situ hybridization (FISH) analysis of the FUS/CREB3L2 translocation [34]. Both of these tumors have some features similar to the reported child’s brain tumor. It is important to emphasize that the tumor had no morphologic or immunophenotypic features of melanoma. The relatively low-grade appearance of the brain tumor presented herein is discrepant with its rapid growth. The rapid increase in size could be explained by expansion of the myxoid extracellular material rather than neoplastic cell proliferation.
The heterozygous state of NRAS in the melanocytic nevus and the brain tumor combined with additional novel somatic mutations suggest cooperative involvement of oncogenic pathways in the brain tumor and especially in the skin lesions (BRAF p.G466V). Only a single sarcoma (a rhabdomyosarcoma) has been described with the NRAS p.G13R mutation [35]. Somatic mosaics of NRAS mutations in almost identical protein regions (G12D, G12S, G13D) have been described in relation to juvenile myelomonocytic leukemia [36],[37]. Note that neurofibromin is a major regulator of the NRAS pathway [38]. NF1 is not assessed in the AmpliSeq Cancer Panel and we did not attempt direct sequencing. However, given that the documented abnormalities are distal to neurofibromin signaling, a mutation in NF1 is not necessary to explain this child’s phenotype.
A germline SNP was found in the MET gene of this child (inherited from her father who is not known to have any neoplastic disease). The MET gene encodes the receptor for hepatic growth factor/scatter factor (HGF/SF) and appears to be involved in cell motility, proliferation, and invasiveness [39]. Mice that overexpress HGF/SF overstimulate the MET pathway and develop melanosis in the central nervous system and patterned hyperpigmentation of the skin similar to NCMS [40]. These mice also develop fibrosarcoma and rhabdomyosarcoma [41]. MET has been detected by immunohistochemistry in optic canal nevus cells from a child with NCMS [42]. The p.T1010I variant has been identified in thyroid carcinomas [33], neuroendocrine carcinoma (NEC) of lung [43], a pleomorphic xanthoastrocytoma case [11], and has been implicated as a risk factor for familial colorectal cancer [44]. One early report described this change in MET as capable of altering signaling in NEC [45], however more recently this variation was not shown to alter c-Met phosphorylation in NEC [46], nor does it seem capable of transforming the Ba/F3 pro-B lymphocyte cell line [47]. To summarize, the significance of the MET SNP in this child is unclear.
Hemimegalencephaly has been associated with other neurocutaneous syndromes including epidermal nevus syndrome, proteus syndrome, hypomelanosis of Ito, and neurofibromatosis-1 [48],[49]. Recently, de novo somatic mutations with mosaicism in the PI3K-AKT3-mTOR pathway were shown to cause hemimegalencephaly [50]. NRAS is known to have direct interaction with PI3K [21]. Given that this child had no mutation in PI3K, the NRAS mosaicism is likely the explanation for the hemimegalencephaly.
Conclusion
In summary, development of a primary intracerebral mesenchymal neoplasm in a child with NCMS and hemimegalencephaly can likely be explained by specific NRAS mutant mosaicism possibly in combination with a MET germline variation, which together constitute a unique combination. This case highlights the importance of DNA analysis from multiple sites, as well as from parents, in individuals with complex disease states. The presence of an NRAS somatic mosaic supports the hypothesized developmental pathogenesis of NCMS. Further exploration of the role of NRAS and MET in development of the neural crest derived pigment cells will be of interest.
Consent
Parents provided explicit consent for genetic tests and use of photographs.
Abbreviations
- BRAF:
-
B-Raf proto-oncogene, serine/threonine kinase
- DNA:
-
Deoxyribonucleic acid
- FISH:
-
Fluorescent in situ hybridization
- MET:
-
MET proto-oncogene, receptor tyrosine kinase
- MR:
-
Magnetic resonance
- NCMS:
-
Neurocutaneous melanosis
- NRAS:
-
Neuroblastoma RAS viral (v-ras) oncogene homolog
- PCR:
-
Polymerase chain reaction
References
Pavlidou E, Hagel C, Papavasilliou A, Giouroukos S, Panteliadis C: Neurocutaneous melanosis: report of three cases and up-to-date review. J Child Neurol 2008, 23: 1382–1391.
Makkar HS, Frieden IJ: Neurocutaneous melanosis. Semin Cutan Med Surg 2004, 23: 138–144.
Kinsler VA, Anderson G, Latimer B, Natarajan D, Healy E, Moore GE, Sebire NJ: Immunohistochemical and ultrastructural features of congenital melanocytic naevus cells support a stem-cell phenotype. Br J Dermatol 2013, 169: 374–383.
Melanosis, Neurocutaneous; NCMS; MIM#249400. Johns Hopkins University, Baltimore, MD; 2014.
Kinsler VA, Thomas AC, Ishida M, Bulstrode NW, Loughlin S, Hing S, Chalker J, McKenzie K, Abu-Amero S, Slater O, Chanudet E, Palmer R, Morrogh D, Stanier P, Healy E, Sebire NJ, Moore GE: Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J Invest Dermatol 2013, 133: 2229–2236.
Kusters-Vandevelde HV, Willemsen AE, Groenen PJ, Kusters B, Lammens M, Wesseling P, Djafarihamedani M, Rijntjes J, Delye H, Willemsen MA, van Herpen CM, Blokx WA: Experimental treatment of NRAS-mutated neurocutaneous melanocytosis with MEK162, a MEK-inhibitor. Acta Neuropathol Commun 2014, 2: 41.
Etchevers HC: Hiding in plain sight: molecular genetics applied to giant congenital melanocytic nevi. J Invest Dermatol 2014, 134: 879–882.
Bekiesinska-Figatowska M, Szczygielski O, Boczar M, Madzik J, Klepacka T, Michalak E, Romaniuk-Doroszewska A, Uliasz M, Peczkowski P, Sawicka E: Neurocutaneous melanosis in children with giant congenital melanocytic nevi. Clin Imaging 2014, 38: 79–84.
Ramaswamy V, Delaney H, Haque S, Marghoob A, Khakoo Y: Spectrum of central nervous system abnormalities in neurocutaneous melanocytosis. Dev Med Child Neurol 2012, 54: 563–568.
Scattolin MA, Lin J, Peruchi MM, Rocha AJ, Masruha MR, Vilanova LC: Neurocutaneous melanosis: follow-up and literature review. J Neuroradiol 2011, 38: 313–318.
Yang MM, Singhal A, Rassekh SR, Yip S, Eydoux P, Dunham C: Possible differentiation of cerebral glioblastoma into pleomorphic xanthoastrocytoma: an unusual case in an infant. J Neurosurg Pediatr 2012, 9: 517–523.
Rokitansky J: Ein ausgezeichneter Fall von Pigment-mal mit ausgebreiteter Pigmentierung der inneren Hin- und Ruchenmarkshaute. Allg Wien Med Z 1861, 6: 113–116.
Fu YJ, Morota N, Nakagawa A, Takahashi H, Kakita A: Neurocutaneous melanosis: surgical pathological features of an apparently hamartomatous lesion in the amygdala. J Neurosurg Pediatr 2010, 6: 82–86.
De Cock J, Snauwaert J, Van Rompaey W, Morren MA, Demaerel P: A newborn with neurocutaneous melanocytosis and Dandy-Walker malformation. Pediatr Neurol 2014, 50: 276–278.
Frieden IJ, Williams ML, Barkovich AJ: Giant congenital melanocytic nevi: brain magnetic resonance findings in neurologically asymptomatic children. J Am Acad Dermatol 1994, 31: 423–429.
Gerami P, Paller AS: Making a mountain out of a molehill: NRAS, mosaicism, and large congenital nevi. J Invest Dermatol 2013, 133: 2127–2130.
Charbel C, Fontaine RH, Malouf GG, Picard A, Kadlub N, El-Murr N, How-Kit A, Su X, Coulomb-L’Hermine A, Tost J, Mourah S, Aractingi S, Guegan S: NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol 2014, 134: 1067–1074.
Bauer J, Curtin JA, Pinkel D, Bastian BC: Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol 2007, 127: 179–182.
Hamm H: Cutaneous mosaicism of lethal mutations. Am J Med Genet 1999, 85: 342–345.
Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al.: A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 2010, 42: 27–29.
Ross AL, Sanchez MI, Grichnik JM: Molecular nevogenesis. Dermatol Res Pract 2011, 2011: 463184.
Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP: Ras mutations in human melanoma: a marker of malignant progression. J Invest Dermatol 1994, 102: 285–290.
van Elsas A, Zerp SF, van der Flier S, Kruse KM, Aarnoudse C, Hayward NK, Ruiter DJ, Schrier PI: Relevance of ultraviolet-induced N-ras oncogene point mutations in development of primary human cutaneous melanoma. Am J Pathol 1996, 149: 883–893.
Pavey S, Johansson P, Packer L, Taylor J, Stark M, Pollock PM, Walker GJ, Boyle GM, Harper U, Cozzi SJ, Hansen K, Yudt L, Schmidt C, Hersey P, Ellem KA, O'Rourke MG, Parsons PG, Meltzer P, Ringner M, Hayward NK: Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene 2004, 23: 4060–4067.
Sekine S, Nakanishi Y, Ogawa R, Kouda S, Kanai Y: Esophageal melanomas harbor frequent NRAS mutations unlike melanomas of other mucosal sites. Virchows Arch 2009, 454: 513–517.
Dessars B, De Raeve LE, Morandini R, Lefort A, El Housni H, Ghanem GE, Van den Eynde BJ, Ma W, Roseeuw D, Vassart G, Libert F, Heimann P: Genotypic and gene expression studies in congenital melanocytic nevi: insight into initial steps of melanotumorigenesis. J Invest Dermatol 2009, 129: 139–147.
Phadke PA, Rakheja D, Le LP, Selim MA, Kapur P, Davis A, Mihm MC Jr, Hoang MP: Proliferative nodules arising within congenital melanocytic nevi: a histologic, immunohistochemical, and molecular analyses of 43 cases. Am J Surg Pathol 2011, 35: 656–669.
Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, Massi D, Fonsatti E, Staibano S, Nappi O, Pagani E, Casula M, Manca A, Sini M, Franco R, Botti G, Caraco C, Mozzillo N, Ascierto PA, Palmieri G: BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol 2012, 30: 2522–2529.
Zuniga S, Las Heras J, Benveniste S: Rhabdomyosarcoma arising in a congenital giant nevus associated with neurocutaneous melanosis in a neonate. J Pediatr Surg 1987, 22: 1036–1038.
Paulus W, Slowik F, Jellinger K: Primary intracranial sarcomas: histopathological features of 19 cases. Histopathology 1991, 18: 395–402.
Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS: Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer 2006, 119: 2922–2930.
Tun K, Ozen O, Kaptanoglu E, Gurcan O, Beskonakli E, Celasun B: Primary intracranial low-grade fibromyxoid sarcoma (Evans tumor). J Clin Neurosci 2008, 15: 1298–1301.
Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, Janakiraman M, Solit D, Knauf JA, Tuttle RM, Ghossein RA, Fagin JA: Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res 2009, 69: 4885–4893.
Buccoliero AM, Castiglione F, Garbini F, Rossi Degl’Innocenti D, Moncini D, Franchi A, Paglierani M, Simoni A, Baroni G, Daniele D, Sardi I, Giordano F, Mussa F, Arico M, Genitori L, Taddei GL: Primary cerebral myxofibrosarcoma: clinical, morphologic, immunohistochemical, molecular, and ultrastructural study of an infrequent tumor in an extraordinary localization. J Pediatr Hematol Oncol 2011, 33: e279-e283.
Stratton MR, Fisher C, Gusterson BA, Cooper CS: Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res 1989, 49: 6324–6327.
Doisaki S, Muramatsu H, Shimada A, Takahashi Y, Mori-Ezaki M, Sato M, Kawaguchi H, Kinoshita A, Sotomatsu M, Hayashi Y, Furukawa-Hibi Y, Yamada K, Hoshino H, Kiyoi H, Yoshida N, Sakaguchi H, Narita A, Wang X, Ismael O, Xu Y, Nishio N, Tanaka M, Hama A, Koike K, Kojima S: Somatic mosaicism for oncogenic NRAS mutations in juvenile myelomonocytic leukemia. Blood 2012, 120: 1485–1488.
De Filippi P, Zecca M, Lisini D, Rosti V, Cagioni C, Carlo-Stella C, Radi O, Veggiotti P, Mastronuzzi A, Acquaviva A, D'Ambrosio A, Locatelli F, Danesino C: Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br J Haematol 2009, 147: 706–709.
Harrisingh MC, Lloyd AC: Ras/Raf/ERK signalling and NF1. Cell Cycle 2004, 3: 1255–1258.
Boccaccio C, Comoglio PM: Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer 2006, 6: 637–645.
Takayama H, La Rochelle WJ, Anver M, Bockman DE, Merlino G: Scatter factor/hepatocyte growth factor as a regulator of skeletal muscle and neural crest development. Proc Natl Acad Sci U S A 1996, 93: 5866–5871.
Takayama H, LaRochelle WJ, Sharp R, Otsuka T, Kriebel P, Anver M, Aaronson SA, Merlino G: Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A 1997, 94: 701–706.
Takayama H, Nagashima Y, Hara M, Takagi H, Mori M, Merlino G, Nakazato Y: Immunohistochemical detection of the c-met proto-oncogene product in the congenital melanocytic nevus of an infant with neurocutaneous melanosis. J Am Acad Dermatol 2001, 44: 538–540.
Tengs T, Lee JC, Paez JG, Zhao X, LaFramboise T, Giannoukos G, Thomas RK: A transforming MET mutation discovered in non-small cell lung cancer using microarray-based resequencing. Cancer Lett 2006, 239: 227–233.
Neklason DW, Done MW, Sargent NR, Schwartz AG, Anton-Culver H, Griffin CA, Ahnen DJ, Schildkraut JM, Tomlinson GE, Strong LC, Miller AR, Stopfer JE, Burt RW: Activating mutation in MET oncogene in familial colorectal cancer. BMC Cancer 2011, 11: 424.
Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R: c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res 2003, 63: 6272–6281.
Voortman J, Harada T, Chang RP, Killian JK, Suuriniemi M, Smith WI, Meltzer PS, Lucchi M, Wang Y, Giaccone G: Detection and therapeutic implications of c-Met mutations in small cell lung cancer and neuroendocrine tumors. Curr Pharm Des 2013, 19: 833–840.
Tyner JW, Fletcher LB, Wang EQ, Yang WF, Rutenberg-Schoenberg ML, Beadling C, Mori M, Heinrich MC, Deininger MW, Druker BJ, Loriaux MM: MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res 2010, 70: 6233–6237.
Flores-Sarnat L: Hemimegalencephaly: part 1. Genetic, clinical, and imaging aspects. J Child Neurol 2002, 17: 373–384.
Flores-Sarnat L: Neurocutaneous melanocytosis. Handb Clin Neurol 2013, 111: 369–388.
Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG: De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012, 44: 941–945.
Acknowledgements
Dr. Del Bigio holds the Canada Research Chair in Developmental Neuropathology. We thank Dr. Peter Burger (Johns Hopkins University) for his consultation on the brain tumor. We thank Tiana Baskin, Sharon Allen and Susan Janeczko for their technical assistance. We are grateful to the family for their cooperation with these investigations and permission to publish the findings. SY would like to acknowledge the funding support of BrainCare BC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FS – Literature review and initial drafts of manuscript. SY – Genetic analysis and drafting manuscript. PJM – Neurosurgical care of child, editing manuscript. AEC – Clinical care of child, genetic explanation, editing manuscript. MRD – Conceived project, final synthesis of manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Shih, F., Yip, S., McDonald, P.J. et al. Oncogenic codon 13 NRAS mutation in a primary mesenchymal brain neoplasm and nevus of a child with neurocutaneous melanosis. acta neuropathol commun 2, 140 (2014). https://doi.org/10.1186/s40478-014-0140-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-014-0140-8