
Abstract
High-grade Brainstem Glioma (BSG), also known as Diffuse Intrinsic Pontine Glioma (DIPG), is an incurable pediatric brain cancer. Increasing evidence supports the existence of regional differences in gliomagenesis such that BSG is considered a distinct disease from glioma of the cerebral cortex (CG). In an effort to elucidate unique characteristics of BSG, we conducted expression analysis of mouse PDGF-B-driven BSG and CG initiated in Nestin progenitor cells and identified a short list of expression changes specific to the brainstem gliomagenesis process, including abnormal upregulation of paired box 3 (Pax3). In the neonatal mouse brain, Pax3 expression marks a subset of brainstem progenitor cells, while it is absent from the cerebral cortex, mirroring its regional expression in glioma. Ectopic expression of Pax3 in normal brainstem progenitors in vitro shows that Pax3 inhibits apoptosis. Pax3-induced inhibition of apoptosis is p53-dependent, however, and in the absence of p53, Pax3 promotes proliferation of brainstem progenitors. In vivo, Pax3 enhances PDGF-B-driven gliomagenesis by shortening tumor latency and increasing tumor penetrance and grade, in a region-specific manner, while loss of Pax3 function extends survival of PDGF-B-driven;p53-deficient BSG-bearing mice by 33%. Importantly, Pax3 is regionally expressed in human glioma as well, with high PAX3 mRNA characterizing 40% of human BSG, revealing a subset of tumors that significantly associates with PDGFRA alterations, amplifications of cell cycle regulatory genes, and is exclusive of ACVR1 mutations. Collectively, these data suggest that regional Pax3 expression not only marks a novel subset of BSG but also contributes to PDGF-B-induced brainstem gliomagenesis.
Similar content being viewed by others


Introduction
Brainstem Glioma (BSG) is a brain tumor that arises anywhere in the brainstem, and is seen predominately in children. Although BSG accounts for only 15-20% of all pediatric brain tumors, it is the leading cause of death for children with brain tumors [1]. The majority of BSG (80-85%) are high grade, diffuse, and are located in the pons—these are also known as Diffuse Intrinsic Pontine Glioma (DIPG) and have an overall survival of less than one year [1]. The term BSG hereafter will refer to high grade BSG or DIPG. Conventional focal radiation therapy remains the standard-of-care for these tumors, with no documented additional benefit for any alternative treatment [2].
Genetic alterations commonly found in BSG include gains and/or activating mutations in platelet-derived growth factor receptor alpha (PDGFRA), amplification of genes involved in the receptor tyrosine kinase-Ras-phosphoinositide 3-kinase signaling pathway or cell cycle regulation, and a K27M mutation of histone H3.3 or 3.1 [3]-[9]. Recently, mutations in the ACVR1 gene have been discovered in 20-32% of cases [10]-[13], as well as mutations in the PPM1D gene [11],[14]. The most frequent tumor suppressor lost is TP53, with INK4A-ARF less commonly absent at the genomic level but possibly silenced through alternative mechanisms [3],[6],[15],[16]. Previous work on BSG has characterized distinct subsets of the disease, including mesenchymal and oligodendroglial [5], N-Myc and Hedgehog [17], MYCN, silent, and H3-K27M [10] or H3-K27M and wildtype [6]. Together, these classifications highlight the heterogeneity of this disease and the likelihood that effective treatment will require an understanding of the mechanisms driving the growth of each individual subtype.
In addition to the heterogeneity within pediatric BSG, increasing evidence suggests that this disease is biologically distinct not only from adult glioma but also from pediatric supratentorial glioma in terms of genetic alterations and expression signatures [3]-[5]. Gliomas arising in the midline of the central nervous system (including the brainstem) commonly harbor the H3.3/3.1-K27M mutation only, while gliomas that arise in the cerebral cortex may contain an alternative H3.3-G34R/V mutation [6]-[9]. Regional differences in glioma may be a consequence in part of innate differences in the cells from which the tumors arise. Several studies have shown that NSCs from various regions of the brain differ in their capacity to respond to oncogenic stimuli in vitro and in vivo[18],[19]. In addition, human infratentorial and supratentorial low-grade gliomas have distinct expression signatures corresponding to normal astrocytes and NSCs from their respective regions [20], while the expression patterns of G34R/V and K27M mutant gliomas mimic the developmental region in which they arise [8].
One gene found to be expressed at higher levels in infratentorial compared to supratentorial low-grade glioma is paired box 3 (PAX3) [20]. Pax3 is a developmental transcription factor that displays a regionalized expression pattern in the neural tube, neural crest, and somite of the vertebrate embryo, with expression decreasing as differentiation progresses [21]. Early in embryogenesis, Pax3 is transiently expressed in the posterior and dorsal neural tube [22]-[24]. This leads to its expression in the developing CNS to be localized to infratentorial structures such as the brainstem and cerebellum, but not to supratentorial domains such as the cerebral cortex (see P4 in situ hybridization for Pax3 in [25]). Pax3 is not only necessary for the proper specification of these developing tissues [21],[26]-[28], but also for cell survival as homozygous loss of Pax3 in the mouse leads to severe neural crest and neural tube defects, attributable to an induction of p53-dependent apoptosis [29]-[31].
Ectopic expression of Pax3 is found to be pro-tumorigenic in sarcomas and neural crest-derived tumors such as melanoma and neuroblastoma [21]. However its role in glioma is less well-understood. Pax3 is re-expressed and upregulated in human glioma relative to normal brain tissue with its expression increasing concomitant with World Health Organization Grade and worsening prognosis, irrespective of tumor location [32]. Functionally, Pax3 has been implicated in promoting proliferation and invasion and inhibiting apoptosis of glioma cell lines, and promoting growth of glioma subcutaneous xenografts [33].
In this study, we sought to gain insight into the unique attributes of BSG by comparing it to high-grade glioma of the cerebral cortex (CG). In an expression array comparing mouse BSG and CG induced by PDGF-B overexpression and Ink4a-ARF loss [34], we identified Pax3 as being a brainstem-specific marker. Pax3 is regionally expressed in the neonatal mouse brain as well, characterizing a subset of Nestin progenitors in the brainstem but not in the cerebral cortex. Ectopic overexpression of Pax3 in brainstem progenitors inhibits apoptosis in vitro and enhances PDGF-B-induced brainstem gliomagenesis in vivo, in a region-specific manner. In the absence of p53, while Pax3 no longer inhibits apoptosis, it increases proliferation in vitro and maintains its pro-tumorigenic role in vivo. Importantly, the regional expression pattern of Pax3 is mirrored in human glioma, revealing a subset of BSG with high PAX3 expression that associates with alterations in PDGFRA and cell cycle regulatory genes and is exclusive of ACVR1 mutations. These data suggest that regional expression of Pax3 contributes to PDGF-B-induced brainstem gliomagenesis and characterizes a subset of human BSG.
Materials and methods
Mice
Nestin-Tva (Ntv-a) and Ntv-a;Ink4a-ARF−/−[34] and Ntv-a;p53fl/fl mice [35] have been described. Olig2-eGFP-L10a [36],[37] were bred to Ntv-a;Ink4a-ARF−/− mice to generate Ntv-a;Ink4a-ARF−/−;Olig2-eGFP-L10a mice. Pax3-floxed mice [38] were bred to Ntv-a;p53fl/fl mice to generate Ntv-a;p53fl/fl;Pax3fl/fl (Pax3-KO) and control Ntv-a;p53fl/fl;Pax3+/+ mice. Nestin-CFPnuc mice have been previously described, and express the cyan fluorescent protein fused to a nuclear localization signal under the control of the regulatory elements of the Nestin gene [39]. For genotyping, gDNA was isolated from mice using the REDExtract-N-Amp Tissue PCR Kit (Sigma) per the manufacturer's protocol, using previously published primers. All work with mice was done in accordance with the Duke University Animal Care and Use Committee and the Guide for the Care and Use of Laboratory Animals.
RCAS/Tv-a glioma mouse modeling
In the RCAS/Tv-a glioma mouse modeling system, avian retroviruses (produced from RCAS plasmids) infect mouse cells expressing Tv-a (the receptor for RCAS viruses). DF1 cells (ATCC) were cultured and transfected with RCAS plasmids as described [35] using Fugene 6 or X-TremeGENE 9 (Roche). RCAS-Y, RCAS-PDGF-B, and RCAS-Cre plasmids are described [40]. RCAS-Pax3 plasmid was obtained from Andrew Bendall (University of Guelph, Ontario) [41]. RCAS-H3.3-K27M plasmid was generated by the C. D. Allis lab (The Rockefeller University, New York) [42]. Generation of PDGF-B-induced brainstem glioma [34],[35] and cerebral cortex glioma [43] were as described. Briefly, 1×105 DF1 cells transfected with RCAS plasmids and producing RCAS viruses, were injected into the brainstem or cerebral cortex of neonatal Ntv-a pups (postnatal day 2–4) in 1 μL volume using a custom Hamilton syringe. Combinations of viruses were injected at a 1:1 or a 1:1:1 ratio, and RCAS-Y transfected DF1 cells were used as a negative control. Injected mice were monitored daily and euthanized with CO2 upon appearance of signs of brain tumors (enlarged head, ataxia, weight loss up to 25%) or at 12 weeks post-injection in the absence of symptoms.
FACS of Olig2-eGFP-L10a glioma
Brainstem or Cerebral Cortex Gliomas from Ntv-a;Ink4aARF−/−;Olig2-eGFP-L10a mice injected with RCAS-PDGF-B were harvested and dissociated into a single cell suspension as described [35]. Briefly, tissue was digested in papain (Worthington) and DNAse (Sigma Aldrich) at 37°C, followed by three cycles of triturations in ovomucoid (Worthington) and centrifugation at 600 rpm. The resulting cells were passed through a 70 μm strainer, an aliquot of cells was set aside for the unsorted control, and the remainder of cells were sorted using a FACSVantage sorter (BD Bioscience) by the Duke Cancer Institute Flow Cytometry Core into GFP + and GFP- populations. Cell populations were centrifuged, washed in PBS, snap frozen and stored at −80°C for mRNA isolation.
Immunofluorescence
Ntv-a or Nestin-CFPnuc pups were sacrificed at P3 and their whole brains were fixed in 4% PFA in PBS for 24 hours, cryopreserved in 30% sucrose in PBS for 24–48 hours, followed by embedding in OCT on dry ice/ethanol. Blocks were sectioned using either a Shandon or Leica Cryostat into 12 μm thick sections in a sagittal orientation. Sections were rehydrated in PBS-T (0.1% Triton-X100) and blocked in PBS-T with 10% normal goat serum. Primary antibodies were diluted in PBS-T with 1% BSA overnight at 4 degrees, and secondary antibodies were diluted in PBS-T for 1 hour. Antibodies used were anti-PAX3 (DSHB, concentrated form, 1:200), and anti-GFP (Invitrogen, 1:200); AlexaFluor-488 and −594 secondary antibodies (Invitrogen) were used at 1:200. Slides were mounted with Vectashield with Dapi (Vector Laboratories) and imaged using a Zeiss Axio Imager.
In order to quantify PAX3 and Nestin expression, three Nestin-CFPnuc P3 mice were analyzed. High-powered (20x) pictures of the PAX3-expressing cells in each brainstem region (midbrain, dorsal pons, mid pons, and ventral pons) were taken, 1–3 images per region per section, until at least 6 sections were represented for each region. All pictures were taken using a Zeiss Axio Imager and consistent camera settings. For the cerebral cortex, 2 random 20x images were taken in at least 6 sections per mouse. Using Metamorph Premier software, a threshold was set for positive Nestin and PAX3 staining in each image. For each marker, the same threshold was applied to each image across the dorsal, mid, and ventral pons regions for each section. For the midbrain region, the threshold for PAX3 was consistently lowered slightly to accommodate a lower intensity of the PAX3 staining compared to the other regions. The threshold for Nestin staining in the cerebral cortex was set separately, and remained the same for all sections and all mice. There was no positive nuclear PAX3 staining in any cerebral cortex section. The positive nuclei (size > 200) for each marker were then counted using Integrated Morphometry Analysis. A color threshold was then applied to each Nestin/PAX3 overlay (the same threshold was used across regions for each section), and the number of double-positive cells were counted as described above. The cell counts for each region per section were totaled, and the percentage of Nestin + cells that were double positive per region was calculated. The percentages were averaged together across sections of the same mouse to generate a mean percentage for each region per mouse.
Mouse and human expression analysis
Frozen BSG and CG samples, along with brainstem and cerebral cortex from age-matched non tumor-bearing mice of the same genetic background (n = 3 for all groups), were sent to Expression Analysis (EA, Durham, NC) for total mRNA isolation and hybridization to Affymetrix GeneChip Mouse Genome 430 2.0 Arrays. Total mRNA samples isolated from Olig2-eGFP-L10a-positive PDGF-B;Ink4aARF−/− glioma cells were sent to EA for hybridization to the same arrays. Data was analyzed using Partek Genomics Suite (Partek Incorporated) for quality control, Robust Multi-Array (RMA) was used for normalization, and differentially expressed genes were determined by one-way ANOVA using the cutoffs described in the text.
Human BSG and CG samples with expression data available from St. Jude [4],[44] were selected for analysis. Log-scale Robust Multiarray Analysis (RMA) from the affy[45] package in Bioconductor [46] was used for normalization to eliminate systematic differences across the arrays. Differentially expressed genes between brainstem and cerebral cortex glioma samples were identified using the limma[47] package in Bioconductor, which utilizes an empirical Bayes' approach for parameter estimation and significance testing. The False Discovery Rate (FDR) method was employed to control for multiple hypothesis testing. To compare gene expression differences within the brainstem samples, RMA normalization was applied to the arrays where they were then classified based on their normalized PAX3 expression levels (probe 231666_at). Arrays where PAX3 had a normalized expression value > 4 were classified as PAX3-High, while all others were classified as PAX3-Low. Identification of genes differentially expressed between the two groups was calculated using limma as described above. These same classifications of PAX3-High and PAX3-Low were used for associations with genetic alterations, where the genetic alterations were acquired from previously published data [4],[9],[11],[48].
Human BSG and CG samples with expression data available from Necker-Sick Children [5] were selected for an independent analysis. The Agilent microarray data was processed and normalized using the limma[47] package in Bioconductor [46]. The normexp method (normal + exponential convolution model) was used to correct for background intensities. Quantile normalization was used to eliminate systematic differences across all of the arrays. Differentially expressed genes between brainstem and cerebral cortex glioma samples were determined using the methods described above for the first dataset. To compare gene expression differences within the brainstem samples, arrays were classified based on their PAX3 expression levels (probe name: A_23_P502706). Arrays with a PAX3 expression level > 7 were classified as PAX3-High, whereas all others were classified as PAX3-Low. Identification of genes differentially expressed between the two groups was calculated using limma as described above.
In vitro infection of brainstem progenitors with RCAS viruses
Normal brainstem was isolated from Ntv-a or Ntva;p53fl/fl postnatal day 3 (P3) pups and was digested and dissociated as described above for Olig2-eGFP-L10a tumors. Cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C and 5% CO2, and passaged a maximum of 3 times for experiments. Cells were plated in clear 6-well plates for Annexin V assays, clear 96-well plates for BrdU assays, and white-walled, clear-bottomed 96-well plates for Caspase 3/7 assays.
To concentrate RCAS viruses, DF1 cells transfected with RCAS plasmids were passaged a minimum of 6 times from transfection, and then passaged 1:12. After 3 days, virus-containing media was harvested, centrifuged to remove cell debris, filtered through 0.45 μm pores, and concentrated 100-fold using Retro-X Concentrator (Clontech) per the manufacturer's instructions.
Brainstem progenitors were plated and infected with RCAS viruses at 1:100. Assays were conducted 3–5 days post-infection, or were split when confluent and subsequently plated for assays. For BrdU assays, the Cell Proliferation ELISA, BrdU Colorimetric kit (Roche) was utilized, per the manufacturer's instructions using a Molecular Devices Versa Max Tunable Microplate reader. Caspase 3/7 activity was measured using the ApoToxGlo Triplex assay (Promega) per the manufacturer's instructions using a Turner Biosystems Modulus Microplate Reader. To measure the percentage of cells staining positive for Annexin V, the FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen) was utilized per the manufacturer's instructions. For cell counting experiments, 50,000 infected cells were plated in 6 well plates in duplicate. For each time point, 2 wells of each line were trypsinized and counted with a Sceptor 2.0 Cell Counter (Millipore). All experiments were done a minimum of three times on at least three independent preparations of progenitor cells.
Tumor grading and immunohistochemistry
Tumor samples fixed in 10% formalin were embedded in paraffin by the Duke Pathology Core and cut into 5 μm thick sections using a Leica RM2235 microtome. H&E staining was performed using standard protocols. Tumor grading was done using the following criteria: Grade II glioma indicated by increased cellular density only; Grade III glioma indicated by the presence of microvascular proliferation; Grade IV glioma indicated by the presence of pseudopalisading necrosis. Immunohistochemistry (IHC) was performed using an automated processor (Discovery XT, Ventana Medical Systems, Inc.). Anti-PCNA antibody (Calbiochem) was used at 1:2000.
PCR
DNA was isolated from frozen tumors using the DNeasy Blood and Tissue Kit (Qiagen) per the manufacturer's instructions. Detection of the recombined Pax3-floxed allele was performed using previously published primers [38] and 60°C annealing temperature. For RT-PCR (both conventional and real time), cultured cells or snap frozen tumors were processed for mRNA isolation using the RNeasy Mini kit (QIAGEN) per the manufacturer's protocol. cDNA was synthesized from total mRNA using SuperScript II and OligodT primer (Invitrogen). RT-PCR primers for PDGF-B, Forward: 5′-AGTGACCACTCGATCCGCTCCT-3′, Reverse: 5′- TTTGGGGCGTTTTGGCTCGCTG-3′, 55°C annealing temperature. PDGF-B primers amplify both mouse and human PDGF-B cDNA. qRT-PCR reactions were set up with iQ SYBR Green Supermix (BioRad) and run on a Bio-Rad iQ5 Multicolor Real-Time PCR Detection System. Primers for Pax3, Forward: 5′- TCCATCCGACCTGGTGCCAT-3′, Reverse: 5′- TTCTCCACGTCAGGCGTTG, and Actin, Forward: 5′- TATTGGCAACGAGCGGTTCC-3′, Reverse: 5′- GGCATAGAGGTCTTTACGGATGTC-3′; 60°C annealing temperature. Relative gene expression levels were generated using the ΔΔCt method [49] using Actin as the reference gene.
Western blot
Frozen tumors were either grinded with a mortar and pestle on liquid nitrogen followed by lysis in RIPA buffer containing 1x Protease Inhibitor Cocktail (Sigma Aldrich), 10 mM PMSF, 50 mM NaF, 1 mM NaVO4, and 1 mM DTT or lysed and homogenized in the same buffer using a Glas-Col Variable Speed-Reversible Homogenizer set to a motor speed of 80 followed by sonication using a Brason Sonifier 250. Cleared and denatured lysates were run on a NuPAGE 4-12% Bis-Tris gradient gel (Invitrogen), and transferred onto nitrocellulose membrane using the iBlot (Invitrogen). Antibodies were diluted in Odyssey Blocking Buffer (Li-Cor) with 0.2% Tween-20. Blots were imaged using the Odyssey (Li-Cor) per the manufacturer's instructions. Antibodies used were: anti-PAX3 (DSHB, concentrated form, 1:1000) and anti-ACTIN (Santa Cruz Biotechnology, 1:500). Secondary antibodies from Li-Cor were used at 1:10,000 for IRDye800CW or 1:20,000 for IRDye680LT.
Statistical analysis
Statistics were calculated using GraphPad Prism 5 software. In vitro assays were performed a minimum of 3 times using cells from at least 3 independent P3 litters. Data are represented as the mean with SEM, using student's paired t-test. qRT-PCR for Pax3 in mouse tissue was analyzed using student's unpaired t-test. In vivo analyses were performed using log-rank test (for survival) and Fisher's exact test (for tumor penetrance and grade). For analyses of human data, unpaired t-test was used for the age at diagnosis of PAX3-Low versus PAX3-High tumors, and Fisher's exact test was used to test for significance of the association between high Pax3 expression and genetic alterations.
Results
Mouse brainstem glioma and cerebral cortex glioma have distinct gene expression signatures
In order to identify regional differences in glioma, and in particular unique characteristics of those arising in the brainstem, we generated mouse Brainstem Glioma (BSG) and Cerebral Cortex Glioma (CG) by injecting RCAS-PDGF-B virus-producing cells into the brainstem or cerebral cortex, respectively, of postnatal day 2-4 (P2-4) Nestin-Tv-a(Ntv-a);Ink4aARF−/− mice (Figure 1a) [34],[43]. The gene expression profiles of the resulting tumors were first compared to their normal tissue counterparts. Using an FDR-adjusted p-value <0.05 and fold-change ≥2.0, 7,869 genes were differentially expressed between normal cortex (NC) and CG (Figure 1b, Additional file 1: Table S1), while 7,263 genes were altered in BSG compared to normal brainstem (NBS) (Figure 1b, Additional file 2: Table S2). Of the latter group, 2,547 genes were specific to the brainstem gliomagenesis process and were not altered during gliomagenesis in the cerebral cortex (Figure 1b). In addition, the comparison between BSG and CG revealed only 23 genes differentially expressed between these two types of tumors (Figure 1b-c, Additional file 3: Table S3), including upregulation of Irx5 and Pax3 and downregulation of Bmp4 and Foxg1. We were interested in genes that functionally contribute to the brainstem gliomagenesis process, and so when these 23 genes were intersected with the 2,547 BSG-associated genes, 10 probes, representing 8 brainstem-specific gliomagenesis genes, were identified (Figure 1b, Table 1).

Regional differences in PDGF-B-driven mouse glioma. a Glioma was initiated in the mouse by injecting RCAS-PDGF-B virus into the brainstem or cerebral cortex of neonatal Ntv-a;Ink4a-ARF−/− mice. Shown is a sagittal section of a wildtype postnatal day 4 brain, stained with H&E, indicating the location of brainstem and cerebral cortex injections. Scale bar is 1 mM. b-c Expression profiling was conducted on the resulting Brainstem Glioma (BSG) and Cerebral Cortex Glioma (CG) and compared with age-matched normal brainstem (NBS) and normal cerebral cortex (NC), n = 3 for each. b Venn diagram showing the intersection of genes differentially expressed between NBS and BSG (green circle), between NC and CG (blue circle), and between BSG and CG (red circle). c Hierarchical clustering of 23 genes differentially regulated between BSG and CG. FDR-adjusted p-value <0.05 and fold-change ≥2.0.
The expression array comparing Brainstem and Cerebral Cortex Gliomas described above was conducted with whole tumor tissue. Therefore it is possible that non-neoplastic cells contributed to some of the expression differences. To investigate this, we utilized the Olig2-eGFP-L10a reporter strain [36],[37], in which the eGFP-L10a fusion protein is expressed under the control of the Olig2 promoter. As Olig2 expression marks all tumor cells of our glioma model [34], this effectively labels tumor cells with GFP. Ntv-a;Ink4a-ARF−/−;Olig2-eGFP-L10a mice were injected with RCAS-PDGF-B into the brainstem or cerebral cortex to generate BSG or CG, respectively, within 4–6 weeks from injection (Additional file 4: Figure S1a). Tumors were harvested, dissociated, and sorted into GFP-positive and -negative populations. The GFP-positive fractions were analyzed for gene expression differences by microarray, and 118 genes were found differentially regulated between Olig2-BSG and Olig2-CG tumor cells (Additional file 4: Figure S1b, Additional file 5: Table S4). Importantly, of the 8 brainstem-specific gliomagenesis genes in Table 1, the genes highlighted in bold were also differentially regulated between Olig2-BSG and Olig2-CG cells, and presumably play a role in BSG tumor cell biology.
Pax3 expression characterizes mouse brainstem glioma
Among the 8 brainstem-specific gliomagenesis genes identified in this screen, 3 independent probes for the developmental transcription factor paired box 3 (Pax3) showed upregulation in BSG over CG and over normal brainstem (Table 1, Figure 2a). This differential expression pattern was confirmed via qRT-PCR (Figure 2b). Pax3 was undetectable by qRT-PCR in normal cerebral cortex or CG tissue (n = 3 for each, data not shown). Importantly, Pax3 was also differentially expressed between Olig2-BSG and -CG cells, with increased expression in BSG relative to CG (Figure 2c, Additional file 5: Table S4). To determine whether Pax3 expression is confined to the Olig2-tumor cell compartment of BSG, qRT-PCR for Pax3 was conducted on the sorted GFP-positive and -negative cells, relative to unsorted tumor cells. This revealed that Pax3 expression is primarily found within the Olig2-tumor cell compartment of BSG (Figure 2d). Pax3 mRNA was undetectable by qRT-PCR in all compartments of Olig2-eGFP-L10a CG (n = 3, data not shown).

Pax3 is expressed in mouse Brainstem Glioma. a Log expression level of Pax3 for one probe from the expression array in Figure 1. b qRT-PCR for Pax3 in normal brainstem (NBS) and brainstem glioma (BSG) tissue, n = 3 for each. Relative Pax3 expression is normalized to Actin. p = 0.01, unpaired t-test. c Log expression level of Pax3 for one of three probes from the Olig2-eGFP-L10a glioma expression array from Additional file 4: Figure S1b. d qRT-PCR for Pax3 in unsorted (Uns), GFP+, and GFP- fractions of Olig2-eGFP-L10a BSG (n = 5). Pax3 expression levels are normalized to Actin and represented as a percentage of unsorted cells. p = 0.04 Uns vs. GFP+, p = 0.002 GFP+ vs. GFP-, paired t-test. e Western blot for PAX3 (53 kDa) in the following mouse tissues from left to right: P3 cerebellum (CB), P3 cerebral cortex (NC), and P3 brainstem (NBS); adult normal cerebral cortex (NC), Cerebral Cortex Glioma (CG), adult normal brainstem (NBS), and Brainstem Glioma (BSG). ACTIN (43 kDa) is shown as a loading control. f Western blot for PAX3 (53 kDa) in PDGF-B;H3.3-K27M;p53-deficient Cerebral Cortex Glioma (CG) and Brainstem Glioma (BSG). ACTIN (43 kDa) is shown as a loading control.
PAX3 protein can also be detected at higher levels in mouse BSG than CG by western blot (Figure 2e). To determine whether high expression of PAX3 occurs in p53-deficient BSG, we injected Ntv-a;p53fl/fl mice with DF1 cells producing RCAS-PDGF-B and RCAS-Cre viruses [35]. In the tumors that arose in these mice, we found PAX3 protein expression at levels comparable to those found in Ink4aARF-deficient tumors (Figure 2e). In addition, to investigate whether the regional expression pattern of Pax3 is relevant in the context of the commonly occurring H3.3-K27M histone mutation, we injected Ntv-a;p53fl/fl mice with DF1 cells producing RCAS-PDGF-B, RCAS-H3.3-K27M, and RCAS-Cre viruses [42]. As shown in Figure 2f, we see high levels of PAX3 protein in PDGF-B;H3.3-K27M;p53-deficient gliomas initiated in the brainstem, but not in the cerebral cortex. Collectively, these data indicate that Pax3 is a brainstem-specific marker of mouse PDGF-B-driven glioma in the context of Ink4aARF-deficiency, p53-deficiency, as well as H3.3-K27M expression.
Pax3 is regionally expressed in the neonatal mouse brain
High Pax3 expression characterizes PDGF-B-driven mouse BSG while its expression is lower in glioma arising in the cerebral cortex. We speculated that enhanced PDGF signaling induced by PDGF-B ligand might upregulate Pax3 in the brainstem and be responsible for its high expression level. However, when brainstem progenitor cells isolated from Ntv-a P3 mice were infected in vitro with RCAS-PDGF-B virus, Pax3 mRNA levels did not increase (Figure 3a).

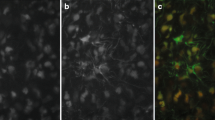
Regional expression of Pax3 in the neonatal mouse brain. a qRT-PCR for Pax3 in RCAS-PDGF-B- versus RCAS-Y-infected brainstem progenitor cells. Pax3 levels are normalized to Actin. b-f Immunofluorescence for PAX3 (red) and DAPI (blue) (upper panels), and PAX3 (red) and Nestin-CFP (green) (lower panels) in P3 Nestin-CFPnuc brain; 20x magnification, scale bar is 50 μM in b dorsal pons (white arrows point to Nestin+/PAX3+ cells), c ventral pons, d 4th ventricle roof, e midbrain, f cerebral cortex. g The percentage of Nestin+ cells in each of the indicated brain regions that also express PAX3, quantified as described in the Methods section.
As Pax3 is a developmental transcription factor whose expression normally decreases as differentiation proceeds, we next investigated whether Pax3 is expressed at the time of tumor initiation in the brainstem, which in our mouse model is P2-4. By Western blot, PAX3 protein was detected in wildtype P3 brainstem but not cerebral cortex (Figure 2e, cerebellum is shown as a positive control), consistent with its expression pattern in glioma. To further characterize the expression pattern of Pax3 in the developing brain, we conducted immunofluorescence for PAX3 protein in the P3 mouse brain and found several regions of expression within and around the developing brainstem. Distinct clusters of cells immunoreactive for PAX3 were found in the dorsal, mid, and ventral pons (Figure 3b-c, Additional file 6: Figure S2a-b, upper panels). In addition, we found PAX3-positive cells lining the roof of the 4th ventricle and radiating up and out from there into portions of the midbrain, which lies just anterior to the mouse pons (Figure 3d-e, upper panels). PAX3-positive cells were also found occasionally lining the floor of the 4th ventricle (Additional file 6: Figure S2c, upper panels). By contrast, there were no PAX3-positive cells detected by immunofluorescence in the cerebral cortex (Figure 3f, upper panel).
We next investigated whether Pax3 expression characterizes any Nestin-progenitors, the targeted cell-of-origin in our glioma mouse model, in the P3 mouse brainstem. We conducted co-immunofluorescence for cyan fluorescent protein (CFP) and PAX3 in the P3 brain of Nestin-CFPnuc mice [39] which express CFP fused to a nuclear localization signal under the control of the Nestin promoter. PAX3 expression was found in a subset of Nestin progenitors in the dorsal pons, the roof and floor of the 4th ventricle, and the midbrain (Figures 3b, d, e, and Additional file 6: Figure S2c, lower panels, and quantified in Figure 3g). Conversely, we found very little to no co-expression of Nestin and PAX3 in the ventral or mid pons and none in the cerebral cortex (Figure 3c, f, and S2b, lower panels, quantified in Figure 3g). Thus the regional expression pattern of Pax3, and its co-expression with Nestin, in the neonatal mouse brain correlates with its expression in glioma.
Pax3 inhibits apoptosis of brainstem progenitor cells
To investigate the functional role of Pax3 in brainstem progenitor cells, the brainstem region (including the midbrain and pons) from P3 Ntv-a mice was isolated, dissociated, and cultured in vitro. The Nestin-expressing progenitor cells were infected with RCAS-Y, RCAS-Pax3, RCAS-PDGF-B, or a 1:1 combination of RCAS-Pax3 and RCAS-PDGF-B (Figure 4a and Additional file 7: Figure S3a). RCAS-Pax3 alone, or in combination with PDGF-B, did not directly increase proliferation of brainstem progenitors based on BrdU incorporation (Additional file 7: Figure S3b). However, Pax3 inhibited basal apoptosis as evidenced by caspase 3/7 activation (Figure 4b). Similarly, Pax3 overexpression reduced the percentage of cells staining positive for Annexin V, a marker for early apoptosis, when compared to RCAS-Y-infected cells (Figure 4c).

Pax3 inhibits apoptosis of brainstem progenitor cells. Brainstem progenitor cells from P3 Ntv-a mice were cultured in vitro and infected with the indicated RCAS viruses. a qRT-PCR for Pax3 in the infected progenitor cells, normalized to Actin. Levels for Pax3-infected cells are expressed relative to RCAS-Y-infected cells, p = 0.007 paired t-test, and levels for PDGF-B + Pax3-infected cells are expressed relative to PDGF-B-infected cells, p = 0.03 paired t-test. b Caspase 3/7 activation of Pax3-infected cells is normalized to that of RCAS-Y-infected cells, p = 0.03 paired t-test, while caspase 3/7 activation of PDGF-B + Pax3-infected cells is normalized to that of PDGF-B-infected cells, p = 0.02 paired t-test. c Annexin V staining of RCAS-Pax3- versus RCAS-Y-infected progenitor cells. The percentage of Pax3-infected cells that were AnnexinV+/PI- was normalized to the percentage of RCAS-Y-infected cells that were AnnexinV+/PI-. p = 0.005, paired t-test.
Pax3 enhances PDGF-B-induced brainstem gliomagenesis in vivo
In order to determine if Pax3-induced inhibition of apoptosis is sufficient to induce glioma in vivo, P3 Ntv-a mice were injected in the brainstem with RCAS-Pax3, RCAS-PDGF-B, or a 1:1 mixture of both viruses. While PDGF-B overexpression alone led to asymptomatic low-grade glioma in 5 out of 20 mice, Pax3 overexpression alone did not lead to tumor formation (Figure 5a-c). When compared to PDGF-B injection alone, the addition of Pax3 to PDGF-B significantly reduced survival (Figure 5a) and increased tumor penetrance to 20 out of 25 mice (p = 0.0003, Figure 5b). Importantly, addition of Pax3 to PDGF-B overexpression caused a subset of tumors to progress to high-grade glioma based on the presence of microvascular proliferation (0/5 PDGF-B versus 5/20 PDGF-B + Pax3 high-grade, p = 0.056, Figure 5b-c). The Pax3-induced high-grade gliomas harbored increased proliferation compared to the low-grade tumors induced by PDGF-B alone based on PCNA staining (Figure 5d). The pro-tumorigenic role of Pax3 in glioma is brainstem-specific, as co-injection into the cortex of both PDGF-B and Pax3 did not significantly reduce survival or increase tumor penetrance (p = 1.0) or grade (p = 1.0) compared to injection of PDGF-B alone (Additional file 8: Figure S4). Therefore, in the presence of enhanced PDGF-B signaling, Pax3 is sufficient to generate aggressive high-grade BSG, a feat that PDGF-B alone cannot achieve in the brainstem.

Pax3 enhances PDGF-B-induced Brainstem Gliomagenesis. a Kaplain-Meier survival curve of Ntv-a mice injected with DF1 cells expressing RCAS-Pax3, RCAS-PDGF-B, or RCAS-PDGF-B + RCAS-Pax3. b Mice from (a) were sacrificed at the onset of tumor symptoms or at 12 weeks in the absence of symptoms. Brains were harvested, fixed in formalin, and analyzed for the presence of tumors using hematoxylin and eosin (H&E) staining, and the tumors graded as described in Materials and Methods. Shown is the percentage of mice in each group with no tumor, grade II glioma, and grade III glioma. c-d Representative H&E staining (c) and IHC for proliferating cell nuclear antigen, PCNA (d) of normal brainstem (top row), PDGF-B-induced grade II BSG (middle row), and PDGF-B + Pax3-induced grade III BSG (bottom row). c Magnification is 10x in the left panels and 40x in the right panels. White arrow in lower-left panel indicates microvascular proliferation, which is enlarged in the 40x panel to its right. Scale bar for 10x is 100 μM, 40x is 25 μM. d 40x magnification; Scale bar is 25 μM.
Pax3 promotes proliferation and gliomagenesis in the brainstem independent of p53
As the majority of human BSG are p53-deficient, we investigated the role of Pax3 in our BSG mouse model in the absence of this tumor suppressor. Brainstem progenitor cells were isolated from P3 Ntv-a;p53fl/fl mice and infected with RCAS-Cre to knock-out p53 along with either RCAS-Y or RCAS-Pax3. In the absence of p53, Pax3 overexpression no longer inhibited caspase 3/7 activation (Figure 6a). However, in these p53-deficient progenitor cells, Pax3 overexpression did lead to a significant increase in cell proliferation, indicated by BrdU incorporation (Figure 6b) and cell counting (Figure 6c-d).

Pax3 promotes proliferation in vitro and brainstem gliomagenesis in vivo in the absence of p53. a-d Brainstem progenitor cells from P3 Ntv-a;p53fl/fl mice were cultured in vitro, infected with the indicated RCAS viruses, and assayed for (a) Caspase 3/7 activity at 72 hours after plating, normalized to Cre + RCAS-Y, (b) BrdU incorporation at 72 hours after plating, normalized to Cre + RCAS-Y, p = 0.0007 paired t-test, and (c-d) cell number at the indicated time points after plating. c One representative experiment conducted in one progenitor line. d The fold change in Cre + Pax3 cells over Cre + RCAS-Y cells at each time point compiled from multiple progenitor lines, p = 0.03 (day 4), p = 0.005 (day 5), p < 0.0001 (day 6), paired t-test. e Kaplan-Meier survival curve of Ntv-a;p53fl/fl mice (Control) and Ntv-a;p53fl/fl;Pax3fl/fl mice (Pax3-KO) injected with RCAS-PDGF-B + RCAS-Cre into the brainstem at P2-4.
In order to test whether Pax3 is functionally required for the initiation or progression of p53-deficient BSG in vivo, we crossed conditional Pax3 knockout mice [38] into our p53-deficient BSG model [35]. Neonatal Ntv-a;p53fl/fl;Pax3fl/fl mice (Pax3-KO) and Ntv-a;p53fl/fl;Pax3+/+ mice (Control), were injected with RCAS-PDGF-B and RCAS-Cre. Although both groups of mice succumbed to high-grade BSG, the Pax3-KO mice displayed a significant 33% increase in survival (median survival 38 days versus 28.5 days, Figure 6e). Recombination of the Pax3-floxed allele was confirmed via PCR of gDNA and loss of PAX3 protein expression was confirmed via Western blot in Pax3-KO tumors (Additional file 9: Figure S5a-b). The increased survival of Pax3-KO mice suggests that Pax3 functionally contributes to brainstem gliomagenesis, although it is ultimately not required as tumors can quickly evade the loss of Pax3.
High Pax3 expression defines a novel subset of human BSG
To correlate our observations regarding Pax3 expression and function in mouse glioma with human disease, pediatric BSG (all of which were characterized as DIPG) and CG samples were utilized from previously published gene expression profiles, one comprised of post-treatment autopsies [4],[44], and one comprised of pre-treatment biopsies [5]. The BSG and CG profiles from each dataset were compared, generating lists of significantly differentially expressed genes (FDR-adjusted p-value <0.05, Additional file 10: Table S5 and Additional file 11: Table S6). PAX3 was upregulated in BSG relative to CG in both datasets, 3.7-fold in the autopsy samples (Figure 7a), and 1.8-fold in the biopsy samples (Figure 7b).

High Pax3 expression characterizes a subset of human Brainstem Glioma. a-b Previously published expression profiles of human BSG and CG samples: post-treatment autopsy samples from St. Jude (a) and pre-treatment biopsy samples from Necker-Sick Children (b) were analyzed for gene expression differences as described in Materials and Methods. Log expression level of PAX3 in BSG versus CG. Red circles indicate the 11/28 (a) and 11/25 (b) of BSG samples with high PAX3 expression. c Table showing PAX3-High and PAX3-Low tumors from the dataset in (a) and their genetic alterations. Black squares indicate a sample is positive for that alteration. PDGFRA alteration includes PDGFRA gain, detected either by SNP array or FISH, or mutation (single nucleotide variants, indels, and structural variants), detected by whole-genome sequencing. d Age at diagnosis of PAX3-Low versus PAX3-High human BSG. e Schematic of the human brain showing the regional differences in Pax3 expression in glioma.
Interestingly, high PAX3 expression characterized approximately 40% of the BSG samples in both datasets (11/28, Figure 7a, and 11/25, Figure 7b). To glean information regarding the tumors with high PAX3 expression, we first compared gene expression profiles of the PAX3-Low versus PAX3-High BSG samples from both datasets. Besides PAX3, there were very few genes that met the criteria for significance (adjusted p-value < 0.05, Additional file 12: Table S7 and Additional file 13: Table S8), and none that shed any additional light on the biology of the PAX3-High subset. We next compared the genetic alterations occurring in PAX3-Low versus PAX3-High BSG samples from Figure 7a [4],[9],[11],[48]. As is shown in Figure 7c, Table 2, and Additional file 14: Table S9, high PAX3 expression significantly associated with wildtype ACVR1, PDGFRA amplification or mutation, and CDK4/6-CCND1/2/3 amplification. In addition, PPM1D and H3.1-K27M mutations were found exclusively in PAX3-Low tumors (27% and 18%, respectively), although these relationships were not significant due to their low frequency. The majority of PAX3-High tumors also contained TP53 alterations (82%) and the H3.3-K27M mutation (82%). Lastly, compared to children with PAX3-Low BSG, those harboring PAX3-High BSG were significantly older at diagnosis (Figure 7d). Collectively, these data describe a novel subset of human BSG with high Pax3 expression that are commonly characterized by increased PDGF signaling, and highlight an important regional difference between pediatric gliomas arising in the cerebral cortex (supratentorial) and the brainstem (infratentorial) (Figure 7e).
Discussion
Brainstem Glioma (BSG) is a distinct disease, biologically and clinically, from glioma of the cerebral cortex (CG). While decades of research has provided insight into the cellular, molecular, and genetic alterations occurring in CG and guided therapeutic strategies for that disease, the field of BSG research is still in its infancy. In an effort to identify unique characteristics of BSG, we compared gene expression profiles of mouse PDGF-B-induced BSG and CG, and identified 23 genes that are differentially regulated between gliomas arising in the two regions. Among those genes, 8 were associated with the gliomagenesis process in the brainstem but not in the cerebral cortex—these genes we identify as "brainstem-specific gliomagenesis genes". Of these genes, Pax3, Irx5, and Chl1 were also differentially regulated between the Olig2-compartments of BSG and CG suggesting that they represent biological differences within the tumor cells. As Olig2 characterizes the majority of human BSG, particularly the oligodendroglial (PDGFRA) and H3-K27M subgroups [5],[8],[50],[51], as well as marks a candidate cell-of-origin for BSG [52], the expression profile of these cells has important implications for at least a subset of the human disease.
Due to the controlled nature of this experiment, the gene expression signatures identified in this study can be primarily attributed to regional characteristics of the brainstem and cerebral cortex. It has not been previously reported that gliomas induced with the same genetic alterations in different regions of the mouse brain have distinct gene expression. We have found mouse BSG to harbor a distinct expression pattern of developmental homeobox genes and transcription factors, including Pax3, Irx5, Foxg1, and Lhx2. Overall, these observations are similar to those made by others in comparing human infratentorial to supratentorial low-grade glioma [20], and BSG to other pediatric high-grade glioma [5]. The latter study did not include PAX3 in their list of genes upregulated in BSG, however, potentially due to the inclusion of thalamic gliomas in their high-grade glioma group, some of which could harbor high PAX3 expression.
Herein is the first report of Pax3 expression in BSG. Importantly, we find regional expression of Pax3 in mouse PDGF-B-driven glioma in the context of Ink4aARF-loss, p53-loss, and overexpression of H3.3-K27M that is mirrored in human glioma. PAX3 mRNA is expressed at higher levels in BSG compared to CG in two independent sets of human samples, which include both pre-treatment biopsies and post-treatment autopsies. The analyses also revealed that approximately 40% of human BSG harbor relatively high PAX3 expression and that these tumors associate with increased PDGFRA signaling and alterations of cell-cycle regulatory genes, both of which characterize our PDGF-B-driven BSG mouse model. The majority of PAX3-High tumors also contain TP53 alterations and the H3.3-K27M mutation, genetic alterations that also coexist with Pax3 expression in our mouse model. Collectively, these data suggest that PAX3-High tumors may be part of the oligodendroglial and the H3-K27M subgroups previously described to harbor PDGFRA alterations [5],[10]. In addition, PPM1D, H3.1-K27M, and ACVR1 mutations occur only in PAX3-Low tumors. ACVR1 mutations have also been reported to be mutually exclusive of PDGFRA alterations [11],[13], although as we did not observe a link between PDGF signaling and PAX3 expression, we believe the exclusivity between high PAX3 and ACVR1 mutation to be independent of PDGFRA. Consistent with ACVR1 mutations occurring in younger patients [11], Pax3-High (and thus ACVR1 wildtype) tumors are associated with older patients in this study.
The regional expression pattern of Pax3 in glioma correlates with its expression in the developing mouse brain, pointing to the possibility that the expression level of Pax3 in different regions of the developing brain dictate its expression level in gliomas arising in those regions. It is plausible that Pax3 is highly expressed in BSG because the gliomagenesis process prevents its normal downregulation, although this has not been directly tested. In support of this, however, several populations of Pax3-positive progenitor cells exist throughout the P3 mouse brainstem. A subset of these cells co-express the stem/progenitor cell marker Nestin, particularly in the dorsal pons and midbrain, locations in the mouse in which Pax3-High BSG are localized. As Nestin cells are the targeted cell-of-origin for the BSG model used here, it is possible that Nestin+/Pax3+ progenitors give rise to Pax3-High glioma in this mouse model. Although a Nestin+ cell is a candidate cell-of-origin for human glioma [53], the cell type(s) and the location of the cell(s) giving rise to human BSG specifically are still in question. The majority of human BSG are at diagnosis localized primarily to the ventral pons, suggesting a potential ventral location for the cell-of-origin [52]. This would be in contradiction to a dorsal Nestin+/Pax3+ cell-of-origin as discussed here, and represents a limitation of this model. However we do not yet know the identity or location of any Nestin+/Pax3+ progenitors in the developing human pons, and as there are significant differences between mouse and human brainstem anatomy, we cannot be certain that the dorsal Nestin+/Pax3+ progenitors identified here in the mouse correlate with a dorsal location in the human pons. Further investigation into the expression of Pax3 along with other glioma markers in the developing mouse and human brainstem will be the focus of future studies to hopefully clarify this issue.
Investigation into the function of Pax3 in the brainstem revealed that ectopically expressed Pax3 is not oncogenic in the absence of any other genetic alterations. While in vitro, Pax3 overexpression inhibits basal apoptosis, this effect is insufficient to induce tumor formation in vivo. When combined with the pro-proliferative effects of increased PDGF signaling, a common event found in PAX3-High human BSG, ectopic Pax3 enhances gliomagenesis, increasing tumor frequency and promoting progression to a high-grade malignancy. Although we are unable to determine the percentage of tumor cells expressing both ectopic PDGF-B and Pax3 in this model, the survival advantage associated with both seen here suggests that the tumorigenesis process would select for doubly infected cells.
Pax3-induced inhibition of apoptosis is seemingly depen-dent on p53, as deletion of p53 in brainstem progenitors in vitro abolishes the anti-apoptotic effects of Pax3. This is in accordance with literature describing a direct interaction between Pax3 and p53 proteins leading to degradation of p53 [30],[54]. Interestingly, in the absence of p53, Pax3 promotes the proliferation of brainstem progenitors, which may explain how deletion of Pax3 in p53-deficient BSG delays gliomagenesis in vivo. These pro-survival effects of Pax3 suggest that it can cooperate with other oncogenic drivers, either through inhibition of p53-dependent apoptosis or by promoting proliferation, functions that it provides to other types of cancer cells, including glioblastoma cell lines [33],[55]-[57]. It should be noted that the in vitro experiments described here were conducted in the presence of serum, which induces differentiation. As Pax3 function is most likely dependent upon differentiation status, studying the effects of Pax3 in the context of stem cell conditions should be a focus of future research.
Collectively, our functional data suggest that Pax3 is able to promote the initiation as well as the progression of brainstem gliomagenesis when combined with other genetic alterations. As Pax3 has been found expressed in cerebral cortex glioma [32], infratentorial low-grade glioma [20], as well as high grade BSG as shown here, it appears that Pax3 is expressed in glioma (albeit at different levels) regardless of the tumor grade or location, and may play a functional role in all scenarios dependent on the context of additional genetic alterations. We find here that Pax3 increases the frequency of both low- and high-grade BSG in the context of PDGF signaling, supporting its role as an oncogene in both low- and high-grade brainstem lesions. Although results herein show a lack of Pax3 upregulation and function in cerebral cortex gliomas of mice, Chen et al. showed that Pax3 levels correlate with increasing WHO grade of supratentorial gliomas [32] and subsequently Xia et al. showed that Pax3 functionally promotes survival of GBM cell lines and xenografts originally isolated from supratentorial tumors [33]. Based on our direct comparison between BSG and CG, we would expect the levels of Pax3 expression in these supratentorial gliomas to be lower than in BSG. The functional discrepancies could be explained by varying contexts of genetic alterations, differences between mouse and human tumor biology, or an inhibitory effect on Pax3 expression or function of the cerebral cortex stroma in mice.
In summary, the data presented here show that mouse BSG and a subset of human BSG highly express Pax3, distinguishing these tumors from gliomas arising in the cerebral cortex. This expression pattern of Pax3 is mirrored in the developing brain, which may account for the gene's regional specificity in glioma. We have identified a subset of human BSG with high PAX3 expression that associates with PDGFRA alterations and amplification of cell-cycle regulatory genes, and is exclusive of ACVR1 mutations. Functional studies show that in addition to being a regional marker for BSG, Pax3 also inhibits apoptosis and promotes proliferation of brainstem progenitor cells, and contributes to PDGF-B-induced brainstem gliomagenesis in vivo. Important complements to these studies in the future will be to investigate the function of Pax3 in human BSG cells, both in cell lines and xenografts, as well as in epigenetically-induced models to study how the presence of the H3.3-K27M mutation contributes to or alters Pax3 function. Further investigation into the mechanisms upstream and downstream of Pax3 will increase our understanding of this subset of BSG and may lead to the identification of targets or pathways that could be exploited for therapeutic purposes.
Additional files
References
Hargrave D, Bartels U, Bouffet E: Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 2006, 7(3):241–248. doi:10.1016/s1470–2045(06)70615–5
Grimm SA, Chamberlain MC (2013) Brainstem Glioma: A Review. Curr Neurol Neurosci Rep 13(5): doi:10.1007/s11910–013–0346–3
Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C: Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor and Poly (ADP-ribose) Polymerase As Potential Therapeutic Targets. J Clin Oncol 2010, 28(8):1337–1344. doi:10.1200/jco.2009.25.5463
Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, Da Silva NS, Onar-Thomas A, Baker JN, Gajjar A, Ellison DW, Baker SJ: Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol 2011, 29(30):3999–4006. doi:10.1200/JCO.2011.35.5677
Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier M-P, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L, Roujeau T, Dessen P, Richon C, Lazar V, Le Teuff G, Sainte-Rose C, Geoerger B, Vassal G, Jones C, Grill J: Mesenchymal Transition and PDGFRA Amplification/Mutation Are Key Distinct Oncogenic Events in Pediatric Diffuse Intrinsic Pontine Gliomas. PLoS One 2012, 7(2):e30313. doi:10.1371/journal.pone.0030313
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, Von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C: K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 2012, 124(3):439–447. doi:10.1007/s00401–012–0998–0
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, et al.: Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482(7384):226–231. doi:10.1038/nature10833. nature10833
Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones David TW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu X-Y, Fontebasso Adam M, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann Martin U, Van Meter T, Frühwald Michael C, Hauch H, Pekrun A, Radlwimmer B, Niehues T, Von Komorowski G, et al.: Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22(4):425–437. doi:10.1016/j.ccr.2012.08.024
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Baker SJ: Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012, 44(3):251–253. doi:10.1038/ng.1102. ng.1102
Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, Barszczyk M, Chornenkyy Y, Bourgey M, Bourque G, Montpetit A, Cordero F, Castelo-Branco P, Mangerel J, Tabori U, Ho KC, Huang A, Taylor KR, Mackay A, Bendel AE, Nazarian J, Fangusaro JR, Karajannis MA, et al.: Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 2014, 46(5):451–456. doi:10.1038/ng.2936. ng.2936
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, Lu C, Nagahawatte P, Hedlund E, Rusch M, Pounds S, Lin T, Onar-Thomas A, Huether R, Kriwacki R, Parker M, Gupta P, Becksfort J, Wei L, Mulder HL, Boggs K, Vadodaria B, Yergeau D, et al.: The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 2014, 46(5):444–450. doi:10.1038/ng.2938. ng.2938
Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, De Torres C, Cruz O, Mora J, Entz-Werle N, Ingram WJ, Monje M, Hargrave D, Bullock AN, Puget S, Yip S, Jones C, Grill J: Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 2014, 46(5):457–461. doi:10.1038/ng.2925. ng.2925
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, Johann P, Tomita T, Goldman S, Nagib M, Bendel A, Goumnerova L, Bowers DC, Leonard JR, Rubin JB, Alden T, Browd S, Geyer JR, Leary S, Jallo G, Cohen K, Gupta N, Prados MD, et al.: Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 2014, 46(5):462–466. doi:10.1038/ng.2950
Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, Yang S, Jones S, Wang S, Zhou W, Zhu H, Killela PJ, Zhang J, Wu Z, Li G, Hao S, Wang Y, Webb JB, Friedman HS, Friedman AH, McLendon RE, He Y, Reitman ZJ, Bigner DD, Yan H: Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet 2014, 46(7):726–730. doi:10.1038/ng.2995
Sure U, Ruedi D, Tachibana O, Yonekawa Y, Ohgaki H, Kleihues P, Hegi ME: Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. J Neuropathol Exp Neurol 1997, 56(7):782–789. 10.1097/00005072-199756070-00004
Wakabayashi T, Natsume A, Hatano H, Fujii M, Shimato S, Ito M, Ohno M, Ito S, Ogura M, Yoshida J: P16 Promoter Methylation in the Serum as a Basis for the Molecular Diagnosis of Gliomas. Neurosurgery 2009, 64(3):455–462. doi:10.1227/01.neu.0000340683.19920.e3
Saratsis AM, Kambhampati M, Snyder K, Yadavilli S, Devaney JM, Harmon B, Hall J, Raabe EH, An P, Weingart M, Rood BR, Magge SN, MacDonald TJ, Packer RJ, Nazarian J: Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathol 2014, 127(6):881–895. doi:10.1007/s00401–013–1218–2
Lee Da Y, Gianino SM, Gutmann DH: Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell 2012, 22(1):131–138. doi:10.1016/j.ccr.2012.05.036
Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH: Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev 2010, 24(20):2317–2329. doi:10.1101/gad.1957110
Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD, Albin MG, Emnett RJ, Loeser S, Watson MA, Nagarajan R, Gutmann DH: Distinct Genetic Signatures among Pilocytic Astrocytomas Relate to Their Brain Region Origin. Cancer Research 2007, 67(3):890–900. doi:10.1158/0008–5472.can-06–0973
Robson EJD, He S-J, Eccles MR: A PANorama of PAX genes in cancer and development. Nature Reviews Cancer 2006, 6(1):52–62. Doi: 10.1038/nrc1778
Bang AG, Papalopulu N, Goulding MD, Kintner C: Expression of Pax-3 in the lateral neural plate is dependent on a Wnt-mediated signal from posterior nonaxial mesoderm. Dev Biol 1999, 212(2):366–380. doi:10.1006/dbio.1999.9319
Bang AG, Papalopulu N, Kintner C, Goulding MD: Expression of Pax-3 is initiated in the early neural plate by posteriorizing signals produced by the organizer and by posterior non-axial mesoderm. Development 1997, 124(10):2075–2085.
Tanabe Y, Jessell TM: Diversity and pattern in the developing spinal cord. Science 1996, 274(5290):1115–1123. 10.1126/science.274.5290.1115
Allen Developing Mouse Brain Atlas. 2013.
Monsoro-Burq A-H, Wang E, Harland R: Msx1 and Pax3 Cooperate to Mediate FGF8 and WNT Signals during Xenopus Neural Crest Induction. Dev Cell 2005, 8(2):167–178. doi:10.1016/j.devcel.2004.12.017
Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM: Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc Res 2000, 47(2):314–328. 10.1016/S0008-6363(00)00098-5
Olaopa M, Zhou HM, Snider P, Wang J, Schwartz RJ, Moon AM, Conway SJ: Pax3 is essential for normal cardiac neural crest morphogenesis but is not required during migration nor outflow tract septation. Dev Biol 2011, 356(2):308–322. doi:10.1016/j.ydbio.2011.05.583
Epstein DJ, Vekemans M, Gros P: Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell 1991, 67(4):767–774. 10.1016/0092-8674(91)90071-6
Wang XD, Morgan SC, Loeken MR: Pax3 stimulates p53 ubiquitination and degradation independent of transcription. PLoS One 2011, 6(12):e29379. doi:10.1371/journal.pone.0029379
Pani L, Horal M, Loeken MR: Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes Dev 2002, 16(6):676–680. doi:10.1101/gad.969302
Chen J, Xia L, Wu X, Xu L, Nie D, Shi J, Xu X, Ni L, Ju S, Wu X, Zhu H, Shi W: Clinical Significance and Prognostic Value of PAX3 Expression in Human Glioma. J Mol Neurosci 2012, 47(1):52–58. doi:10.1007/s12031–011–9677–1
Xia L, Huang Q, Nie D, Shi J, Gong M, Wu B, Gong P, Zhao L, Zuo H, Ju S, Chen J, Shi W: PAX3 is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Brain Research 2013, 1521: 68–78. doi:10.1016/j.brainres.2013.05.021
Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, Hiner RL, Gall S, Huse JT, Jabado N, MacDonald TJ, Holland EC: Preclinical Evaluation of Radiation and Perifosine in a Genetically and Histologically Accurate Model of Brainstem Glioma. Cancer Research 2010, 70(6):2548–2557. doi:10.1158/0008–5472.can-09–2503
Barton KL, Misuraca K, Cordero F, Dobrikova E, Min HD, Gromeier M, Kirsch DG, Becher OJ: PD-0332991, a CDK4/6 Inhibitor, Significantly Prolongs Survival in a Genetically Engineered Mouse Model of Brainstem Glioma. PLoS One 2013, 8(10):e77639. doi:10.1371/journal.pone.0077639
Dougherty JD, Fomchenko EI, Akuffo AA, Schmidt E, Helmy KY, Bazzoli E, Brennan CW, Holland EC, Milosevic A: Candidate pathways for promoting differentiation or quiescence of oligodendrocyte progenitor-like cells in glioma. Cancer Res 2012, 72(18):4856–4868. doi:10.1158/0008–5472
Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N: Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 2008, 135(4):749–762. doi:10.1016/j.cell.2008.10.029
Koushik SV, Chen H, Wang J, Conway SJ: Generation of a conditionalloxP allele of thePax3 transcription factor that enables selective deletion of the homeodomain. Genesis 2002, 32(2):114–117. doi:10.1002/gene.10051
Encinas JM, Vaahtokari A, Enikolopov G: Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci U S A 2006, 103(21):8233–8238. doi:10.1073/pnas.0601992103
Pitter KL, Galban CJ, Galban S, Tehrani OS, Li F, Charles N, Bradbury MS, Becher OJ, Chenevert TL, Rehemtulla A, Ross BD, Holland EC, Hambardzumyan D: Perifosine and CCI 779 co-operate to induce cell death and decrease proliferation in PTEN-intact and PTEN-deficient PDGF-driven murine glioblastoma. PLoS One 2011, 6(1):e14545. doi:10.1371/journal.pone.0014545
Bendall AJ, Ding J, Hu G, Shen MM, Abate-Shen C: Msx1 antagonizes the myogenic activity of Pax3 in migrating limb muscle precursors. Development 1999, 126(22):4965–4976.
Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD: Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340(6134):857–861. doi:10.1126/science.1232245
Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC: PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 2001, 15(15):1913–1925. doi:10.1101/gad.903001
Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, Baker SJ: Integrated Molecular Genetic Profiling of Pediatric High-Grade Gliomas Reveals Key Differences With the Adult Disease. J Clin Oncol 2010, 28(18):3061–3068. doi:10.1200/jco.2009.26.7252
Gautier L, Cope L, Bolstad BM, Irizarry RA: affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20(3):307–315. doi:10.1093/bioinformatics/btg405
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004, 5(10):R80. doi:10.1186/gb-2004–5-10-r80
Gentleman R: Bioinformatics and computational biology solutions using R and Bioconductor. Statistics for biology and health. Springer Science+Business Media, New York; 2005.
Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, Broniscer A, Wetmore C, Jones C, Ellison DW, Baker SJ: Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 2013, 73(20):6219–6229. doi:10.1158/0008–5472
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001, 25(4):402–408. doi:10.1006/meth.2001.1262
Ballester LY, Wang Z, Shandilya S, Miettinen M, Burger PC, Eberhart CG, Rodriguez FJ, Raabe E, Nazarian J, Warren K, Quezado MM: Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol 2013, 37(9):1357–1364. doi:10.1097/PAS.0b013e318294e817
Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, Sarkaria J, Zhang Z: The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013, 27(9):985–990. doi:10.1101/gad.217778.113
Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MSB, Fisher PG, Weissman IL, Rowitch DH, Vogel H, Wong AJ, Beachy PA: Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci 2011, 108(11):4453–4458. doi:10.1073/pnas.1101657108
Chen J, McKay RM, Parada LF: Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 2012, 149(1):36–47. 10.1016/j.cell.2012.03.009
Underwood TJ, Amin J, Lillycrop KA, Blaydes JP: Dissection of the functional interaction between p53 and the embryonic proto-oncoprotein PAX3. FEBS Letters 2007, 581(30):5831–5835. doi:10.1016/j.febslet.2007.11.056
He SJ: Transfection of melanoma cells with antisense PAX3 oligonucleotides additively complements cisplatin-induced cytotoxicity. Mol Cancer Ther 2005, 4(6):996–1003. Doi: 10.1158/1535–7163.mct-04–0252
Scholl FA, Kamarashev J, Murmann OV, Geertsen R, Dummer R, Schafer BW: PAX3 is expressed in human melanomas and contributes to tumor cell survival. Cancer Res 2001, 61(3):823–826.
Bernasconi M, Remppis A, Fredericks WJ, Rauscher FJ 3rd, Schafer BW: Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc Natl Acad Sci U S A 1996, 93(23):13164–13169. 10.1073/pnas.93.23.13164
Acknowledgements
We would like to thank Christine Hoeman and Francisco Cordero for mouse PDGF-B;H3.3-K27M;p53-deficient glioma tumors or lysates, Kyle Halvorson for illustration of Figure 7e, Andrew Bendall for the RCAS-Pax3 plasmid, Grigori Enikolopov for Nestin-CFPnuc transgenic mice, Mike Cook and Bin Li for Flow Cytometry, Yasheng Gao for expertise in microscopy, and Dan Wechsler and Robert Wechsler-Reya for critical comments.
Financial support
OJB is supported by the Damon Runyon Cancer Research Foundation, U.S. Department of Defense, Rory David Deutsch Foundation, and Pediatric Brain Tumor Foundation; SJB is supported by NIH grant CA096832; SJC is supported by NIH grant HL60714.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interest.
Electronic supplementary material
40478_2014_134_MOESM1_ESM.xlsx
Additional file 1: Table S1.: Differentially expressed genes between mouse normal cortex (NC) and Cerebral Cortex Glioma (CG), FDR-adjusted p-value < 0.05, fold-change ≥ 2. (XLSX 884 KB)
40478_2014_134_MOESM2_ESM.xlsx
Additional file 2: Table S2.: Differentially expressed genes between mouse normal brainstem (NBS) and Brainstem Glioma (BSG), FDR-adjusted p-value < 0.05, fold-change ≥ 2. (XLSX 811 KB)
40478_2014_134_MOESM3_ESM.xlsx
Additional file 3: Table S3.: Differentially expressed genes between mouse Brainstem Glioma (BSG) and Cerebral Cortex Glioma (CG), FDR-adjusted p-value < 0.05, fold-change ≥ 2. (XLSX 42 KB)
40478_2014_134_MOESM4_ESM.pdf
Additional file 4: Figure S1.: Regional differences in Olig2-Glioma cell compartments. a Kaplan-Meier survival curve of Ntv-a;Ink4a-ARF−/−;Olig2-eGFP-L10a mice injected with RCAS-PDGF-B into the brainstem or cerebral cortex at P2-4 to generate Brainstem Glioma (BSG) and Cerebral Cortex Glioma (CG), respectively. b Gliomas from (a) were harvested, dissociated, and sorted into GFP + and GFP- compartments by FACS and the GFP + BSG and CG samples (n = 3 for each) were compared using expression profiling. Shown is hierarchical clustering of 118 genes differentially regulated between BSG and CG Olig2-cells. p <0.01 and fold change ≥2.0. (PDF 92 KB)
40478_2014_134_MOESM5_ESM.xlsx
Additional file 5: Table S4.: Differentially expressed genes between Olig2-eGFP-L10a gliomas initiated in the brainstem (BS) and the cerebral cortex (CX), p-value < 0.01 and fold change ≥2.0. (XLSX 65 KB)
40478_2014_134_MOESM6_ESM.pdf
Additional file 6: Figure S2.: Regional expression of Pax3 in the neonatal mouse brain. a Immunofluorescence for PAX3 (red) in P3 Ntv-a pons. Nuclei are stained with DAPI (blue). 10x magnification, scale bar is 100 μM. White boxes indicate dorsal, mid, and ventral pons populations of PAX3-expressing cells. 4th ventricle and Cerebellum (Cb) are indicated for reference. b-c Immunofluorescence for PAX3 (red) and DAPI (blue), upper panels; PAX3 (red) and Nestin-CFP (green), lower panels. 20x magnification, scale bar is 50 μM. b mid pons, c 4th ventricle floor: left panels are representative of the majority of sections analyzed in which the Nestin-progenitors lining the ventricle are negative for PAX3. Right panels are representative of rare sections in which a subset of the Nestin-progenitors lining the ventricle expresses PAX3. (PDF 4 MB)
40478_2014_134_MOESM7_ESM.pdf
Additional file 7: Figure S3.: Normal brainstem progenitors isolated from P3 Ntv-a mice were infected with RCAS-Y, RCAS-Pax3, RCAS-PDGF-B, or RCAS-Pax3 + RCAS-PDGF-B. a mRNA from infected cells was isolated and analyzed for PDGF-B expression by RT-PCR. PCR primers for PDGF-B amplify endogenous mouse PDGF-B as well as the human RCAS-PDGF-B. b BrdU incorporation of normal brainstem progenitors from Ntv-a P3 mice infected with the RCAS viruses as indicated below the graph (RCAS-Y vector was used to control for the total amount of virus). Data is represented as fold-change over RCAS-Y. (PDF 55 KB)
40478_2014_134_MOESM8_ESM.pdf
Additional file 8: Figure S4.: Ntv-a mice were injected into the cerebral cortex with DF1 cells expressing RCAS-Pax3, RCAS-PDGF-B, or RCAS-PDGF-B + RCAS-Pax3 on P2-4, and monitored for signs and symptoms of brain tumors. a Kaplain-Meier survival curve; p = 0.5, PDGF-B vs. PDGF-B + Pax3. b Mice from the experiment in (a) were sacrificed at the onset of tumor symptoms, or at 12 weeks in the absence of symptoms, and the brains of all mice were analyzed for the presence of glioma using hematoxylin and eosin (H&E) staining, and their tumors graded as described in Materials and Methods. Shown is the percentage of mice in each group with no tumor, grade II, III, and IV glioma. (PDF 53 KB)
40478_2014_134_MOESM9_ESM.pdf
Additional file 9: Figure S5.: Deletion of Pax3 in p53-deficient Brainstem Glioma. Ntv-a;p53fl/fl mice (Control) and Ntv-a;p53fl/fl;Pax3fl/fl mice (Pax3-KO) were injected with RCAS-PDGF-B + RCAS-Cre into the brainstem at P2-4. Representative Control and Pax3-KO tumors were analyzed for recombination of the Pax3-floxed allele by PCR of gDNA (a) and for PAX3 protein expression by Western blot (b). (PDF 90 KB)
40478_2014_134_MOESM10_ESM.xlsx
Additional file 10: Table S5.: Differentially expressed genes between human Brainstem Glioma (Brainstem) and Cerebral Cortex Glioma (Cortical) from post-treatment autopsy samples, FDR-adjusted p-value < 0.05. (XLSX 2 MB)
40478_2014_134_MOESM11_ESM.xlsx
Additional file 11: Table S6.: Differentially expressed genes between human Brainstem Glioma (Brainstem) and Cerebral Cortex Glioma (Cortical) from pre-treatment biopsy samples, FDR-adjusted p-value < 0.05. (XLSX 1 MB)
40478_2014_134_MOESM12_ESM.xlsx
Additional file 12: Table S7.: Differentially expressed genes between PAX3-High and PAX3-Low Brainstem Glioma post-treatment autopsy samples, FDR-adjusted p-value < 0.05. (XLSX 42 KB)
40478_2014_134_MOESM13_ESM.xlsx
Additional file 13: Table S8.: Differentially expressed genes between PAX3-High and PAX3-Low Brainstem Glioma pre-treatment biopsy samples, FDR-adjusted p-value < 0.05. (XLSX 53 KB)
40478_2014_134_MOESM14_ESM.xlsx
Additional file 14: Table S9.: Genetic alterations associated with Pax3 expression in human Brainstem Glioma samples. (XLSX 41 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Misuraca, K.L., Barton, K.L., Chung, A. et al. Pax3 expression enhances PDGF-B-induced brainstem gliomagenesis and characterizes a subset of brainstem glioma. acta neuropathol commun 2, 134 (2014). https://doi.org/10.1186/s40478-014-0134-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-014-0134-6