
Abstract
Mitochondria are energy-generated organelles and take an important part in biological metabolism. Mitochondria could be transferred between cells, which serves as a new intercellular communication. Mitochondrial transfer improves mitochondrial defects, restores the biological functions of recipient cells, and maintains the high metabolic requirements of tumor cells as well as drug resistance. In recent years, it has been reported mitochondrial transfer between cells of bone marrow microenvironment and hematological malignant cells play a critical role in the disease progression and resistance during chemotherapy. In this review, we discuss the patterns and mechanisms on mitochondrial transfer and their engagement in different pathophysiological contexts and outline the latest knowledge on intercellular transport of mitochondria in hematological malignancies. Besides, we briefly outline the drug resistance mechanisms caused by mitochondrial transfer in cells during chemotherapy. Our review demonstrates a theoretical basis for mitochondrial transfer as a prospective therapeutic target to increase the treatment efficiency in hematological malignancies and improve the prognosis of patients.
Similar content being viewed by others

Introduction
Mitochondria are highly dynamic double-membraned organelles found in the majority of eukaryotic cells [1]. They contain circular DNA and independently carry out cellular processes such as gene transcription and protein translation. Mitochondria play a crucial role in the cellular metabolic pathways, such as supporting cellular activities through generating ATP and controlling the production of nucleotides, cholesterol, and heme [2]. Aside from energy generation, they perform a variety of other functions such as controlling programmed cell death [3] and regulating cell proliferation. They enable interaction with other organelles, store calcium ions, and control the dynamic balance of cellular calcium ion concentration [4].
Mitochondrial malfunction has been involved in a broad spectrum of human illnesses. Since energy production is closely linked to mitochondria, mitochondrial dysfunction may be the initiating link in many diseases such as neuronal degeneration. For example, oxidative stress can lead to impairment of mitochondrial structure and function, and consequently synaptic damage and neuronal apoptosis, thus promoting the progression of Alzheimer’s disease [5]. Also, the imbalance in dynamic mitochondrial fission has been found in cardiovascular diseases such as atherosclerosis [6] and metabolic diseases [7].
Mitochondria were formerly assumed to be permanently housed in their somatic cell, however, recently they have been reported to be transported between cells [8,9,10]. This phenomenon is known as intercellular mitochondrial transfer and constitutes one of the eukaryotic cells’ innate survival systems. This is a novel mechanism for intercellular communication. More recently, increased evidence proved that mitochondrial transfer could occur between many cell lines. Mesenchymal stem cells (MSCs), for example, transport mitochondria to multiple cells, involving epithelial cells, macrophages, and tumor cells [11,12,13]. Another example is the discovery that macrophages could obtain mitochondria from nearby adipocytes, a process that identifies a transcriptionally different macrophage subpopulation [14]. Macrophages also act as donors to deliver mitochondria to cardiomyocytes by endocytosis, triggering ferroptosis and thus causing cardiomyocyte damage [15].
Mitochondrial intercellular transfer enhances mitochondrial integration to the endogenous web of recipient cells, leading to a significant change in the bio-energetic state and other functions in the receptor ones, and also generates changes associated with cell differentiation, cell survival, and even drug resistance [16]. Therefore, mitochondrial transfer has emerged as an excellent therapeutic strategy. Replacing nonfunctional mitochondria with healthy ones has the potential to revert mitochondrial malfunction in some mitochondrial diseases, thus restoring the bioenergy requirements of impaired cells [17]. Moreover, transport of healthy mitochondria rejuvenates the impaired cells like epithelial cells [18], neurons [19], and cardiomyocytes [20].
Recently, mitochondrial transfer has been observed in several kinds of hematological malignancies involving acute myeloid leukemia, acute lymphoblastic leukemia, and multiple myeloma. In these hematological diseases, mitochondrial transfer seems to promote tumor progression and result in chemotherapy resistance [20,21,22], implying that mitochondria make an appealing and biologically reasonable therapeutic target in hematological malignancies. Therefore, clarifying the functions and mechanisms of mitochondrial transfer would provide novel therapeutic approaches for leukemia and other diseases.
Mechanisms of mitochondrial transfer
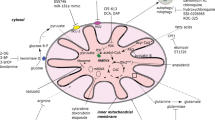
There have been a few mechanisms described to mediate transcellular mitochondrial transfer. Among them, tunneling nanotubes (TNTs) are the primary means of mitochondrial transport, and multiple TNTs are able to be built between cells to interconnect, constituting a complex regulatory network for intercellular substance and signaling [23]. Other transfer mechanisms are already discovered, such as extracellular vesicles, gap junctions, and cell fusion (Fig. 1). Understanding the mechanisms which mediate mitochondrial transfer is of great importance to elucidate the regulatory processes and functional outcomes.

Three main mechanisms of mitochondrial transfer. The main influence factors and outcomes of TNTs, EVs and GJs are shown in the figure
TNTs
TNTs appear to represent a novel route of intercellular communication, indicating the ability to swap organelles for mammalian cells [8]. As is known, TNTs are open-ended tubes/channels that join the cytoplasm in two or as many interactive cells. TNTs transport lipid droplets, proteins, ions, RNAs, organelles, and pathogens bidirectionally over ‘long’ distances (150 mm) [8, 23, 24]. TNTs can be engaged in a variety of physiological and pathological events, like immune reactions, cellular proliferation and apoptosis, pathogen transport, and angiogenesis [25, 26]. To date, TNTs have been identified in different organisms and tissues, for example, MSCs [27], macrophages [28], neuronal cells [29], cardiomyocytes [30], T-cells [31], and renal proximal tubular epithelial cells [32].
Formation of TNTs
TNT formulation is mediated by two main pathways. The former mechanism relies extremely on cell mobility. When two cells approach each other spatially and then separate after close contact, the remaining membrane pores form TNTs [30, 32]. The process could be temporarily modulated since the continuous intercellular contact occurs within a few minutes. The second occurs when donor cells extend and integrate directly onto the cell membrane of target cells via membrane protrusions (MPs) that contain actin filaments, independent of cell fluidity or close contact. TNT generation, however, is relied on the actin protein in both methods.
TNTs have a cytoskeleton consisting mainly of cell membranes as well as fibrillar actin and microtubulin [33]. Besides fibrillar actin, there are also microtubules or cytokeratin filaments in TNTs of several cell lines [33,34,35]. These skeleton proteins aid in the active movement of cargoes and mitochondria between cells.
A number of molecular mediators of actin-driven TNT synthesis have been reported. [36,37,38,39]. Leukocyte-specific transcript 1(LST1, a small adaptor protein expressed in leukocytes [40]), for example, stimulates the development of TNTs via a RelA-reliant principle. When LST1 is recruited to the stromal membrane, it interacts with filamin by activated RelA (RelA-GTP) [39, 41]. In addition, a myeloid-specified protein, M-sec (a protein capable of modifying the cytoskeleton), is associated with the synthesis of thin TNTs [37]. It has been demonstrated that M-sec expression is required for TNT-mediated freight transport [36, 40, 42]. In reality, increased expression of M-sec stimulates the formulation of TNTs, while knock-out of M-sec reduces the production of TNTs by up to one-third [36, 43]. Nevertheless, the definite mechanism of its participation in TNT formation needs further investigation.
Several other pathways have been reported to affect the synthesis of TNTs. Under oxidative stress, the cellular expression of p53 is elevated and activation of AKT-PI3K-mTOR signal pathway is achieved, controlling the protrusion and growth of nanotubes [44]. Meanwhile, cytokinesis control protein 42 homolog (CDC 42) has also been proven to be essential in the elongation of TNTs after cell membrane bulging in donor cells [37].
Molecular actors of TNTs
Mitochondrial transfer is regulated by actin motors and Milton adaptor proteins [44, 45]. Miro1 protein (encoded by RHOT1) is an intracellular calcium-sensitive bridging protein concerning the modulation of mitochondria homeostasis and transmission. Miro1 binds mitochondria to KLF 5 kinesin under assistance of other auxiliary proteins such as TRAK 1, TRAK2 and Myo 10. They shape a motor adaptor complex when combined [20], thus facilitating mitochondrial transport in TNTs and regulating their motion. Meanwhile, these structures stabilize and preserve mitochondria against degradation [46, 47].
It is known that Miro1 functions in neurons as well as among other kinds of cells [48,49,50,51]. Miro1 plays a leading role in mitochondrial location and transfers in remote trafficking mediated by tubulin cytoskeleton [52]. When Miro1-overexpressied MSCs were co-cultured with injured epithelial cells, there was an enhanced development of TNTs between the two cells along with an increasing number of mitochondria transport [13]. The MSCs exhibited increased rescue potential, which contributed to the repair of epithelial damage. However, knockout of Miro1 with shRNA had the opposite effect, showing a significant decrease in mitochondrial donation [33]. All these data indicate that Miro1 expression regulates mitochondrial transfer [20].
Miro2 protein (encoded by RHOT2) is also the main connector for intracellular transport of mitochondria. In the presence of Miro1, it seems somewhat redundant for Miro2 in that it does not fully compensate for the loss of Miro1. Surprisingly, the two Miro proteins modulate kinesin-based motion, with Miro2 playing a dominating part in short-distance transmission [53] and more involved in modulating the connection of mitochondria and the actin cytoskeleton.
Extracellular vesicles (EV)
In general, a diverse population of vesicles are discharged from the internal into extracellular environment, and the vesicles are named EVs. They are globular structures enclosed by a phospholipid bilayer membrane which encases many cellular proteins, nucleic acids, chemicals, and structural components [53,54,55]. Based on the source and molecular structure, EVs include exosomes (30–100 nm in diameter), microvesicles (MVs) (100 nm to 1 μm in diameter) or apoptotic bodies (50 nm-2 μm in diameter) [56]. Because of their quick removal by macrophages, apoptotic bodies are rarely examined [57].
EVs can carry multiple substances over long distance to alter the fate of host cells and organismal functions [56, 58]. Besides TNT-mediated transport, mitochondria could be packed in EVs [59, 60], and transferred between cells. The imported mitochondria can avoid lysosomal destruction after being endocytosed by the target cell and coexist with the new host cell. This is an effective method for moving functional loads from one cell to a different one [61, 62]. These findings add an innovative way of engagement to the multidimensional communication network, as well as a new signal transport mechanism [63].
Many types of cells, such as astrocytes and MSCs, can secrete mitochondria-carrying EVs [20, 63, 64] and transfer them to epithelial cells, immune cells and neurons [65]. It has shown that MSCs could modulate macrophages by EVs-containing mitochondria, enhancing their respiratory and phagocytic function in clinically related lung damage models [66]. Furthermore, another study reported that EVs from hormone therapy-resistant breast cancer patients contained high levels of mitochondrial DNA (mtDNA). MSCs, as the mesenchymal component of tumors, could transfer mitochondria via EVs towards neoplastic cells and promote the oxidative phosphorylation (OXPHOS), which facilitated cancer cell proliferation as well as metastasis; on another aspect, tumor-derived exosomes affected the differentiation of MSCs by regulating diverse signaling pathways [67]. It has been recently discovered that the release of microvesicles from astrocytes is controlled by a calcium-reliant mechanism concerning CD38 and circular ADP ribose signaling [64]. Via this progress, astrocytes transported their mitochondria to neurons affected by stroke as a “rescue” phenomenon. In the opposite case, EVs can be utilized to clear impaired-depolarized mitochondria from bone marrow mesenchymal stem cells (BMSCs) and be exported to nearby macrophages [68]. This transport recycled the mitochondria and secreted exosomes, and allowed macrophages to tolerate the transferred damaged mitochondria. Besides, another study showed that thermogenically stressed brown adipocytes prevent the defeat of the thermogenic procedures by releasing oxidatively damaged mitochondria via EVs. When reabsorbed into parental brown adipocytes, EVs containing mitochondria have been cleared by phagocytic activity of macrophages, which contributed to the protection of cellular physiology [69]. Mitochondrial uptake via EVs vectors has been described in various physiological states and diseases. Though CD38 is expressed on various types of leukemia including CLL [70] and MM [71], the specific role of CD38 in mitochondrial transport through EVs by leukemia cells in bone marrow microenvironment is unclear.
Gap junctions (GJs)
GJs are another important ways for mitochondrial transfer, though there are fewer studies about GJs compared with TNTs due to that TNTs are easily observed in in vitro co-culture systems [20]. GJs consist of gap junction protein (connexin, Cx), connexon and gap junctional channels (GJCs), which join the cytoplasm of two separate cells and serve as an important channel for material exchange and signal communication between neighboring cells. The six identical Cxs on the cell membrane form a tubular structure around the linker. Two linkers on neighboring cell membrane connect end-to-end to form GJs. And the GJCs allow ions (Na+, K+, Ca2+, etc.), second messengers (e.g., IP3, cAMP, cGMP),and other small molecules to be exchanged across cells [72]. Therefore, GJs could regulate the intracellular mechanisms of signaling and autophagy. In vertebrate cells, Cx is a homologous transmembrane protein encoded by a multigene family. Over 20 Cx isoforms have already been defined, among which Cx43 is the most broadly expressed as well as thoroughly investigated. Cx43 is engaged in a range of physiological activities, for example, substance exchange, vesicle transport, and mitochondrial respiration [73]. Studies have shown that Cx43 is essential in mitochondrial uptake during lung and brain injury responses [73, 74] and also regulates CXCL12 excretion of MSCs [75].
In a study examining the protected effects of mitochondrial exchange of BMSCs in lung tissue, BMSCs release vesicles encapsulating mitochondria to reach alveolar epithelial cells via GJs, which are subsequently taken up by endocytosis [76]. This mitochondrial transfer mediated by Cx43 markedly reduced Cx43-mediated mitochondrial transfer markedly reduces LPS-induced acute lung damage, which includes enhanced leukocytosis, protein leakage, suppression of surfactant secretion, and high fatality rates [77]. Moreover, it has been demonstrated that GJs could mediate mitochondria transmission out of MSCs to the damaged motor neurons [78]. Also, Cx43 and Cx32 could create heterotypic GJs from MSCs to neurons, in which Cx43 was displayed on MSCs instead of motor neurons; whereas, Cx32 was expressed exactly the opposite way of Cx4317. GJs play important roles in mediating mitochondrial transfer via Cx43 and other proteins.
Cell fusion
Except for TNTs, EVs and GJs, cell fusion is another proposed method of mitochondrial transport in cell-to-cell communication. As cell fusion is rarely found in higher eukaryotes under normal physiologic conditions, it is not the main mechanism for mitochondrial transfer.
Cell fusion is the process whereby the membranes of two or several cells unit together and partake organelles and cytoplasmic components. The process may be triggered when injury and inflammation occur [79]. Particularly, cell fusion modulates the stem cells potential and takes a crucial part in both regeneration and tumorigenesis [80]. Cell fusion is either transient or perpetual. After permanent cell fusion occurs, hybrid cells share cytoplasmic components and selectively lose donor cell nuclei, and recipient cell nuclei are reprogrammed to exhibit tissue-specific stem/progenitor cell properties [81]. As for transient cell fusion, it permits a temporary intercellular exchange of substances and signals, including transfer of donor cell mitochondria [82]. Fusion of MSCs and terminally differentiated somatic cells mediates reprogramming of the latter and contributes to tissue regeneration [83].
As for the form of cell fusion, it occurs both by partial cell fusion formed by TNTs and by full cell fusion. When co-cultured human MSC with transgenic mouse cardiomyocytes, the transfer of mitochondria can be identified at full fusion of MSCs with cardiomyoblasts with mitochondrial damage [84]. Yet, mitochondria have as well been shown to be transported through TNTs between the two cells, enabling cells in a state of oxidative stress to exchange damaged mtDNA during fusion and maintain the mitochondrial biogenesis of aerobic respiration. Likewise, primary glioblastoma cells acquired mitochondria through phagocytosis of tumor-activated stromal cells (TASC), extracting TASC cytosolic components for themselves [85]. Therefore, mitochondrial transfer could be mediated by cell fusion in physiological and pathological states.
Trigger signals
Early signals triggering mitochondrial transfer
Stress conditions, like infections or inflammations, hypoxia, x-rays, and ultraviolet, can trigger early signals for mitochondrial transfer [27, 33, 85,86,87,88,89,90,91]. Stressed cells emit early stimulation signals, and donor cells receive them from the local microenvironment and then undergo massive synthesis of mitochondria, which select different pathways for transfer in a specific environment. When severe tissue injury occurs, many mitochondria-related components including mtDNA and extracellular ATP are liberated outside the injured cells in the form of damage associated molecular patterns(DAMPs), which accumulate around the injured tissue or cross through capillaries and merge into the bloodstream [92, 93]. Besides DAMPs, reactive oxygen species(ROS) from cells in stress or inflammatory states also stimulates mitochondrial trafficking from donor to recipient cells [13]. By releasing high levels of ROS as a distress signal, stressed cells acquire mitochondria from other cells so as to down-regulate intracellular oxidative stress [94]. Moreover, factors that govern mitochondrial transfer include mitochondrial autophagy [94, 95], KIF5B, glucose [96], ATP [65], TNF-α [12], and the microenvironment of BMSCs.
Functionally normal mitochondria are able to be transmitted from MSCs into receptor cells, while injured mitochondria in recipient cells under stress can also be transferred to MSCs via TNTs and be cleared by mitophagy [95]. In this case, mitochondria of damaged cells work as danger-signaling organelles [97]. Through the bidirectional transfer of TNTs, stressed cells can also transfer substances such as ROS to MSCs, as well as retrograde signaling like AMP/ATP and NAD+/NADH in stressed cells [20]. On the one hand, the retrograde signaling in TNTs acts as an early stimulatory signal to stimulate mitochondrial biosynthesis and transfer within MSCs via upregulating the expression level of the protein PGC-1α related to mitochondrial biosynthesis [98]. On the other hand, it can stimulate the function of MSCs against apoptosis and cell damage repair [99]. Therefore, it is possible to avoid apoptosis and promote cell survival after receiving stress signals and transferring components such as mitochondria. In this light, TNTs might be viewed as a facility for increasing cell survival under stress [100].
Nuclear factor kappa B (NF-κB) signaling
NF-κB signaling presents in most cells, and is involved in inflammatory and other diseases [101]. Dysregulated NF-κB signaling causes chronic inflammation and autoimmune diseases. TNF-α-NF-κB-TNFaIP2 signaling pathway has been found to be engaged in TNT synthesis among MSCs and cardiomyocytes, indicating inflammation is essential to influence the efficiencies of mitochondrial transfer in MSCs [12]. TNF-α could activate NF-κB pathway and undergo phosphorylation, thereby stimulating the expression of TNFaIP2 protein. The upregulated protein provokes the accumulation of F-actin, resulting in improved mitochondrial transport between MSCs and impaired cells. Sc-514, an NF-κB inhibitor, greatly lowers TNT generation, indicating that NF-κB signaling regulates TNT synthesis [12]. In addition, another study also discovered that NF-κB activation participated in the modulation and synthesis of TNTs, while cytarabine alone or in combination with daunorubicin inhibited NF-κB and downregulated TNTs [102]. All these data offer further support to the NF-κB pathway to participate in TNT formulation.
Extracellular mitochondria
Although most studies demonstrated that mitochondrial transfer occurs via intercellular mechanisms, more and more evidence indicate that mitochondria could be freed to the extracellular environment and then transmitted to donor cells [103]. As previously noted, mitochondria could be exited in extracellular environment in integral and free form (freeMitos), or be surrounded with membranes, like within vesicles, or as cell-free circulating mtDNA (ccf-mtDNA) [104]. These various kinds of mitochondria perform diverse roles varying from promoting restoration effects to serving as signals on interaction with other cells.
It has been proven that cells could leave some elastic fibers behind during migration, and little vesicles grow at the top or intersection of the elastic fiber, which are named migrasomes [105]. During cell migration, some intracellular material is continuously transported to the migratory body by constricting the duct of these fibers. And then the contracted fibers are broken, leading to the release of migrators and engulfment by cells around the extracellular space. Furthermore, mitochondria were unexpectedly found present in extracellular migrators, and these mitochondria showed an unhealthy and damaged state. Overall, the study discovered a previously unknown phenomenon in cells, mitocytosis (mitochondrial exocytosis), which could clean out damaged mitochondria and maintain mitochondrial homeostasis in cells.
These external mitochondrial interactions with other cells open up a novel field of research where mitochondria transcend the roles as cellular powerhouses and act as signal organelles [105,106,107]. Elucidating the effects of extracellular mitochondria and their various formats helps develop new therapies for better health and define innovative disease-related biomarkers.
Biological functions
Mitochondrial transfer may preserve metabolic homeostasis and cell-to-cell interaction in the microenvironment in response to metabolic and oxidative stressors. It alters the functional status of recipient cells, and takes a critical part in the progression and viability of neoplastic cells as well as in the repair of damaged cells.
Metabolic communication
Through TNT-mediated mitochondrial transport, the energy metabolism of recipient cells has been changed, with the increased production of OXPHOS and ATP and thereby maintaining metabolic homeostasis in a variety of cells [108].
As is reported, mitochondrial transmission from MSCs to adenocarcinoma cells has caused a series of changes of the latter in energy metabolism, including decreased extracellular lactate and ROS, and increase in extracellular ATP, membrane potential and oxygen consumption, indicating a complete recovery of mitochondrial activity after being co-cultured. Similarly, osteosarcoma cells, after co-culture with MSCs, displayed the higher mitochondrial activity by boosting intracellular ATP and oxygen consumption rates [109]. Also, damaged astrocytes can receive mitochondria of MSCs, thus restoring their energy productivity and cell proliferation [110]. The heat-stress-producing brown adipocytes are demonstrated to release EVs containing oxidatively damaged mitochondrial fractions and are cleared by macrophages, thereby maintaining thermogenesis as well as cellular metabolic homeostasis [69]. It is also shown that adipocytes transport intracellular mitochondria to macrophages to regulate the homeostasis of white adipose tissue and could be inhibited in obese conditions [14]. In a preclinical acute respiratory distress syndrome model, evidence suggests that mitochondrial transport from MSCs towards macrophages takes a critical part in immune response [111]. And, direct co-culture of MSCs with phagocytic cells enhances the OXPHOS and intracellular ATP activity of recipient cells, and then enhances their phagocytic activity and immune response, thus promoting the improvement of the repair process.
Regulation of tumor microenvironment and chemo-resistance
The progression of tumors requires interaction between tumor cells and neoplastic microenvironments. In normal conditions, mitochondrial transfer can induce cellular reprogramming. It has been demonstrated that co-culture of fully-differentiated cardiomyocytes and MSCs caused the mitochondrial transfer from MSCs into cardiomyocytes, promoting a progenitor-like state of the latter [82]. As for cancers, tumor cells can motivate stromal cells to increase the range of pathways that support cancer cell proliferation, hence promoting tumor growth. It has been found that a vital molecular signal could be transmitted from tumor cells to stromal cells by TNTs, causing consequent generation of survival-promoting molecules including cytokines [112]. Moreover, mitochondrial transfer may facilitate metabolic reprogramming of tumor [113]. Tumor cells utilize mitochondrial transport to reconstitute the tumor microenvironment, resulting in enhanced intracellular mitochondrial function, thereby causing an elevated proliferation and aggressive phenotype of these cells. For example, in the bladder cancer model, the invasion of tumor cells has been enhanced in vitro and in vivo by mitochondria transport [114].
Meanwhile, mitochondrial transmission has been shown to mediate chemotherapy resistance [115] in many tumors including breast, ovarian, and bladder cancer. Under co-cultured conditions, the breast or ovarian cancer model indicated the presence of TNTs between tumor cells and BMSCs. Mostly, mitochondria migrated from BMSCs to cancer cells, enhancing the chemo-resistance to the DNA-damaging drug doxorubicin [10]. Besides, after transferring healthy mitochondria into breast cancer cells with dysfunctional mitochondria, normal aerobic respiration of cancer cells was restored and cell invasiveness was enhanced [116]. However, the opposite result is observed in artificial-isolated mitochondria transferred from normal breast epithelial to breast cancer cells [117]. The viability and tumorigenicity of the tumor cells have been impaired as a result of mitochondrial transplantation related apoptosis mediated by AIF, the increased parkin protein as well as reduced fragmented mitochondria [118].
Tissue repair
Mitochondrial transfer could promote wound healing and might be implemented in the repair of impaired tissue. Mitochondrial transfer has been proven to take a protective part in the vascular system. Mitochondria transferred between endothelial progenitor cells via a TNT pattern could promote regeneration and repair of damaged myocardium [119]. In addition, when co-culture human adipose stem cells and murine cardiomyocytes, it can be found that the intercellular F-actin junctions were formed which was associated with mitochondrial transfer [82]. The transferred mitochondria are helpful in the recovery of cellular status through partial cell fusion. Another study transplanted BMSCs to rats via the carotid artery, and the results suggested the intercellular mitochondrial transmission between MSCs and injured endothelial cells prevented oxidative damage and apoptosis under cerebral ischemia-reperfusion conditions [120], further confirming its protective role in the damaged cerebral microvascular system.
Intercellular mitochondrial transfer is also reported to be involved in the maintenance of homeostatic of lung tissue. Mitochondrial transfer was observed from BMSCs to alveolar epithelial cells in a murine model of acute lung damage treated with LPS, and resulted in increased alveolar ATP levels [76]. This mitochondrial transfer exerted a protective effect on alveolar epithelial cells, thereby repairing airway damage and improving lung inflammation [33]. In astrocyte/neuronal co-culture systems, neurons damaged by rotenone were related to reversal of neurodegeneration and axonal pruning after internalization of mitochondria [121]. In the murine model of transitory focal cerebral ischemia, healthy mitochondria were released from astrocytes and absorbed by neurons, and restored their aerobic cellular respiration and proliferation [110]. These discoveries indicate an effect for mitochondrial transfer in neurological recovery after stroke, involved in the maintenance of neural integrity, with potential in the treatment of neurodegenerative diseases.
In serious corneal injury situations, corneal stem cell transplantation has become an emerging tactic for corneal regeneration and scar formation prevention. The rate of bioenergetic parameters such as ATP production and maximal respiration rate were inferior in rotenone-treated corneal epithelial cells in comparison to healthy corneal epithelial cells. All these indices showed significant improvement in corneal epithelial cells co-cultured with MSCs, suggesting that mitochondrial transmission in MSCs is a crucial mechanism of MSC treatment for corneal impairment [13]. In addition, when co-cultured BMSCs with renal tubular epithelial cells under high-glucose exposure in a diabetic nephropathy model, the results showed that BMSCs imported their mitochondria into injured renal proximal tubular epithelial cells by TNTs, which significantly inhibited the apoptosis of the latter [18]. All these data show that mitochondrial transfer is an important and novel strategy for tissue repair.
Mitochondrial transfer in hematopoiesis and hematological malignancies
Hematopoiesis
Mitochondrial transfer has been observed in normal hematopoiesis. Upon exposure to bacterial infection, hematopoietic stem cells (HSCs) expanded quickly in response to stress stimulation and the immune system underwent a rapid granulocytic response associated with acute bacterial infection. At this time, mitochondria were transferred from BMSCs to HSCs via GJs under the regulation of ROS. As a result, the mitochondrial mass of HSCs increased, leading to a metabolic shift from glycolysis to OXPHOS. Mechanistically, ROS-induced oxidative stress regulated phosphatidylinositol 3-kinase (PI3K) activation to mediate the opening of connexin channels, thereby permitting transport to occur. Here, HSCs act as recipients of transfer to acquire mitochondria in response to acute bacterial infection [21].
In another discovery, mitochondrial transfer was found to occur in the opposite direction, i.e., from hematopoietic stem and progenitor cells (HSPCs) to MSCs. After radiation prior to bone marrow transplantation (BMT), the mitochondria of MSCs were dramatically impaired. HSPCs transferred healthy mitochondria to the stromal microenvironment (ME) via HSPC Cx43-mediated cell contact, thereby improving mitochondrial activity in recipient MSCs. As a result, supportive stromal ME was restored and hematopoietic compartment reconstruction was improved. Understanding the mechanisms that regulate stromal recovery after myeloablative stress is highly clinically relevant for optimizing BMT procedures and emphasizing the importance of adjuvant non-HSC accelerated hematopoietic transplantation [122].
Moreover, after injecting erythroblast island (EBI) macrophages into mice that suffered from multiple modes of anemia stress, it could be found that mitochondrial transfer occurred from EBI macrophages to early erythroblasts via direct uptake. At the early erythropoietic stage, mitochondrial transfer alters the bioenergetic profile of recipient cells through CD47-Sirpα interactions, promoting their proliferation, protein synthesis, and energy generation, thereby driving the recovery of erythroid lineage cells in response to stress. This finding provides evidence for a supportive role of splenic and bone marrow EBI macrophages in erythropoiesis during erythrocyte stress and confirms the essential mediating role of mitochondrial transfer [123].
These above findings suggest that mitochondrial transfer occurs in normal hematopoiesis under infection, radiation and stress, and involves various cell types and different directions of transport. These transfers alter the metabolic state and physiological functions of the recipient cells, and facilitate the body’s adaptation to and recovery from stress.
Hematological malignancies
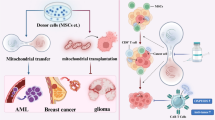
Hematological malignancies define a highly heterogeneous disorder with strongly variant prognoses that continuously relapse after therapies. Hematological malignancies have the leading morbidity and mortality and seriously affect human health [124]. Because of the crucial role of TNTs in communication between cancer cells and BMSCs, mitochondrial transfer is also essential in hematological tumors [125]. The majority of researches on the contribution of mitochondrial transfer in hematological malignancies focus on leukemia and myeloma (Fig. 2).

Mitochondrial transfer in hematological malignancies. Mitochondria are transmitted mainly by TNTs and other forms, leading an increase in survival of hematopoietic malignant cells and chemoresistance to drugs. More details can be seen in the text
Acute myeloid leukemia (AML)
AML is featured by the invasion of clonal and inadequately differentiated hematopoietic cells in bone marrow [126]. Via Warburg effect, a majority of tumor cells convert their ATP production from OXPHOS to glycolysis [127]. In contrast with the Warburg hypothesis, the survival of AML cells is strongly reliant on oxidative phosphorylation for ATP production, and they are constantly adjusting to variations of nutrition and oxygen supply in BM microenvironment [127, 128]. It has been shown that mitochondria and OXPHOS could affect chemotherapeutic drug sensitivity and efficacy in vivo. High OXPHOS signaling and metabolism were identified as key marker of chemo-resistance, and AML cells with this profile were more resistant to Ara-C chemotherapy. Similarly, essential mitochondrial function also contributes to increased AML resistance to Ara-C. Chemo-resistant leukemia cells could initiate tumor regeneration in vivo, exhibiting high OXPHOS genetic characteristics, high ROS levels with an altered intracellular redox state, and maintenance of polarized mitochondria [129].
It has been found that the proteins associated with cell energy metabolism are up-regulated in AML cells after cocultured with BMSCs [130]. Despite the improved metabolism in recipient cells, there is no definitive evidence that mitochondria are indeed transmitted. Moschoi et al. recently demonstrated that MS-5 unidirectionally transferred intact functional mitochondria into AML cells via the endocytic pathway and that mitochondrial transfer increased in a time-dependent manner, increasing ATP production by OXPHOS in AML cells up to 1.5-fold. The endocytosis inhibitors blocked mitochondrial transfer between murine MS-5 and human AML cells. During this process, AML cells became less sensitive to the chemotherapeutic drug Ara-C and the uptake of mitochondria was enhanced under chemotherapeutic conditions [131].
Mitochondrial exchange between BMSCs and AML cells has already been observed in co-culture conditions both by lentiviral transfection and fluorescent staining of BMSCs mitochondria, which was also shown in vivo [22]. A high level of oxidative stress has been found to exist in AML [132, which promotes ROS-driving mitochondrial trafficking from BMSCs to AML cells through TNTs. Specifically, NOX-2-derived superoxide produced from AML cells induces mitochondrial transfer. The antioxidant N-acetylcysteine (NAC) inhibited mitochondrial transfer, while the oxidant hydrogen peroxide (H2O2) further promoted mitochondrial trafficking between BMSCs and AML cells [22]. The elevated intracellular ROS levels in AML in response to chemotherapeutic agents further increased the oxidative stress environment, thereby increasing mitochondrial transfer. Suppression of NOX2 inhibited mitochondrial transport, enhanced AML apoptosis and increased the viability in an in vivo xenograft model. It suggests a unique treatment method for AML that involves tumor-specific reliance on NOX2-drived mitochondrial transfer. However, NOX2 inhibitors are not yet available in clinical trials. In addition, toxicity may be an issue with this strategy, as NOX2 deficiency in humans often ends in death within the first decade of life [133].
As mentioned before, AML cells rely on OXPHOS for ATP production. Another study explored the mechanisms whereby the bone marrow microenvironment promotes secondary resistance of AML cells to OXPHOS repression. They utilized IACS-010759 to treat AML cells, a newly complex I OXPHOS inhibitor, and discovered that direct contact with BMSCs induced the compensation of AML cells by activating mitochondrial respiration and developing tolerance to OXPHOS suppression. In terms of mechanism, suppression of OXPHOS inspired mitochondrial transport from MSCs towards AML cells through TNTs under direct-contact co-culture circumstances. Additionally, in AML cells, this inhibition induced mitochondrial division, an increase in functional mitochondria, and mitochondrial phagocytosis. Moreover, it was demonstrated that mitochondrial transmission of MSCs to AML cells induced by OXPHOS suppression was further strengthened by Ara-C [134]. These findings suggest that AML cells managed to replete functional mitochondria via exogenous transport of MSCs-provided mitochondria and internal fission of healthy mitochondria under OXPHOS restriction. Meanwhile, mitochondrial phagocytosis eliminates damaged mitochondria. These alterations have a major part within the compensatory adjustment of leukemic cells towards the energy stress of bone marrow microenvironment.
Clinical observational trials using Venetoclax combined with hypomethylating agents targeting BCL2, an important modulator in mitochondrial apoptosis pathway, have shown a tolerable safety profile and good overall remission rates in elderly AML patients [135]. Therefore, mitochondria are an appealing and biologically reasonable drug target for AML therapy. Through the utilization of an in vitro co-culture system and an in vivo mouse model, daratumumab, a monoclonal anti-CD38 antibody, has been found to impair the metabolic capacity of AML by blocking mitochondrial transport between BMSCs and the parent cells [136]. This leads to a decrease in AML-derived OXPHOS, thereby inhibiting tumor proliferation and reducing tumor load. Daratumumab should be investigated more as a treatment for mitochondria-dependent tumor development, according to these findings. Another study demonstrated that metformin, the most common medicine for curing type II diabetes, dramatically increased the chemo-sensitivity of AML cells under co-culture with BMSCs by inhibiting mitochondrial transport. Metformin’s chemo-sensitizing impact is mechanistically mediated by inhibiting intercellular contact-dependent mitochondrial transport and mitochondrial OXPHOS in receiver AML cells, with effects comparable to those of the control medicine cytochalasin D, a TNT formation inhibitor. Besides, metformin enhanced the anti-tumor effects of Ara-C in AML cells co-cultured with BMSCs in an immune-deficient mouse xenograft model [137]. This research determines the possible applicability of metformin in the sensitization of AML cells to chemotherapy. Metformin has been used in combination with a few anti-leukemia agents and shown positive therapeutic results. For example, through metformin-triggered mitochondrial membrane depolarization, the Bcl-2 inhibitor ABT-737 enhanced mitochondrial death of leukemic cells [138].
Current studies demonstrate that mitochondria transfer between AML cells and BMSCs via TNTs or endocytic pathways. AML receptor cells have enhanced metabolic capacity and resistance to chemotherapeutic agents. Mitochondrial transfer become a promising target in AML therapy. In combination with existing chemotherapeutic regimens, it can increase the killing power of chemotherapeutic agents on AML cells by inhibiting mitochondrial transport, which is expected to be widely applied in the clinic.
Chronic myeloid leukemia (CML)
CML is a myeloproliferative neoplasm and manifests increased peripheral blood leukocytes. CML is triggered by reciprocal chromosome translocations, which results in a BCR-ABL fusion gene [139]. The annual mortality rate has fallen dramatically [140] since the availability of imatinib [141]. However, some patients fail to obtain an entire cytogenetic response or acquire resistance and progression to a primitive stage. Stromal cells and leukemic cells are found to interact bidirectionally to support leukemogenesis [142]. Leukemia cells cultivated in vitro generally cease to exhibit drug-resistant characteristics, implying a cytoprotective role for the bone marrow microenvironment. In general, intercellular interaction is critical in stroma-mediated drug resistance in CML. The TNT formulation between CML cells and BMSCs facilitates the transmission of mitochondria, cellular vesicles and proteins [143]. It has been found that TNT bulges areas contained many vesicles, which were transmitted between stromal cells and CML cells via TNTs. TNT-mediated vesicles transport and functional proteome were involved in stromal protection of leukemic cells and contributed to survival of leukemic cells. As mentioned above, mitochondrial transfer can increase the resistance of CML cells to certain chemotherapeutic agents, thus protecting the leukemic primitive cells. Similarly, the transport of cell vesicles between stromal cells and leukemic cells was found to protect CML cells against imatinib-induced apoptosis, which increased the chemotherapy resistance of CML cells during imatinib treatment.
In conclusion, mitochondrial trafficking between CML cells and BMSCs by TNTs increases resistance towards chemotherapeutic drugs and decreases the apoptosis rate of CML cells. This could be considered a therapeutic tool when leukemic cells develop drug resistance or when individuals fail to obtain a complete cytogenetic response.
Acute lymphoblastic leukemia (ALL)
T-cell acute lymphoblastic leukemia (T-ALL)
T-ALL is one of the most invasive hematologic malignancies. It originates from the malignant conversion of T-cell progenitors. The cure rate of T-ALL has improved to about 50% in adults with the treatment of high-dose multi-agent chemotherapy [144]. However, lots of T-ALL patients develop primary chemo-resistance and leukemia recurrence. These difficulties continue to be the main obstacles to clinical attempts at curing T-ALL [145].
BMSCs have been shown to have an impact on cellular mitochondrial dynamics of T-ALL and affect the chemo-resistance of leukemia cells. In a study, MSCs were shown to provide protection against chemotherapeutic cell mortality and cytotoxicity in T-ALL under both indirect and direct co-culture conditions [146]. The cells in the direct contact system were more viable and had better pro-survival effects. Considering that mitochondria are a vital resource of ROS, upregulation of mitochondrial ROS level is a viable method to kill cancer cells [147]. And exposing of T-ALL cells to MSCs reduced the mitochondrial ROS level and facilitated pre-glycolytic transfer, which resulted in higher glucose absorption and lactate generation [146]. These findings suggest MSCs could preserve T-ALL cells from chemotherapeutic cytotoxicity and promote ALL cell proliferation and survival, which partly depends on reduced ROS contents in mitochondria as well as a pre-glycolytic metabolic transition. Furthermore, the protective actions couple with a mitochondrial breakage process that is controlled by ERK-mediated activation of DRP1 phosphorylation. Therefore, disrupting leukemic cell/matrix connections and focusing on mitochondrial dynamics could offer an innovative approach for T-ALL treatment that can be employed in conjunction with standard chemotherapeutic drugs. Moreover, it was proposed that T-ALL cells are capable to deliver more mitochondria to MSCs after chemotherapeutic drug-induced oxidative stress, but gain fewer mitochondria from MSCs. The process is medicated by TNTs and ICAM-1, which is helpful for chemo-resistance mediated by cell adhesion [148]. There are many examples of mitochondrial transport from MSCs to others, such as AML cells, breast cancer cells and other malignant cells. However, few have demonstrated mitochondrial trafficking from other cells to MSCs. The divergence in the direction of transfer might be due to their distinct states of metabolism. AML cells have more OXPHOS, while T-ALL cells are more glycolysis upon co-culture [149]. AML cells input mitochondria to meet the need for high-energy metabolism, whereas ALL cells expel mitochondria to decrease cellular ROS. These findings facilitate revealing the signals and mechanisms driving the directionality of transport. However, whether T-ALL cells might also provide mitochondria towards other cells, and what effect this has on tumor progression deserve further exploration.
The important role of mitochondrial metabolism in the maintenance of bioenergetic and metabolic homeostasis makes it an interesting therapeutic target. T-ALL cells have oxidative stress and metabolic disruption, which is a frequent hallmark of cancer cells. The level of ROS in T-ALL cells is much higher than that in normal cells. As excessive ROS causes leukemic cell death, it has been shown that inducing intracellular oxidative stress is an essential anticancer machinery of leukemia chemotherapy [150]. ALL cells display a hyperenergetic profile with high glycolysis and high OXPHOS [151]. Calcium migration to the mitochondria via the inositol 1,4,5-triphosphate receptor (InsP3R) maintains mitochondrial activity, which makes T-ALL cells susceptible to the suppression of InsP3R. Recently, after treatment by Xestospongin B (XeB), the specific inhibitor of InsP3R, higher mitochondrial respiration exhibited by T-ALL cells was attenuated [152]. Long-term therapy with XeB resulted in T-ALL cell death without influencing the normal counterparts. Therefore, inhibition of InsP3R by XeB has been considered as a potential therapy for T-ALL.
In summary, mitochondrial transport from T-ALL cells to MSCs is mainly regulated by TNTs, which reduces the level of mitochondrial ROS in the cells and induces drug resistance. Clinical disruption of the communication between leukemia and stromal cells could provide a novel therapeutic method in combination with conventional chemotherapeutic agents.
B-cell acute lymphoblastic leukemia (B-ALL)
Mitochondrial transfer is also present in B-ALL. It has been shown that primary B-cell precursor ALL (BCP-ALL) cells utilize TNTs to communicate with MSCs. The signaling led to the secreting of pro-survival cytokines, induced stroma-mediated prednisolone resistance, and increased the viability of B-ALL cells. Suppression of TNTs significantly inhibited this process and made BCP-ALL cells re-sensitized to a vital anti-leukemic medicine prednisolone. The discovery of TNT signals in ALL-MSC interaction sheds light on the pathobiology of ALL and brings novel opportunities for developing more efficient treatments [112]. Another study found that isolated MSCs from ALL patients undergoing chemotherapy are typically activated, and carry cancer-associated fibroblast (CAF) phenotype. The phenotype has an altered cytoskeleton and gene expression with high levels of pro-inflammatory cytokine secretion. A subsequent series of experiments demonstrated that primary MSCs and MSC cell line HS27a could be triggered into CAF by clinically relevant concentrations of Ara-C and erythromycin. CAF/activated MSCs transferred mitochondria into B-ALL cells via TNTs, which prevents exogenous ROS-inducing agent-induced B-ALL cells from apoptosis and death [153]. Overall, mitochondrial trafficking decreases the sensibility of B-ALL cells to chemotherapy treatment and protects B-ALL cells from ROS-induced cell death. Moreover, mitochondrial transfer contributes to ALL cells survival and promotes ALL development, and the specific mechanism remains unclear.
Multiple myeloma (MM)
MM is a hematological malignant tumor that features abnormal clonal plasma cells in bone marrow [154]. The growth of abnormal clonal plasma cells causes damaging bone lesions, acute kidney impairment, anemia and hypercalcemia [155]. The disease generally progresses slowly and remains a malignancy that cannot be completely cured.
It is recognized that MM cells typically utilize a non-mitochondria-based glycolytic process to produce their ATP and be sensitive to glycolytic inhibitors [156]. The dependence of MM cells upon oxidative phosphorylation is triggered by an intercellular transport of mitochondria from adjacent nonmalignant BMSCs to increase cellular respiration, leading to increased proliferation of MM cells [157]. CD38, a typical marker of MM cells, has been linked to mitochondrial transfer medicated by TNTs. For example, the non-malignant transmission of mitochondria from astrocytes to neurons after stroke is governed by CD3864. In MM cells, CD38 was shown to be highly expressed [71], while shRNA-mediated knock-down of CD38 inhibited mitochondrial transport to MM cells and improved survival in animals in vivo. Inhibition of CD38 is a viable therapeutic strategy in MM in the current clinical setting, where treatment combination using drugs targeting CD38 such as daratumumab has shown considerable clinical effect in previously untreated and relapsed MM patients [158]. Thus, these studies provide a scientific basis for the inhibition of mitochondrial transport in MM, and a physiological basis for the choice of proper drugs to be applied in conjunction with mitochondrial transport blockers.
It was reported that myeloma MSCs are less dependent upon mitochondrial metabolism than healthy ones, exhibiting significantly higher glycolytic rates such as decreased NAD+/NADH ratios and increased intracellular lactate [159]. The metabolic reorganization of MM-MSCs may rely upon the hypoxic BM milieu, while MSCs also showed an enhanced proclivity to transport mitochondria to MM cells. This suggests that mitochondrial transport between plasma cells and stromal cells could contribute to the pro-tumor phenotype of MSCs. CXCL12/CXCR4 axis, which is important for normal and MM cells homing in the bone marrow, has been reported to be related to mitochondrial transmission [160]. CXCL12 is a critical modulator in the tumor microenvironment, influencing various oncogenic processes including angiogenesis, osteoclast-genesis, tumor cell immigration and adherence to stromal cells [161]. Furthermore, it was also found that plasma cells promoted CX43 expression of MSCs, leading to the activation of CXCL12 and its receptor CXCR4 on MM cells, which facilitates mitochondrial trafficking between MSCs and plasma cells. The selective suppression of CXCR4 by Plerixafor contributed to a considerable reduction in mitochondrial transport. Furthermore, CXCR4 intracellular expression in myeloma cells from BM specimens showed more co-localization with CD138 + cells in bortezomib-naive patients compared with bortezomib-responsive patients, implying that CXCR4 mediates chemo-resistance in MM. In summary, the experimental results indicate that the CXCL12/CXCR4 axis mediates intercellular coupling, indicating that myeloma microhabitats could be used as targets for improving and developing therapeutic approaches.
In conclusion, the role of mitochondrial trafficking in BM microenvironment is crucial for the pathogenesis, progression and drug resistance of hematological tumors (Table 1). Most of the current studies on mitochondrial transport involve hematological malignancies such as AML and ALL. In fact, mitochondria can also be regarded as potential targets for treatment in other hematological diseases such as CLL. For example, a study found that changing the apoptosis-dependent character of mitochondria restored the susceptibility of CLL cells to chemotherapy [162]. In the future, mitochondrial transfer is expected to be found between different cells of more hematological benign and malignancies.
Conclusion and perspectives
Here, we offer an overview of intercellular mitochondrial transfer, and discuss the modes and mechanisms underlying the process in diverse illnesses. Mitochondrial transfer has multiple different effects. The main reason is that mitochondria with reduced membrane potential can be released from cells of injured tissues, thus maintaining cellular homeostasis. In addition, mitochondria can also rescue metabolically damaged neighboring cells more altruistically, so as to promote the repair of cellular damage in a variety of tissues and apply to the treatment of many diseases. At the same time, cancer cells receive mitochondria from their neighbors to restore energy metabolism and increase proliferation, invasion and metastasis as well as resistance to chemotherapy. The above evidence suggests that mitochondria could be used as therapeutic targets in future therapy. This knowledge raises significant potential for modulating mitochondrial transfer to enhance normal cellular homeostasis and energy dynamics or to disturb the pathologic adaptations of tumor cells.
To date, it has been found that intercellular mitochondrial transfer plays a major role in the tumorigenesis, progression and drug resistance of hematological malignancies like AML and ALL. In other hematological malignancies, mitochondria are also targeted to enhance sensitivity to chemotherapeutic agents. All these findings could pave the way for a novel approach to the treatment of hematological malignancies in days to come. The current treatment by blocking TNTs can inhibit mitochondrial transfer and has worked well. Although the mechanism of mitochondrial transport is still not well understood, it may be related to key molecules such as CD38, motor protein KIF5B, and Cx43. These molecules could provide molecular targets for targeted therapy of hematological malignancies. At the same time, the key challenge relating to mitochondrial transfer therapy is their dual role in normal hematopoiesis and malignant diseases. As mentioned earlier, BMSCs under stress conditions provide mitochondria to HSCs. Therefore, during the targeting mitochondrial transfer therapy, it is of vital importance to consider the specific selection of recipients of mitochondrial transport between HSCs and hematopoietic malignant cells as well as the accurate regulation of the direction of transfer. It is not yet known whether the transfer of mitochondria from stromal cells to HSCs or malignant cells will have any effect during chemotherapy. Moreover, in normal hematopoiesis and malignant diseases, the differences in the intensity or types of the stress, as well as the diversities in the BM stromal microenvironment may have an effect on the bias of mitochondrial transfer. Therefore, when blocking mitochondrial transfer for hematological malignancies therapy, we should try to avoid the impact of off-target effects and make effort to develop more specific targeting molecules.
Nonetheless, additional research is required to cross knowledge gaps, eliminate clinical application challenges, and address ethical concerns related to the therapy.
Data availability
The datasets used during the current study are available from the corresponding author upon reasonable request.
References
Frey,T. G.&Mannella,C. A.The internal structure of mitochondria.Trends Biochem Sci25,319–324(2000).
Gammage PA, Frezza C. Mitochondrial DNA: the overlooked oncogenome? BMC Biol. 2019;17:53.
Bahat A, MacVicar T, Langer T. Metabolism and innate immunity meet at the mitochondria. Front Cell Dev Biol. 2021;9:720490.
Annesley,S. J.&Fisher,P. R.Mitochondria in health and disease.Cells8,680(2019).
Alavi Naini,S. M.&Soussi-Yanicostas,N.TauHyperphosphorylationanddxidativestress,acriticalviciouscircleinneurodegenerativetauopathies?Oxid Med Cell Longev2015,151979(2015).
Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. 2018;50:121–7.
Weiss H, et al. The mitochondrial Atp8 mutation induces mitochondrial ROS generation, secretory dysfunction, and β-cell mass adaptation in conplastic B6-mtFVB mice. Endocrinology. 2012;153:4666–76.
Rustom A, Saffrich R, Markovic I, Walther P, Gerdes H-H. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–10.
Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A. 2006;103:1283–8.
Pasquier,J.et al.Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance.J Transl Med11,94(2013).
Feng Y, et al. Human bone marrow mesenchymal stem cells rescue endothelial cells experiencing chemotherapy stress by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2019;28:674–82.
Zhang Y, et al. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-α yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Reports. 2016;7:749–63.
Jiang,D.et al.Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage.Cell Death Dis7,e2467(2016).
Brestoff,J. R.et al.Intercellular mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity.Cell Metab33,270–282.e8(2021).
Chen,J.et al.Macrophages induce cardiomyocyte ferroptosis via mitochondrial transfer.Free Radic Biol Med190,1–14(2022).
Zampieri,L. X.,Silva-Almeida,C.,Rondeau,J. D.&Sonveaux,P.Mitochondrial transfer in cancer: a comprehensive review.Int J Mol Sci22,3245(2021).
Tan,Y. L.et al.Mesenchymal stromal cell mitochondrial transfer as a cell rescue strategy in regenerative medicine: a review of evidence in preclinical models. Stem Cells Transl Med11,814–827(2022).
Konari,N.,Nagaishi,K.,Kikuchi,S.&Fujimiya,M.Mitochondria transfer from mesenchymal stem cells structurally and functionally repairs renal proximal tubular epithelial cells in diabetic nephropathy in vivo.Sci Rep9,5184(2019).
Yamaoka,S.,Nakajima,M.,Fujimoto,M.&Tsutsumi,N.MIRO1 influences the morphology and intracellular distribution of mitochondria during embryonic cell division in arabidopsis.Plant Cell Rep30,239–244(2011).
Paliwal,S.,Chaudhuri,R.,Agrawal,A.&Mohanty,S.Regenerative abilities of mesenchymal stem cells through mitochondrial transfer.J Biomed Sci25,31(2018).
Mistry,J. J.et al.ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection.Proc Natl Acad Sci U S A116,24610–24619(2019).
Marlein,C. R.et al.NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts.Blood130,1649–1660(2017).
Abounit,S.&Zurzolo,C.Wiring through tunneling nanotubes–from electrical signals to organelle transfer.J Cell Sci125,1089–1098(2012).
Sahinbegovic,H.et al.Intercellular mitochondrial transfer in the tumor microenvironment.Cancers (Basel)12,1787(2020).
Sisakhtnezhad,S.&Khosravi,L.Emerging physiological and pathological implications of tunneling nanotubes formation between cells.Eur J Cell Biol94,429–443(2015).
Cselenyák,A.,Pankotai,E.,Horváth,E. M.,Kiss,L.&Lacza,Z.Mesenchymal stem cells rescue cardiomyoblasts from cell death in an in vitro ischemia model via direct cell-to-cell connections.BMC Cell Biol11,29(2010).
Plotnikov,E. Y.,Khryapenkova,T. G.,Galkina,S. I.,Sukhikh,G. T.&Zorov,D. B.Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture.Exp Cell Res316,2447–2455(2010).
Eugenin,E. A.,Gaskill,P. J.&Berman,J. W.Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking.Cell Immunol254,142–148(2009).
Gousset,K.et al.Prions hijack tunnelling nanotubes for intercellular spread.Nat Cell Biol11,328–336(2009).
Koyanagi,M.,Brandes,R. P.,Haendeler,J.,Zeiher,A. M.&Dimmeler,S.Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes?Circ Res96,1039–1041(2005).
Sowinski,S.et al.Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission.Nat Cell Biol10,211–219(2008).
Domhan,S.et al.Intercellular communication by exchange of cytoplasmic material via tunneling nano-tube like structures in primary human renal epithelial cells.PLoS ONE6,e21283(2011).
Ahmad,T.et al.Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy.EMBO J33,994–1010(2014).
Wang,X.&Gerdes,H.-H.Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells.Cell Death Differ22,1181–1191(2015).
Onfelt,B.,Purbhoo,M. A.,Nedvetzki,S.,Sowinski,S.&Davis,D. M.Long-distancecallsbetweencellsconnectedbytunnelingnanotubules.Sci STKE2005,pe55(2005).
Schiller,C.et al.LST1 promotes the assembly of a molecular machinery responsible for tunneling nanotube formation.J Cell Sci126,767–777(2013).
Hase,K.et al.M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex.Nat Cell Biol11,1427–1432(2009).
Gousset,K.,Marzo,L.,Commere,P.-H.&Zurzolo,C.Myo10 is a key regulator of TNT formation in neuronal cells.J Cell Sci126,4424–4435(2013).
Wang,Y.,Cui,J.,Sun,X.&Zhang,Y.Tunneling-nanotube development in astrocytes depends on p53 activation.Cell Death Differ18,732–742(2011).
Fabisik,M.et al.Regulation of Inflammatory Response by Transmembrane Adaptor Protein LST1.Front Immunol12,618332(2021).
Veranic,P.et al.Different types of cell-to-cell connections mediated by nanotubular structures.Biophys J95,4416–4425(2008).
Ohta,Y.,Suzuki,N.,Nakamura,S.,Hartwig,J. H.&Stossel,T. P.The small GTPase RalA targets filamin to induce filopodia.Proc Natl Acad Sci U S A96,2122–2128(1999).
Dash,C.,Saha,T.,Sengupta,S.&Jang,H. L.Inhibition of tunneling nanotubes between cancer cell and the endothelium alters the metastatic phenotype.Int J Mol Sci22,6161(2021).
Qin,Y.et al.The functions, methods, and mobility of mitochondrial transfer between cells.Front Oncol11,672781(2021).
Hirokawa,N.&Takemura,R.Molecular motors and mechanisms of directional transport in neurons.Nat Rev Neurosci6,201–214(2005).
Saxton,W. M.&Hollenbeck,P. J.The axonal transport of mitochondria.J Cell Sci125,2095–2104(2012).
Pilling,A. D.,Horiuchi,D.,Lively,C. M.&Saxton,W. M.Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons.Mol Biol Cell17,2057–2068(2006).
López-Doménech,G.et al.Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites.Cell Rep17,317–327(2016).
Oeding,S. J.et al.Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX.J Cell Sci131,jcs219469(2018).
Kittler,J.Regulationofmitochondrialtrafficking,functionandqualitycontrolbythemitochondrialGTPasesMiro1andMiro2.Springerplus4,L33(2015).
Kalinski,A. L.et al.Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition.J Cell Biol218,1871–1890(2019).
Nahacka,Z.,Novak,J.,Zobalova,R.&Neuzil,J.Miro proteins and their role in mitochondrial transfer in cancer and beyond.Front Cell Dev Biol10,937753(2022).
López-Doménech,G.et al.Miro proteins coordinate microtubule‐ and actin‐dependent mitochondrial transport and distribution.EMBO J37,321–336(2018).
Foo,J. B.et al.Comparingthetherapeuticpotentialofstemcellsandtheirsecretoryproductsinregenerativemedicine.Stem Cells Int2021,2616807(2021).
Liau,L. L.et al.The potential of mesenchymal stromal cell as therapy in neonatal diseases.Front Pediatr8,591693(2020).
Zappulli,V.,Friis,K. P.,Fitzpatrick,Z.,Maguire,C. A.&Breakefield,X. O.Extracellular vesicles and intercellular communication within the nervous system.J Clin Invest126,1198–1207(2016).
Pitt,J. M.,Kroemer,G.&Zitvogel,L.Extracellular vesicles: masters of intercellular communication and potential clinical interventions.J Clin Invest126,1139–1143(2016).
Maas,S. L. N.,Breakefield,X. O.&Weaver,A. M.Extracellular vesicles: unique intercellular delivery vehicles.Trends Cell Biol27,172–188(2017).
Boyiadzis,M.&Whiteside,T. L.The emerging roles of tumor-derived exosomes in hematological malignancies.Leukemia31,1259–1268(2017).
Amari,L.&Germain,M.Mitochondrial extracellular vesicles - origins and roles.Front Mol Neurosci14,767219(2021).
Nawaz,M.&Fatima,F.Extracellular vesicles, tunneling nanotubes, and cellular interplay: synergies and missing links.Front Mol Biosci4,50(2017).
Zaborowski,M. P.,Balaj,L.,Breakefield,X. O.&Lai,C. P.Extracellular vesicles: composition, biological relevance, and methods of study.Bioscience65,783–797(2015).
Simeone,P.et al.Extracellular vesicles as signaling mediators and disease biomarkers across biological barriers.Int J Mol Sci21,2514(2020).
Hayakawa,K.et al.Transfer of mitochondria from astrocytes to neurons after stroke.Nature535,551–555(2016).
Torralba,D.,Baixauli,F.&Sánchez-Madrid,F.Mitochondria know no boundaries: mechanisms and functions of intercellular mitochondrial transfer.Front Cell Dev Biol4,107(2016).
Morrison,T. J.et al.Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer.Am J Respir Crit Care Med196,1275–1286(2017).
Sansone,P.et al.Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer.Proc Natl Acad Sci U S A114,E9066–E9075(2017).
Phinney,D. G.et al.Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs.Nat Commun6,8472(2015).
Rosina,M.et al.Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue.Cell Metab34,533–548.e12(2022).
Malavasi,F.et al.CD38 and chronic lymphocytic leukemia: a decade later.Blood118,3470–3478(2011).
Almeida,J.et al.High-sensitive immunophenotyping and DNA ploidy studies for the investigation of minimal residual disease in multiple myeloma.Br J Haematol107,121–131(1999).
Beyer,E. C.&Berthoud,V. M.Gap junction gene and protein families: connexins, innexins, and pannexins.Biochim Biophys Acta Biomembr1860,5–8(2018).
Sorgen,P. L.,Trease,A. J.,Spagnol,G.,Delmar,M.&Nielsen,M. S.Protein–protein interactions with connexin 43: regulation and function.Int J Mol Sci19,1428(2018).
Norris,R. P.Transfer of mitochondria and endosomes between cells by gap junction internalization.Traffic22,174–179(2021).
Schajnovitz,A.et al.CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions.Nat Immunol12,391–398(2011).
Islam,M. N.et al.Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury.Nat Med18,759–765(2012).
Zhang,L.,Liu,Q.,Hu,H.,Zhao,L.&Zhu,K.Progress in mesenchymal stem cell mitochondria transfer for the repair of tissue injury and treatment of disease.Biomed Pharmacother153,113482(2022).
Li,H.et al.Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction.Theranostics9,2017–2035(2019).
Aguilar,P. S.et al.Genetic basis of cell-cell fusion mechanisms.Trends Genet29,427–437(2013).
Huang,P.-J.et al.Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains.Cell Transpl25,913–927(2016).
Alvarez-Dolado,M.et al.Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes.Nature425,968–973(2003).
Acquistapace,A.et al.Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer.Stem Cells29,812–824(2011).
Pesaresi,M.,Sebastian-Perez,R.&Cosma,M. P.Dedifferentiation, transdifferentiation and cell fusion: in vivo reprogramming strategies for regenerative medicine.FEBS J286,1074–1093(2019).
Ma,Z.et al.Mesenchymal stem cell-cardiomyocyte interactions under defined contact modes on laser-patterned biochips.PLoS ONE8,e56554(2013).
Salaud,C.et al.Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells.Biochem Biophys Res Commun533,139–147(2020).
Zhu,D.et al.Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes.J Cell Sci118,3695–3703(2005).
Desir,S.et al.Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells.Oncotarget7,43150–43161(2016).
Osswald,M.et al.Brain tumour cells interconnect to a functional and resistant network.Nature528,93–98(2015).
Reindl,J.,Shevtsov,M.,Dollinger,G.,Stangl,S.&Multhoff,G.Membrane Hsp70-supported cell-to-cell connections via tunneling nanotubes revealed by live-cell STED nanoscopy.Cell Stress Chaperones24,213–221(2019).
Lou,E.et al.Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma.PLoS ONE7,e33093(2012).
Kabaso,D.,Lokar,M.,Kralj-Iglič,V.,Veranič,P.&Iglič,A.Temperature and cholera toxin B are factors that influence formation of membrane nanotubes in RT4 and T24 urothelial cancer cell lines.Int J Nanomedicine6,495–509(2011).
Zhang,Q.et al.Circulating mitochondrial DAMPs cause inflammatory responses to injury.Nature464,104–107(2010).
Kimura,S.,Hase,K.&Ohno,H.The molecular basis of induction and formation of tunneling nanotubes.Cell Tissue Res352,67–76(2013).
Chuang,Y.-C.et al.MitochondrialtransferfromWharton’sJellymesenchymalstemcelltoMERRFcybridreducesoxidativestressandimprovesmitochondrialbioenergetics.Oxid Med Cell Longev2017,5691215(2017).
Rodriguez,A.-M.,Nakhle,J.,Griessinger,E.&Vignais,M.-L.Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury.Cell Cycle17,712–721(2018).
Yuan,Y.et al.Mitochondrial transfer from mesenchymal stem cells to macrophages restricts inflammation and alleviates kidney injury in diabetic nephropathy mice via PGC-1α activation.Stem Cells39,913–928(2021).
Mahrouf-Yorgov,M.et al.Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties.Cell Death Differ24,1224–1238(2017).
LeBleu,V. S.et al.PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis.Nat Cell Biol16,992–1003,1–15(2014).
Murray,L. M. A.&Krasnodembskaya,A. D.Concise review: intercellular communication via organelle transfer in the biology and therapeutic applications of stem cells.Stem Cells37,14–25(2019).
Matejka,N.&Reindl,J.Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects.Radiat Oncol14,218(2019).
Li,Q.&Verma,I. M.NF-kappaB regulation in the immune system.Nat Rev Immunol2,725–734(2002).
Omsland,M.,Bruserud,Ø.,Gjertsen,B. T.&Andresen,V.Tunneling nanotube (TNT) formation is downregulated by cytarabine and NF-κB inhibition in acute myeloid leukemia (AML).Oncotarget8,7946–7963(2017).
Velarde,F.et al.Mesenchymal stem cell-mediated transfer of mitochondria: mechanisms and functional impact.Cell Mol Life Sci79,177(2022).
Valenti,D.,Vacca,R. A.,Moro,L.&Atlante,A.Mitochondria can cross cell boundaries: an overview of the biological relevance, pathophysiological implications and therapeutic perspectives of intercellular mitochondrial transfer.Int J Mol Sci22,8312(2021).
Jiao,H.et al.Mitocytosis, a migrasome-mediated mitochondrial quality-control process.Cell184,2896–2910.e13(2021).
Otsu,K.et al.Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells.Blood113,4197–4205(2009).
Miliotis,S.,Nicolalde,B.,Ortega,M.,Yepez,J.&Caicedo,A.Forms of extracellular mitochondria and their impact in health.Mitochondrion48,16–30(2019).
Caicedo,A.et al.MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function.Sci Rep5,9073(2015).
Ym,C.et al.Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations.PLoS ONE7,(2012).
Babenko,V. A.et al.Miro1 enhances mitochondria transfer from multipotent mesenchymal stem cells (MMSC) to neural cells and improves the efficacy of cell recovery.Molecules23,687(2018).
Jackson,M. V.et al.Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the In vitro and in vivo models of ARDS.Stem Cells34,2210–2223(2016).
Polak,R.,deRooij,B.,Pieters,R.&denBoer,M. L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment.Blood126,2404–2414(2015).
Ge,X.et al.Fumarate inhibits PTEN to promote tumorigenesis and therapeutic resistance of type2 papillary renal cell carcinoma.Mol Cell82,1249–1260.e7(2022).
Lu,J.et al.Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells.Oncotarget8,15539–15552(2017).
Hekmatshoar,Y.,Nakhle,J.,Galloni,M.&Vignais,M.-L.The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance.Biochem J475,2305–2328(2018).
Kheirandish-Rostami,M.et al.Mitochondrial characteristics contribute to proliferation and migration potency of MDA-MB-231 cancer cells and their response to cisplatin treatment.Life Sci244,117339(2020).
Elliott,R. L.,Jiang,X. P.&Head,J. F.Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity.Breast Cancer Res Treat136,347–354(2012).
Chang,J.-C.et al.Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer.J Exp Clin Cancer Res38,30(2019).
Pacak,C. A.et al.Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function.Biol Open4,622–626(2015).
Liu,K.et al.Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer.Microvasc Res92,10–18(2014).
Cheng,X.-Y.et al.Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria.Transl Neurodegener9,13(2020).
Golan,K.et al.Bone marrow regeneration requires mitochondrial transfer from donor Cx43-expressing hematopoietic progenitors to stroma.Blood136,2607–2619(2020).
Yang,C.et al.Mitochondria transfer mediates stress erythropoiesis by altering the bioenergetic profiles of early erythroblasts through CD47.J Exp Med219,e20220685(2022).
Auberger,P.,Tamburini-Bonnefoy,J.&Puissant,A.Drug resistance in hematological malignancies.Int J Mol Sci21,6091(2020).
Gerdes,H.-H.,Rustom,A.&Wang,X.Tunneling nanotubes, an emerging intercellular communication route in development.Mech Dev130,381–387(2013).
Döhner,H.,Weisdorf,D. J.&Bloomfield,C.D. Acute myeloid leukemia.N Engl J Med373,1136–1152(2015).
Vander Heiden,M. G.,Cantley,L. C.&Thompson,C. B.Understanding the Warburg effect: the metabolic requirements of cell proliferation.Science324,1029–1033(2009).
Skrtić,M.et al.Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia.Cancer Cell20,674–688(2011).
Farge,T.et al.Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism.Cancer Discov 7,716–735(2017).
Derdak,Z.et al.The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells.Cancer Res68,2813–2819(2008).
Moschoi,R.et al.Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy.Blood128,253–264(2016).
Robinson,A. J.,Davies,S.,Darley,R. L.&Tonks,A.Reactive oxygen species rewires metabolic activity in acute myeloid leukemia.Front Oncol11,632623(2021).
Pepping,J. K.et al.Myeloid-specific deletion of NOX2 prevents the metabolic and neurologic consequences of high fat diet.PLoS ONE12,e0181500(2017).
Saito,K.et al.Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition.Blood Adv5,4233–4255(2021).
DiNardo,C. D.et al.Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia.Blood133,7–17(2019).
Mistry,J. J.et al.Daratumumab inhibits acute myeloid leukemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells.Haematologica106,589–592(2021).
You,R.et al.Metformin sensitizes AML cells to chemotherapy through blocking mitochondrial transfer from stromal cells to AML cells.Cancer Lett532,215582(2022).
Velez,J.et al.Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport.Oncotarget7,51435–51449(2016).
Rowley,J. D.Letter:A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining.Nature243,290–293(1973).
Alvarez,R. H.,Kantarjian,H.&Cortes,J. E.The biology of chronic myelogenous leukemia: implications for imatinib therapy.Semin Hematol44,S4-14(2007).
Druker,B. J.et al.Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia.N Engl J Med344,1031–1037(2001).
Duarte,D.,Hawkins,E. D.&Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment.Blood131,1507–1511(2018).
Kolba,M. D.et al.Tunneling nanotube-mediated intercellular vesicle and protein transfer in the stroma-provided imatinib resistance in chronic myeloid leukemia cells.Cell Death Dis10,817(2019).
Pui,C.-H.,Robison,L. L.&Look,A. T.Acute lymphoblastic leukaemia.Lancet371,1030–1043(2008).
Bassan,R.&Hoelzer,D.Modern therapy of acute lymphoblastic leukemia.J Clin Oncol29,532–543(2011).
Cai,J.et al.ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells.Cell Death Dis7,e2459–e2459(2016).
Zhang,L.et al.Catechins induced acute promyelocytic leukemia cell apoptosis and triggered PML-RARα oncoprotein degradation.J Hematol Oncol7,75(2014).
Wang,J.et al.Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells.J Hematol Oncol11,11(2018).
Suganuma,K.et al.Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation.Leuk Lymphoma51,2112–2119(2010).
Tong,L.,Chuang,C.-C.,Wu,S.&Zuo,L.Reactive oxygen species in redox cancer therapy.Cancer Lett367,18–25(2015).
Hlozkova,K.et al.Metabolic profile of leukemia cells influences treatment efficacy of L-asparaginase.BMC Cancer20,526(2020).
Cruz,P.et al.Inhibition of InsP3R with Xestospongin B reduces mitochondrial respiration and induces selective cell death in T Cell acute lymphoblastic leukemia Cells.Int J Mol Sci22,651(2021).
Burt,R.et al.Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress.Blood134,1415–1429(2019).
Deng,J.et al.BCL2-BH4 antagonist BDA-366 suppresses human myeloma growth.Oncotarget7,27753–27763(2016).
Cowan,A. J.et al.Diagnosis and management of multiple myeloma: a review.JAMA327,464–477(2022).
Dalva-Aydemir,S.et al.Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin.Clin Cancer Res21,1161–1171(2015).
Marlein,C. R.et al.CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma.Cancer Res79,2285–2297(2019).
Mateos,M.-V.et al.Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma.N Engl J Med378,518–528(2018).
Giallongo,C.et al.CXCL12/CXCR4 axis supports mitochondrial trafficking in tumor myeloma microenvironment.Oncogenesis11,6(2022).
Ren,Z.et al.The CXCL12gamma chemokine immobilized by heparan sulfate on stromal niche cells controls adhesion and mediates drug resistance in multiple myeloma.J Hematol Oncol14,11(2021).
Bouyssou,J. M. C.,Ghobrial,I. M.&Roccaro,A. M.Targeting SDF-1 in multiple myeloma tumor microenvironment.Cancer Lett380,315–318(2016).
TenHacken,E.et al.Splicingmodulationsensitizeschroniclymphocyticleukemiacellstovenetoclaxbyremodelingmitochondrialapoptoticdependencies.JCI Insight3,e121438,121438(2018).
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82370173, 32241005).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.D.X.; original draft preparation, G.X.D. and C.C.; methodology, L.J.T. and J.W.B.; Figure design, L.W.C, W.Y.H. and Y.X.Y.; review and editing, M.D.X., G.X.D., C.C., L.W.C., W.Y.H., Y.X.Y., L.J.T., J.H.X., J.W.B., W.H.Y. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no conflict of interest.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, X., Can, C., Liu, W. et al. Mitochondrial transfer in hematological malignancies. Biomark Res 11, 89 (2023). https://doi.org/10.1186/s40364-023-00529-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00529-x