
Abstract
In the last few decades, YAP has been shown to be critical in regulating tumor progression. YAP activity can be regulated by many kinase cascade pathways and proteins through phosphorylation and promotion of cytoplasmic localization. Other factors can also affect YAP activity by modulating its binding to different transcription factors (TFs). Programmed cell death (PCD) is a genetically controlled suicide process present with the scope of eliminating cells unnecessary or detrimental for the proper development of the organism. In some specific states, PCD is activated and facilitates the selective elimination of certain types of tumor cells. As a candidate oncogene correlates with many regulatory factors, YAP can inhibit or induce different forms of PCD, including apoptosis, autophagy, ferroptosis and pyroptosis. Furthermore, YAP may act as a bridge between different forms of PCD, eventually leading to different outcomes regarding tumor development. Researches on YAP and PCD may benefit the future development of novel treatment strategies for some diseases. Therefore, in this review, we provide a general overview of the cellular functions of YAP and the relationship between YAP and PCD.
Similar content being viewed by others
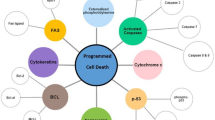


Background
The transcriptional coactivator Yes-associated protein (YAP) is amplified in human cancer and acts as an oncogene in various cancers [1,2,3]; its abundance and activity are increased in many types of cancer, including gastric cancer, colorectal cancer, liver cancer and breast cancer, in which it plays an important role in initiation, progression, metastasis, and drug resistance [4,5,6,7]. YAP and its paralog transcriptional coactivator with PDZ-binding motif (TAZ) are the major downstream effectors of the Hippo pathway, which controls organ size and cell fate during embryonic development [8]. Phosphorylated YAP can be sequestered in the cytoplasm and then degraded by the ubiquitin–proteasome system. Unphosphorylated YAP can enter liquid droplets and then translocate to the nucleus, where it has a series of functions [7, 9]. Nuclear YAP participates in complex and only partially understood molecular cascades that are responsible for the oncogenic responses by regulating multiple processes, in addition to the abovementioned processes, and by acting as an important mediator of cancer immunity [10] and cancer cell interactions [11].
It has also been discovered that YAP plays an important role in the regulation of programmed cell death (PCD) in cancer [12,13,14]. Different forms of PCD, including apoptosis, autophagy, ferroptosis and pyroptosis, have been characterized [15,16,17,18,19]. Understanding the connection between YAP and PCD will promote the development of more effective strategies for the treatment of some diseases, especially cancer. In this review, we discuss the regulation of YAP and the connections between YAP and PCD.
Synthesis and regulatory system of YAP
YAP, a small 350 kb amplicon encoding a 65 kDa protein on human chromosome 11q22, was identified in a screen for gene copy number alterations in mouse mammary tumors [20, 21]. YAP protein, which is highly conserved across different species, has a N-terminal TEAD-binding domain (TBD) followed by one or two WW domains and a C-terminal transactivation domain [22,23,24]. The WW domain is a protein–protein interaction module with two signature tryptophan residues spaced 38 to 40 amino acids apart [22, 25, 26]. However, as YAP lacks a DNA-binding motif, it regulates gene transcription by binding transcription factors (TFs) such as TEAD1–4, p73 and ZEB1 through its TBD and WW domains [27,28,29,30,31,32,33,34,35,36,37]. The YAP–TEAD complex regulates the expression of many oncogenes (Fig. 1), such as CTGF, CYR61, MYC and NUAK2 [28, 38,39,40]. NUAK2 participates in a feed-forward loop that enhances YAP activity [38]. In addition, YAP–TEAD transcriptional activity can be regulated by different proteins through many mechanisms. For example, the SS18-SSX fusion protein and MRTF potentiate TEAD-YAP activity [41, 42], whereas TIAM1 and RUNX3 reduce YAP–TEAD transcriptional activity [31, 43].

Signaling pathways and genes controlling YAP expression and its regulatory system. YAP can be affected by many factors. when YAP is phosphorylated in the cytoplasm by some factors like Hippo pathway, AKT, c-Abl, NLK, MYPT1 and PRP4K, it finally can be degraded, YAP can also be enhanced by OTUB2 so that it can enter in nucleus easily. Active YAP localizes in the nucleus and combines with TEAD, p73 or ZEB1 at its TED domain thus affect other factors. YAP-TEAD co-activator could enhance CTGF, CYR6, MYC and NUAK2, which participates in a feed-forward loop that enhances YAP activity by inhibit Hippo pathway. YAP-TEAD co-activator could also be inhibited by TRPS1, TIAM1 and RUNX3, and be enhanced by MRTF and SS18-SSX. Aurora a kinase can phosphorylate YAP in nucleus thus reduce the activity of it
On the other hand, the translocation of YAP between the nucleus and cytoplasm has emerged as an important means of regulating YAP activity [34, 44, 45]. Active YAP localizes in the nucleus. YAP can be negatively regulated by its circular RNA circYAP, which suppresses the assembly of translation initiation machinery [46]. In addition, YAP protein activity can be modulated by posttranslational modification. OTUB2 activates YAP protein by direct deubiquitination and stabilization [47]. The Hippo signaling pathway is the main regulator of YAP activity, which is accomplished by modulating YAP phosphorylation [48,49,50,51,52]. Through this signaling pathway, YAP can be phosphorylated at five conserved HXRXXS motifs, including the serine 127 (S127) and S397 sites [51]. Additionally, other pathways can regulate YAP activity though Hippo pathway. For instance, MYPT1 activates the Hippo pathway, resulting in YAP suppression [53]. SRC phosphorylates Hippo pathway kinases, thus promoting YAP activity [54]. PRP4K phosphorylates YAP at S111 and S250 to regulate this protein in parallel with the Hippo pathway [55]. The STARD13-correlated ceRNA network leads to the nuclear–cytoplasmic translocation of YAP through two independent pathways, the Hippo pathway and Rho-GTPase/F-actin signaling [56]. AKT and c-Abl phosphorylate YAP at S127 and Y357, respectively, thus suppressing some transcriptional responses [57, 58]. NLK phosphorylates YAP at S128 to evoke YAP redistribution into the nucleus [59]. YAP interacts with Aurora A kinase in the nucleus, which leads to specific phosphorylation of YAP, and then potentiates YAP-mediated transforming ability [60]. Therefore, in the future, it will be important to examine the contribution of different TFs to the regulation of YAP phosphorylation, nuclear localization and to determine the underlying mechanism in greater detail. Meanwhile, the effector molecules downstream of YAP should also be identified.
YAP and PCD
PCD is a normal physiological phenomenon. Under normal circumstances, PCD can help eliminate senescent and dangerous cells, such as cancer cells and infected cells. However, dysregulation of cell death often has negative consequences. And there is a crosstalk among the four major forms of PCD, apoptosis, autophagy, ferroptosis and pyroptosis [15,16,17,18,19]. PCD is typically dysregulated in certain diseases, especially in cancer, and this dysregulation provides necessary conditions for tumor progression. Many oncogenes and tumor suppressor genes are linked to tumorigenesis through PCD, and some proteins and RNAs can simultaneously regulate these different forms of cancer cell death [61]. Previous research has identified YAP-related regulatory networks in cancer. Among these networks and pathways, the evolutionarily conserved Hippo pathway plays an important role in cell growth and organ size control during embryonic development. As the center of this complex network, YAP is necessary for PCD controlled by the Hippo pathway, but YAP is also regulated by many other factors and regulates additional factors to affect PCD (Table 1); these findings prompts researchers to further investigate the relationship between YAP and PCD.
YAP and apoptosis
Apoptosis is initiated in cells in response to diverse physiological and pathological stimuli that ultimately leads to the activation of apoptotic pathways [73]. The dysregulation of apoptosis is one of the classical hallmarks of the maintenance and regulation of tumor growth [74,75,76]. YAP shows antiapoptotic effects in various cells; for example, YAP reduces apoptosis by upregulating Jag-1 to activate Notch signaling in hepatocellular carcinoma cells, and enhancing BCAR4 expression in breast cancer, respectively [62, 63]. In addition, the ubiquitination of YAP by Fbxw7 and its subsequent proteasomal degradation enhance apoptosis in hepatocellular carcinoma cells, while restoring YAP expression partially abrogates Fbxw7-induced cell apoptosis and growth arrest in vitro and in vivo [64]. YAP also enhances the ability of cancer cells to resist apoptosis induced by chemotherapy. Hepatocellular carcinoma patients with a poor response to transarterial chemoembolization (TACE) had higher YAP protein levels than responsive patients, indicating that YAP enhances the chemoresistance of Hepatocellular carcinoma by blocking apoptosis [5]. Additionally, Ciamporcero et al. found that YAP knockdown sensitizes urothelial carcinoma cells to chemotherapy and radiation via the increased accumulation of DNA damage and apoptosis [65]. These studies show that the overproduction of YAP can enhance the resistance of cancer cells to apoptosis, and when YAP activity is inhibited, the increase in apoptosis sensitizes cancer cells to chemotherapy.
In cholangiocarcinoma, YAP and TEADs prevent apoptosis induced by cytotoxic drugs together, but YAP knockdown sensitizes cholangiocarcinoma cells to drug-induced apoptosis [66]. Related research on hypoxic cancer cells showed that YAP can be activated by GPRC5A, a novel hypoxia-induced protein that protects cancer cells from apoptosis during oxygen deprivation, leading to the downregulation of proapoptotic target genes and the increased survival of hypoxic cancer cells. This study further showed that the apoptosis induced by GPRC5A depletion under hypoxic conditions could be rescued by constitutively active YAP [67]. YAP prevents apoptosis in acute pancreatitis, possibly by modulating the inflammatory response [77]. Taken together, the data indicate that YAP acts as a core factor in regulating cancer apoptosis through various mechanisms, and in the presence of apoptotic stimuli, YAP seems to enhance the ability of cells to mitigate apoptosis and survive.
Interestingly, YAP can also function as a proapoptotic factor. When cells experience stress due to severe DNA damage, Akt and c-Abl phosphorylate YAP, thus suppressing the induction of proapoptotic gene expression and ultimately inducing apoptosis [57, 58]. YAP can trigger apoptosis by binding p73 instead of TEAD and thereby upregulating the proapoptotic gene BAX [78,79,80]. These data contradict the reported function of YAP as an oncogene, and it remains unknown whether targets of the YAP–p73 complex other than BAX are relevant to promoting apoptosis and whether the engagement of other TFs by YAP can elicit a proapoptotic transcriptional program. Elucidation of the precise mechanisms by which YAP modulates apoptosis might yield new therapeutic targets.
YAP and autophagy
Autophagy is a process by which intracellular damaged or excessive organelles and cytoplasmic proteins are degraded through a lysosome-mediated process [81,82,83]. This evolutionarily conserved process enables the recycling of nonessential cytosolic materials to maintain cellular homeostasis and overcome metabolic stress and nutrient deprivation. A close association between autophagy and cancer has been established, and autophagy plays an important role in cell survival and death [82, 84,85,86,87]. Excessive or prolonged autophagy can elicit cell death. Some studies showed that YAP is regulated by autophagy and thus affects cell fate [88,89,90,91]. Impaired autophagy is a driver of nuclear YAP localization and promotes YAP activity to enhance tissue remodeling and carcinogenesis in hepatocellular carcinoma cells [92, 93]. Conversely, triple-negative breast cancer utilizes autophagy as a mechanism to promote YAP nuclear entry, thus promoting cell invasion and metastasis [94]. As mentioned above, YAP requires autophagy to sustain the transformed characteristics of cancer cells. And YAP is also involved in the lysosomal degradation of autophagosomes and the fusion of autophagosomes with lysosomes [95]. Therefore, YAP is both upstream and downstream of autophagy. YAP increases autolysosome degradation, thereby enhancing cellular autophagic flux to protect breast cancer cells from nutrient deprivation-induced apoptosis [68]. YAP upregulation endows hepatocellular carcinoma cells with multi-drug resistance via the RAC1–ROS–mTOR pathway, resulting in the repression of autophagy [69]. YAP can inhibit autophagy in human colorectal cancer cells by transcriptionally upregulating Bcl-2, which consequently promotes colorectal cancer progression [70]. Zhao et al. found that in glioblastoma, YAP enhances HMGB1-mediated autophagy to promote glioma progression [71]. The relation between YAP and autophagy is not consistent, as it depends on other factors. Pavel et al. found that the response of YAP to autophagy can differ due to α-catenin levels: high basal α-catenin levels enable YAP to positively regulate autophagy, while low α-catenin levels lead to a negative correlation between YAP and autophagy [96]. Since autophagy can provide nutrients to cells and remove toxic particles autonomously, this biological process is critical for cell survival, especially under nutrient-deprived conditions, such as in the tumor microenvironment. YAP-related autophagy seems to be the first step in inducing pathways and processes that prevent cancer cells from dying [68]. In the future, autophagy-related death may be controlled by regulating YAP to prevent tumor progression, The mechanism underlying the relationship between YAP and autophagy needs to be explored further.
YAP and ferroptosis
Ferroptosis is distinct from the above forms of cell death; since it was first proposed in 2012, it has gradually become a widely researched topic [97,98,99]. Ferroptosis, which is driven by cellular metabolism and iron-dependent lipid peroxidation, has been implicated in cancer growth and drug resistance [100,101,102,103]. Epithelial-to-mesenchymal transition (EMT) is believed to generate cancer stem cells, resulting in metastatic spread and contributing to clinical therapeutic resistance [104]. YAP was recently reported to be necessary for promoting EMT [105, 106]; however, it has been confirmed that EMT can improve the sensitivity of cells to ferroptosis [107, 108], perhaps indicating that YAP promotes ferroptosis through EMT. In later studies, researchers found that the sensitivity to ferroptosis is highly influenced by cell contacts and cellular density [109]. The activation of TFs in the Hippo pathway (such as YAP and TAZ) promotes ferroptosis in cancer cells by regulating the expression of ferroptosis modulators, such as ACSL4, TFRC, EMP1 and ANGPTL4 [72]. In addition, ovarian and renal cancer cells were found to be sensitive to ferroptosis induced by erastin treatment and cystine deprivation when they grew at low density or as individual cells. However, when these same ferroptosis-sensitive cells were grown to confluence or in a high-density situation (like three-dimensional (3D) spheres), they become highly resistant to ferroptosis [110]. As a sensor of cell density, TAZ can regulate cellular sensitivity to ferroptosis through the Hippo pathway in ovarian cancer and renal cell carcinoma, instead of YAP [109, 111]. Wu et al. reported an explanation for this phenomenon. In epithelial cells and mesothelioma, cell–cell contacts inhibit ferroptosis partly through the cadherin 1-mediated inhibition of YAP transcriptional activity. Cells at high density had high levels of ECAD, which activates Merlin and subsequently regulates YAP activity by activating the Hippo pathway, thus influencing the sensitivity to ferroptosis. In addition, HCT116 cells lacking YAP were no longer sensitized to ferroptosis by RNAi-mediated Merlin knockdown, demonstrating that Merlin suppresses ferroptosis by inhibiting YAP activity [72]. There have been few studies on YAP and ferroptosis, the mechanism underlying the relationship between YAP and ferroptosis needs to be explored further. In addition, autophagy is closely related to ferroptosis, and YAP is an important regulator of autophagy. The relationship between these two processes and YAP will be discussed below.
YAP and pyroptosis
Pyroptosis, also known as inflammatory necrosis, is a kind of PCD characterized by the continuous expansion of cells until the cell membrane ruptures, leading to the release of cell contents, which activate a strong inflammatory response. YAP expression is higher in triple-negative breast cancer than in other types of breast cancer, and YAP promotes the metastasis of triple-negative breast cancer cells [112,113,114]. Studies have also found that PD-L1 expression is higher in triple-negative breast cancer than in other types of breast cancer [115, 116], and studies in other types of cancer have identified mutual regulation between PD-L1 and YAP [117]. For example, in BRAF inhibitor-resistant melanoma cells, YAP induces the expression of PD-L1, which participates in tumor escape [118]. Researchers have found that under hypoxia, PD-L1 accumulates in the nucleus, whereby it can regulate the expression and activation of gasdermin C, the main executor of pyroptosis, to promote this form of cell death [119]. Therefore, YAP may regulate pyroptosis by regulating various factors at the protein and mRNA levels. In macrophages, flagellin secreted by Salmonella activates the Nod-like receptor family CARD domain-containing protein 4 (NLRC4) inflammasome, leading to the production of IL-1β and pyroptosis of infected cells. In contrast, in Salmonella-infected B cells, infection induces YAP phosphorylation and promotes the interaction of YAP with Hck, thus preventing the transcriptional activation of NLRC4 and blocking B cell death. Although the outcomes of cellular Salmonella infection are variable, YAP activation can lead to pyroptosis [120]. Cui et al. found that in pancreatic ductal adenocarcinoma, MST1 could suppress the proliferation, migration, invasion, and spheroid formation of pancreatic ductal adenocarcinoma cells via caspase 1-induced pyroptosis mediated by reactive oxygen species (ROS) [121]. Whether MST1, a core component of the Hippo pathway, can influence pyroptosis by affecting YAP activation is worth studying.
Crosstalk between YAP and PCD
There are also various connections between different types of PCD. Although the forms of PCD differ from each other in the mechanism of death, they are related. For example, autophagy is necessary during ferroptosis. As a selective form of autophagy, ferroptosis is initiated by the degradation of ferritin, which triggers labile iron overload, lipid peroxidation, membrane damage, and cell death [122]. Yang et al. discovered a type of autophagy called “clockophagy”, which involves the selective degradation of the core circadian clock protein ARNTL and can facilitate the induction of ferroptosis [123]. Gao et al. identified multiple autophagy-related genes as positive regulators of ferroptosis and showed that ferroptosis is an autophagic cell death process [124]. Thus, ferroptosis and autophagy are closely linked. Furthermore, autophagy enhances labile iron availability and potentiates the activity of ferroptotic stimuli in glutamic-oxaloacetic transaminase 1(GOT1)-knockdown cells, as GOT1 inhibition represses mitochondrial metabolism and promotes a catabolic state [125]. Meanwhile, research on the dependence of breast cancer cells on exogenous glutamine for growth showed that YAP can induce GOT1 [126]. These data indicated that YAP could weaken the link between autophagy and ferroptosis, thereby protecting cells. Taken together, the evidence suggests that YAP plays an important role in the link between autophagy and ferroptosis.
Recent studies have revealed that autophagy can facilitate apoptosis in different ways. Autophagy promotes apoptosis through the degradation of antiapoptotic and cell survival factors; for instance, the autophagic degradation of caveolin-1 contributes to apoptosis in rat hippocampal astrocytes [127]. The product of the autophagy-related gene Atg5 is required for the formation of autophagosomes. The calpain-mediated cleavage of Atg5 generates a proapoptotic protein that associates with the antiapoptotic molecule Bcl-xL to stimulate intrinsic apoptosis [128]. However, the presence of YAP alters this relationship; in particular, YAP-induced autophagy always inhibits apoptosis to elicit cancer cell survival. In an immunohistochemical analysis of clinical thyroid papillary carcinoma tissue microarrays, YAP silencing was accompanied by decreases in Beclin1 and Atg5 expression [129]. In addition, YAP prompted an increase in autophagy by enhancing autolysosome degradation; thus, YAP plays a protective role in cisplatin-resistant human ovarian cancer cells by reducing apoptosis [130]. When breast cancer cells are exposed to nutrient deprivation, YAP increases autolysosome degradation, thereby enhancing autophagic flux in breast cancer cells and protecting these cells from apoptosis [68]. As stated above, the relationship between YAP and autophagy is complex, but YAP ultimately regulate autophagy and then affect cell survival by inhibiting other forms of PCD.
Apoptosis is also connected to ferroptosis. In a study of acute lung injury (ALI), ferroptosis contributed to intestinal ischemia/reperfusion-induced ALI in vivo, and inhibitor of apoptosis-stimulating protein of p53 (iASPP) was found to inhibit ferroptosis and alleviate intestinal ischemia/reperfusion-induced ALI through the Nrf2/HIF-1/TF signaling pathway [131]. In a study on bladder cancer, researchers demonstrated the presence of cross-talk between YAP and Nrf2 [132]; therefore, although there is not sufficient evidence to prove its role in the relationship between apoptosis and ferroptosis, YAP is very likely involved.
YAP can prevent further apoptosis by regulating the inflammatory response [77], with which pyroptosis is strongly associated [133]; these data suggest that YAP can connect these two types of PCD. However, there is a lack of research in this area. In summary, YAP plays a key role in the regulation of various forms of PCD and their relationships, and the specific mechanism is not yet clear and worthy of further in-depth study. In addition, researchers need to focus on the involvement of YAP in the relationships among different types of PCD.
Conclusion
Since the discovery of YAP several decades ago, the biological functions and consequences of YAP have gradually been revealed. YAP is an important downstream effector of the Hippo pathway and a transcriptional cofactor involved in the transcription of many known important tumor factors. As such, YAP is an important factor that should be studied in relation to tumorigenesis, including tumor development and metastasis, as well as the prevention of drug resistance. Numerous studies have shown that YAP is involved in cancer cell death. Our review summarizes the knowledge of PCD and YAP, explores the important regulatory role of YAP in PCD. As stated above, YAP not only regulates individual types of PCD but also is likely a bridging factor that connects different types of PCD. As an oncogene, YAP is supposed to promote cancer progression and inhibit PCD, however, YAP is found to promote PCD such as apoptosis [134] and ferroptosis [72] in some studies. There is great value in discovering the specific underlying mechanism. Moreover, the role of YAP in pyroptosis remains unclear but will be further explored in future research. Research on the association between YAP and PCD could help to establish a systematic tumorigenesis mechanism, guide the development of new cancer treatments, and aid in the discovery of treatments for other inflammatory-related diseases.
Availability of data and materials
Not applicable.
References
Kushner MH, Ory V, Graham GT, et al. Loss of ANCO1 repression at AIB1/YAP targets drives breast cancer progression. EMBO Rep 2019:e48741. https://doi.org/10.15252/embr.201948741.
Kumar D, Nitzan E, Kalcheim C. YAP promotes neural crest emigration through interactions with BMP and Wnt activities. Cell Commun Signal. 2019;17(1):69. https://doi.org/10.1186/s12964-019-0383-x.
Lefort S, Tan S, Balani S, et al. Initiation of human mammary cell tumorigenesis by mutant KRAS requires YAP inactivation. Oncogene. 2020;39(9):1957–68. https://doi.org/10.1038/s41388-019-1111-0.
Dong L, Lin F, Wu W, Liu Y, Huang W. Verteporfin inhibits YAP-induced bladder cancer cell growth and invasion via Hippo signaling pathway. Int J Med Sci. 2018;15(6):645–52. https://doi.org/10.7150/ijms.23460.
Chen M, Wu L, Tu J, et al. miR-590-5p suppresses hepatocellular carcinoma chemoresistance by targeting YAP1 expression. EBioMedicine. 2018;35:142–54. https://doi.org/10.1016/j.ebiom.2018.08.010.
Han H, Qi R, Zhou J, et al. Regulation of the Hippo Pathway by Phosphatidic Acid-Mediated Lipid-Protein Interaction. Mol Cell. 2018;72(2):328–40 e8. https://doi.org/10.1016/j.molcel.2018.08.038.
Shen J, Cao B, Wang Y, et al. Hippo component YAP promotes focal adhesion and tumour aggressiveness via transcriptionally activating THBS1/FAK signalling in breast cancer. J Exp Clin Canc Res. 2018;37(1):175. https://doi.org/10.1186/s13046-018-0850-z.
Moya I, Castaldo S, Van den Mooter L, et al. Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science. 2019;366(6468):1029–34. https://doi.org/10.1126/science.aaw9886.
Cai D, Feliciano D, Dong P, et al. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat Cell Biol. 2019;21(12):1578–89. https://doi.org/10.1038/s41556-019-0433-z.
Wang Z, Wang F, Ding X, et al. Hippo/YAP signaling choreographs the tumor immune microenvironment to promote triple negative breast cancer progression via TAZ/IL-34 axis. Cancer Lett. 2022;527:174–90. https://doi.org/10.1016/j.canlet.2021.12.016.
Shen Q, Hill T, Cai X, et al. Physical confinement during cancer cell migration triggers therapeutic resistance and cancer stem cell-like behavior. Cancer Lett. 2021;506:142–51. https://doi.org/10.1016/j.canlet.2021.01.020.
Zhang X, Li F, Cui Y, Liu S, Sun H. Mst1 overexpression combined with Yap knockdown augments thyroid carcinoma apoptosis via promoting MIEF1-related mitochondrial fission and activating the JNK pathway. Cancer Cell Int. 2019;19:143. https://doi.org/10.1186/s12935-019-0860-8.
Yosefzon Y, Soteriou D, Feldman A, et al. Caspase-3 Regulates YAP-Dependent Cell Proliferation and Organ Size. Mol Cell. 2018;70(4):573–87 e4. https://doi.org/10.1016/j.molcel.2018.04.019.
Seo J, Kim M, Hong H, et al. MK5 Regulates YAP Stability and Is a Molecular Target in YAP-Driven Cancers. Cancer Res. 2019;79(24):6139–52. https://doi.org/10.1158/0008-5472.Can-19-1339.
Wang M, Wang XF, Li YM, et al. Cross-talk between autophagy and apoptosis regulates testicular injury/recovery induced by cadmium via PI3K with mTOR-independent pathway. Cell Death Dis. 2020;11(1):46. https://doi.org/10.1038/s41419-020-2246-1.
Fitzwalter BE, Towers CG, Sullivan KD, et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev Cell. 2018;44(5):555–65 e3. https://doi.org/10.1016/j.devcel.2018.02.014.
Zou Y, Sarem M, Xiang S, Hu H, Xu W, Shastri V. Autophagy inhibition enhances Matrine derivative MASM induced apoptosis in cancer cells via a mechanism involving reactive oxygen species-mediated PI3K/Akt/mTOR and Erk/p38 signaling. BMC Cancer. 2019;19(1):949. https://doi.org/10.1186/s12885-019-6199-7.
Park E, Chung S. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822. https://doi.org/10.1038/s41419-019-2064-5.
Zhang Z, Guo M, Li Y, et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16(8):1482–505. https://doi.org/10.1080/15548627.2019.1687985.
Overholtzer M, Zhang J, Smolen G, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103(33):12405–10. https://doi.org/10.1073/pnas.0605579103.
Sudol M, Bork P, Einbond A, et al. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem. 1995;270(24):14733–41. https://doi.org/10.1074/jbc.270.24.14733.
Chen H, Sudol M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A. 1995;92(17):7819–23. https://doi.org/10.1073/pnas.92.17.7819.
Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18(9):2551–62. https://doi.org/10.1093/emboj/18.9.2551.
Chen L, Chan S, Zhang X, et al. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev. 2010;24(3):290–300. https://doi.org/10.1101/gad.1865310.
Bedford M, Chan D, Leder P. FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 1997;16(9):2376–83. https://doi.org/10.1093/emboj/16.9.2376.
Chen H, Einbond A, Kwak S, et al. Characterization of the WW domain of human yes-associated protein and its polyproline-containing ligands. J Biol Chem. 1997;272(27):17070–7. https://doi.org/10.1074/jbc.272.27.17070.
Gao Y, Yang Y, Yuan F, et al. TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis. 2017;6(9):e383. https://doi.org/10.1038/oncsis.2017.83.
Tan B, Yang M, Singh S, et al. LncRNA NORAD is repressed by the YAP pathway and suppresses lung and breast cancer metastasis by sequestering S100P. Oncogene. 2019;38(28):5612–26. https://doi.org/10.1038/s41388-019-0812-8.
Zhao B, Kim J, Ye X, Lai ZC, Guan KL. Both TEAD-binding and WW domains are required for the growth stimulation and oncogenic transformation activity of yes-associated protein. Cancer Res. 2009;69(3):1089–98. https://doi.org/10.1158/0008-5472.CAN-08-2997.
Pajtler KW, Wei Y, Okonechnikov K, et al. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat Commun. 2019;10(1):3914. https://doi.org/10.1038/s41467-019-11884-5.
Elster D, Tollot M, Schlegelmilch K, et al. TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat Commun. 2018;9(1):3115. https://doi.org/10.1038/s41467-018-05370-7.
Li Z, Zhao B, Wang P, et al. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24(3):235–40. https://doi.org/10.1101/gad.1865810.
Downward J, Basu S. YAP and p73: a complex affair. Mol Cell. 2008;32(6):749–50. https://doi.org/10.1016/j.molcel.2008.12.002.
Nishioka N, Inoue K, Adachi K, et al. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell. 2009;16(3):398–410. https://doi.org/10.1016/j.devcel.2009.02.003.
Lehmann W, Mossmann D, Kleemann J, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun. 2016;7:10498. https://doi.org/10.1038/ncomms10498.
Cao X, Pfaff S, Gage F. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22(23):3320–34. https://doi.org/10.1101/gad.1726608.
Papaspyropoulos A, Bradley L, Thapa A, et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat Commun. 2018;9(1):424. https://doi.org/10.1038/s41467-017-02786-5.
Gill M, Christova T, Zhang Y, et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun. 2018;9(1):3510. https://doi.org/10.1038/s41467-018-05939-2.
Mason D, Collins J, Dawahare J, et al. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J Cell Biol. 2019;218(4):1369–89. https://doi.org/10.1083/jcb.201806065.
Murakami S, Nemazanyy I, White S, et al. A Yap-Myc-Sox2-p53 Regulatory Network Dictates Metabolic Homeostasis and Differentiation in Kras-Driven Pancreatic Ductal Adenocarcinomas. Dev Cell. 2019;51(1):113–28.e9. https://doi.org/10.1016/j.devcel.2019.07.022.
Kim T, Hwang D, Lee D, Kim JH, Kim SY, Lim DS. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. 2017;36(4):520–35. https://doi.org/10.15252/embj.201695137.
Isfort I, Cyra M, Elges S, et al. SS18-SSX-Dependent YAP/TAZ Signaling in Synovial Sarcoma. Clin Cancer Res. 2019;25(12):3718–31. https://doi.org/10.1158/1078-0432.Ccr-17-3553.
Diamantopoulou Z, White G, Fadlullah MZH, et al. TIAM1 Antagonizes TAZ/YAP Both in the Destruction Complex in the Cytoplasm and in the Nucleus to Inhibit Invasion of Intestinal Epithelial Cells. Cancer Cell. 2017;31(5):621–34 e6. https://doi.org/10.1016/j.ccell.2017.03.007.
Calvo F, Ege N, Grande-Garcia A, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–46. https://doi.org/10.1038/ncb2756.
Deng Y, Lu J, Li W, et al. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat Commun. 2018;9(1):4564. https://doi.org/10.1038/s41467-018-07022-2.
Wu N, Yuan Z, Du KY, et al. Translation of yes-associated protein (YAP) was antagonized by its circular RNA via suppressing the assembly of the translation initiation machinery. Cell Death Differ. 2019;26(12):2758–73. https://doi.org/10.1038/s41418-019-0337-2.
Zhang Z, Du J, Wang S, et al. OTUB2 Promotes Cancer Metastasis via Hippo-Independent Activation of YAP and TAZ. Mol Cell. 2019;73(1):7–21.e7. https://doi.org/10.1016/j.molcel.2018.10.030.
Shu Z, Gao Y, Zhang G, et al. A functional interaction between Hippo-YAP signalling and SREBPs mediates hepatic steatosis in diabetic mice. J Cell Mol Med. 2019;23(5):3616–28. https://doi.org/10.1111/jcmm.14262.
Zhang J, Smolen GA, Haber DA. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008;68(8):2789–94. https://doi.org/10.1158/0008-5472.CAN-07-6205.
Oka T, Mazack V, Sudol M. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J Biol Chem. 2008;283(41):27534–46. https://doi.org/10.1074/jbc.M804380200.
Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61. https://doi.org/10.1101/gad.1602907.
Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283(9):5496–509. https://doi.org/10.1074/jbc.M709037200.
Muñoz-Galván S, Felipe-Abrio B, Verdugo-Sivianes E, et al. Downregulation of MYPT1 increases tumor resistance in ovarian cancer by targeting the Hippo pathway and increasing the stemness. Mol Cancer. 2020;19(1):7. https://doi.org/10.1186/s12943-020-1130-z.
Lamar JM, Xiao Y, Norton E, et al. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem. 2019;294(7):2302–17. https://doi.org/10.1074/jbc.RA118.004364.
Cho YS, Zhu J, Li S, Wang B, Han Y, Jiang J. Regulation of Yki/Yap subcellular localization and Hpo signaling by a nuclear kinase PRP4K. Nat Commun. 2018;9(1):1657. https://doi.org/10.1038/s41467-018-04090-2.
Zheng L, Xiang C, Li X, et al. STARD13-correlated ceRNA network-directed inhibition on YAP/TAZ activity suppresses stemness of breast cancer via co-regulating Hippo and Rho-GTPase/F-actin signaling. J Hematol Oncol. 2018;11(1):72. https://doi.org/10.1186/s13045-018-0613-5.
Basu S, Totty N, Irwin M, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14–3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11(1):11–23. https://doi.org/10.1016/s1097-2765(02)00776-1.
Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008;29(3):350–61. https://doi.org/10.1016/j.molcel.2007.12.022.
Moon S, Kim W, Kim S, et al. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017;18(1):61–71. https://doi.org/10.15252/embr.201642683.
Chang SS, Yamaguchi H, Xia W, et al. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene. 2017;36(9):1265–75. https://doi.org/10.1038/onc.2016.292.
Mao C, Wang X, Liu Y, et al. A G3BP1-Interacting lncRNA Promotes Ferroptosis and Apoptosis in Cancer via Nuclear Sequestration of p53. Cancer Res. 2018;78(13):3484–96. https://doi.org/10.1158/0008-5472.CAN-17-3454.
Tschaharganeh DF, Chen X, Latzko P, et al. Yes-Associated Protein Up-regulates Jagged-1 and Activates the NOTCH Pathway in Human Hepatocellular Carcinoma. Gastroenterology. 2013;144(7):1530-42.e12. https://doi.org/10.1053/j.gastro.2013.02.009.
Zheng X, Han H, Liu GP, et al. LncRNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 2017;36(22):3325–35. https://doi.org/10.15252/embj.201797609.
Tu K, Yang W, Li C, et al. Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer. 2014;13:110. https://doi.org/10.1186/1476-4598-13-110.
Ciamporcero E, Shen H, Ramakrishnan S, et al. YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene. 2016;35(12):1541–53. https://doi.org/10.1038/onc.2015.219.
Marti P, Stein C, Blumer T, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62(5):1497–510. https://doi.org/10.1002/hep.27992.
Greenhough A, Bagley C, Heesom K, et al. Cancer cell adaptation to hypoxia involves a HIF-GPRC5A-YAP axis. EMBO Mol Med. 2018;10(11):e8699. https://doi.org/10.15252/emmm.201708699.
Song Q, Mao B, Cheng J, et al. YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS ONE. 2015;10(3):e0120790. https://doi.org/10.1371/journal.pone.0120790.
Zhou Y, Wang Y, Zhou W, et al. YAP promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1-ROS-mTOR pathway. Cancer Cell Int. 2019;19:179. https://doi.org/10.1186/s12935-019-0898-7.
Jin L, Chen Y, Cheng D, et al. YAP inhibits autophagy and promotes progression of colorectal cancer via upregulating Bcl-2 expression. Cell Death Dis. 2021;12(5):457. https://doi.org/10.1038/s41419-021-03722-8.
Zhao M, Zhang Y, Jiang Y, et al. YAP promotes autophagy and progression of gliomas via upregulating HMGB1. J Exp Clin Cancer Res. 2021;40(1):99. https://doi.org/10.1186/s13046-021-01897-8.
Wu J, Minikes A, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–6. https://doi.org/10.1038/s41586-019-1426-6.
Newton K, Wickliffe K, Maltzman A, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575(7784):679–82. https://doi.org/10.1038/s41586-019-1752-8.
Rogers C, Erkes D, Nardone A, Aplin A, Fernandes-Alnemri T, Alnemri E. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689. https://doi.org/10.1038/s41467-019-09397-2.
Mao M, Chen Y, Jia Y, et al. PLCA8 suppresses breast cancer apoptosis by activating the PI3k/AKT/NF-κB pathway. J Cell Mol Med. 2019;23(10):6930–41. https://doi.org/10.1111/jcmm.14578.
Guha P, Gardell J, Darpolor J, et al. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene. 2019;38(4):533–48. https://doi.org/10.1038/s41388-018-0449-z.
Murakami S, Shahbazian D, Surana R, et al. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene. 2016;36(9):1232–44. https://doi.org/10.1038/onc.2016.288.
Ehsanian R, Brown M, Lu H, et al. YAP dysregulation by phosphorylation or ΔNp63-mediated gene repression promotes proliferation, survival and migration in head and neck cancer subsets. Oncogene. 2010;29(46):6160–71. https://doi.org/10.1038/onc.2010.339.
Strano S, Munarriz E, Rossi M, et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem. 2001;276(18):15164–73. https://doi.org/10.1074/jbc.M010484200.
Howell M, Borchers C, Milgram S. Heterogeneous nuclear ribonuclear protein U associates with YAP and regulates its co-activation of Bax transcription. J Biol Chem. 2004;279(25):26300–6. https://doi.org/10.1074/jbc.M401070200.
Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. https://doi.org/10.1038/cdd.2008.150.
Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9(1):1944. https://doi.org/10.1038/s41467-018-04070-6.
Ulbricht A, Eppler FJ, Tapia VE, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. 2013;23(5):430–5. https://doi.org/10.1016/j.cub.2013.01.064.
Zhang Z, Guo M, Li Y, et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2019:1–24. https://doi.org/10.1080/15548627.2019.1687985.
Jin F, Wang Y, Li M, et al. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017;8(1):e2540. https://doi.org/10.1038/cddis.2016.461.
Feng L, Ma Y, Sun J, et al. YY1-MIR372-SQSTM1 regulatory axis in autophagy. Autophagy. 2014;10(8):1442–53. https://doi.org/10.4161/auto.29486.
Yu X, Luo A, Liu Y, et al. MiR-214 increases the sensitivity of breast cancer cells to tamoxifen and fulvestrant through inhibition of autophagy. Mol Cancer. 2015;14:208. https://doi.org/10.1186/s12943-015-0480-4.
Wang P, Gong Y, Guo T, et al. Activation of Aurora A kinase increases YAP stability via blockage of autophagy. Cell Death Dis. 2019;10(6):432. https://doi.org/10.1038/s41419-019-1664-4.
Yuan P, Hu Q, He X, et al. Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis. 2020;11(2):141. https://doi.org/10.1038/s41419-020-2343-1.
Liang N, Zhang C, Dill P, et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med. 2014;211(11):2249–63. https://doi.org/10.1084/jem.20140341.
Jiang Y, Ji F, Liu Y, et al. Cisplatin-induced autophagy protects breast cancer cells from apoptosis by regulating yes-associated protein. Oncol Rep. 2017;38(6):3668–76. https://doi.org/10.3892/or.2017.6035.
Cho J, Lee Y, Bae S, Chou J. Activation of tumor-promoting pathways implicated in hepatocellular adenoma/carcinoma, a long-term complication of glycogen storage disease type Ia. Biochem Biophys Res Commun. 2020;522(1):1–7. https://doi.org/10.1016/j.bbrc.2019.11.061.
Lee Y, Noon L, Akat K, et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun. 2018;9(1):4962. https://doi.org/10.1038/s41467-018-07338-z.
Chen W, Bai Y, Patel C, Geng F. Autophagy promotes triple negative breast cancer metastasis via YAP nuclear localization. Biochem Biophys Res Commun. 2019;520(2):263–8. https://doi.org/10.1016/j.bbrc.2019.09.133.
Totaro A, Zhuang Q, Panciera T, et al. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc Natl Acad Sci U S A. 2019;116(36):17848–57. https://doi.org/10.1073/pnas.1908228116.
Pavel M, Park S, Frake R, et al. α-Catenin levels determine direction of YAP/TAZ response to autophagy perturbation. Nat Commun. 2021;12(1):1703. https://doi.org/10.1038/s41467-021-21882-1.
Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149(5):1060–72. https://doi.org/10.1016/j.cell.2012.03.042.
Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. https://doi.org/10.1038/cddis.2016.208.
Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165–76. https://doi.org/10.1016/j.tcb.2015.10.014.
Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8. https://doi.org/10.1038/nchembio.2239.
Song X, Zhu S, Chen P, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr Biol. 2018;28(15):2388–99 e5. https://doi.org/10.1016/j.cub.2018.05.094.
Brown C, Amante J, Chhoy P, et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell. 2019;51(5):575–86.e4. https://doi.org/10.1016/j.devcel.2019.10.007.
Xie Y, Zhu S, Song X, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20(7):1692–704. https://doi.org/10.1016/j.celrep.2017.07.055.
Yang J, Antin P, Berx G, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341–52. https://doi.org/10.1038/s41580-020-0237-9.
Shao D, Xue W, Krall E, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158(1):171–84. https://doi.org/10.1016/j.cell.2014.06.004.
Liu M, Zhang Y, Yang J, et al. Zinc-Dependent Regulation of ZEB1 and YAP1 Coactivation Promotes Epithelial-Mesenchymal Transition Plasticity and Metastasis in Pancreatic Cancer. Gastroenterology. 2021;160(5):1771–83.e1. https://doi.org/10.1053/j.gastro.2020.12.077.
Viswanathan V, Ryan M, Dhruv H, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–7. https://doi.org/10.1038/nature23007.
Müller S, Sindikubwabo F, Cañeque T, et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat Chem. 2020;12(10):929–38. https://doi.org/10.1038/s41557-020-0513-5.
Yang W, Ding C, Sun T, et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019;28(10):2501–08.e4. https://doi.org/10.1016/j.celrep.2019.07.107.
Sun T, Chi J. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes Dis. 2021;8(3):241–9. https://doi.org/10.1016/j.gendis.2020.05.004.
Yang W, Huang Z, Wu J, Ding C, Murphy S, Chi J. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res. 2020;18(1):79–90. https://doi.org/10.1158/1541-7786.Mcr-19-0691.
Jiang K, Liu P, Xu H, et al. SASH1 suppresses triple-negative breast cancer cell invasion through YAP-ARHGAP42-actin axis. Oncogene. 2020;39(27):5015–30. https://doi.org/10.1038/s41388-020-1356-7.
Sulaiman A, McGarry S, Li L, et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol Oncol. 2018;12(4):423–40. https://doi.org/10.1002/1878-0261.12167.
Zhang Z, Qiu N, Yin J, et al. SRGN crosstalks with YAP to maintain chemoresistance and stemness in breast cancer cells by modulating HDAC2 expression. Theranostics. 2020;10(10):4290–307. https://doi.org/10.7150/thno.41008.
Qin G, Wang X, Ye S, et al. NPM1 upregulates the transcription of PD-L1 and suppresses T cell activity in triple-negative breast cancer. Nat Commun. 2020;11(1):1669. https://doi.org/10.1038/s41467-020-15364-z.
Maeda T, Hiraki M, Jin C, et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018;78(1):205–15. https://doi.org/10.1158/0008-5472.Can-17-1636.
Nguyen C, Yi C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer. 2019;5(5):283–96. https://doi.org/10.1016/j.trecan.2019.02.010.
Kim M, Kim C, Kim S, et al. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res. 2018;6(3):255–66. https://doi.org/10.1158/2326-6066.Cir-17-0320.
Hou J, Zhao R, Xia W, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22(10):1264–75. https://doi.org/10.1038/s41556-020-0575-z.
Perez-Lopez A, Rosales-Reyes R, Alpuche-Aranda CM, Ortiz-Navarrete V. Salmonella Downregulates Nod-like Receptor Family CARD Domain Containing Protein 4 Expression To Promote Its Survival in B Cells by Preventing Inflammasome Activation and Cell Death. J Immunol. 2013;208(4):1200415. https://doi.org/10.4049/jimmunol.1200415.
Cui J, Zhou Z, Yang H, et al. MST1 Suppresses Pancreatic Cancer Progression via ROS-Induced Pyroptosis. Mol Cancer Res. 2019;17(6):1316–25. https://doi.org/10.1158/1541-7786.Mcr-18-0910.
Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab. 2021;32(7):444–62. https://doi.org/10.1016/j.tem.2021.04.010.
Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. https://doi.org/10.1126/sciadv.aaw2238.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–32. https://doi.org/10.1038/cr.2016.95.
Kremer D, Nelson B, Lin L, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. 2021;12(1):4860. https://doi.org/10.1038/s41467-021-24859-2.
Yang C, Stampouloglou E, Kingston N, Zhang L, Monti S, Varelas X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018;19(6):e43577. https://doi.org/10.15252/embr.201643577.
Chen Z, Nie S, Qu M, et al. The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 2018;9(7):771. https://doi.org/10.1038/s41419-018-0795-3.
Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8(10):1124–32. https://doi.org/10.1038/ncb1482.
Liu Z, Zeng W, Wang S, et al. A potential role for the Hippo pathway protein, YAP, in controlling proliferation, cell cycle progression, and autophagy in BCPAP and KI thyroid papillary carcinoma cells. Am J Transl Res. 2017;9(7):3212–23.
Xiao L, Shi X, Zhang Y, et al. YAP induces cisplatin resistance through activation of autophagy in human ovarian carcinoma cells. Onco Targets Ther. 2016;9:1105–14. https://doi.org/10.2147/ott.S102837.
Li Y, Cao Y, Xiao J, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27(9):2635–50. https://doi.org/10.1038/s41418-020-0528-x.
Ciamporcero E, Daga M, Pizzimenti S, et al. Crosstalk between Nrf2 and YAP contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic Biol Med. 2018;115:447–57. https://doi.org/10.1016/j.freeradbiomed.2017.12.005.
Silva M. Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J Leukoc Biol. 2010;88(5):885–96. https://doi.org/10.1189/jlb.0410205.
Liu W, Wang F, Li C, et al. Silibinin relieves UVB-induced apoptosis of human skin cells by inhibiting the YAP-p73 pathway. Acta Pharmacol Sin. 2021. https://doi.org/10.1038/s41401-021-00826-x.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
CYF and MMS designed the review. CYF and MMS researched the literature and drafted the manuscript. LY edited the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, Y., Mao, M. & Lu, Y. The biology of YAP in programmed cell death. Biomark Res 10, 34 (2022). https://doi.org/10.1186/s40364-022-00365-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-022-00365-5