
Abstract
Background
Esophageal cancer (ECa) is the 7th most incident cancer and the 6th leading cause of cancer-related death. Most patients are diagnosed with locally advanced or metastatic disease, enduring poor survival. Biomarkers enabling early cancer detection may improve patient management, treatment effectiveness, and survival, are urgently needed. In this context, epigenetic-based biomarkers such as DNA methylation are potential candidates.
Methods
Herein, we sought to identify and validate DNA methylation-based biomarkers for early detection and prediction of response to therapy in ECa patients. Promoter methylation levels were assessed in a series of treatment-naïve ECa, post-neoadjuvant treatment ECa, and normal esophagus tissues, using quantitative methylation-specific PCR for COL14A1, GPX3, and ZNF569.
Results
ZNF569 methylation (ZNF569me) levels significantly differed between ECa and normal samples (p < 0.001). Moreover, COL14A1 methylation (COL14A1me) and GPX3 methylation (GPX3me) levels discriminated adenocarcinomas and squamous cell carcinomas, respectively, from normal samples (p = 0.002 and p = 0.009, respectively). COL14A1me & ZNF569me accurately identified adenocarcinomas (82.29%) whereas GPX3me & ZNF569me identified squamous cell carcinomas with 81.73% accuracy. Furthermore, ZNF569me and GPX3me levels significantly differed between normal and pre-treated ECa.
Conclusion
The biomarker potential of a specific panel of methylated genes for ECa was confirmed. These might prove useful for early detection and might allow for the identification of minimal residual disease after adjuvant therapy.
Similar content being viewed by others


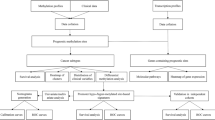
Background
Esophageal cancer (ECa) is the 7th most common malignancy and the 6th cause of cancer-related mortality worldwide [1]. ECa comprises two main histological subtypes: adenocarcinoma and squamous cell carcinoma, the most prevalent type [2]. Although therapeutic improvements have increased ECa survival rates [3], curative-intent treatment options remain limited to surgery, chemotherapy (ChT), and radiotherapy (RT), either alone or in trimodal therapy [4]. Furthermore, most tumors are diagnosed at an advanced stage, entailing very low 5-year survival rates, ranging from 5 to 20% [5, 6]. Intensive research was, thus, carried out to find alternative treatment strategies to prolong life expectancy and quality of life (QoL) for ECa patients. Currently, neoadjuvant treatment with ChT and/or RT is the standard of care for patients with locally advanced disease [2]. Nevertheless, surgery is often associated with decreased QoL due to complications and comorbidities [7, 8].
Epigenetic alterations, including their key players, have emerged as promising biomarkers for ECa. Among those, gene methylation is the most extensively studied epigenetic modification. Although esophageal adenocarcinomas (EA) and esophageal squamous cell carcinomas (ESCC) display different morphological and molecular features, several studies have shown that methylation is an early event in both histotypes, already present in premalignant lesions [9,10,11,12]. Thus, promoter methylation of specific genes might be used to discriminate normal from cancerous esophageal cells, enabling the detection of disease at early stages, increasing the likelihood of curative treatment. Except for CDKN2A and APC, which are commonly aberrant methylated genes in both EA and ESCC, a specific methylome has been reported for each histotype [13]. The importance of these alterations is further highlighted by the numerous reports correlating aberrant DNA methylation with ECa patient prognosis [14, 15].
Methods
Based on the literature evidence, we aimed to identify and validate methylome alterations that might constitute biomarkers for early detection, as well as identification of minimal residual disease after neoadjuvant treatment, eventually precluding the need for esophagectomy. Hence, the studies selected from the literature clearly reported sensitivity and specificity for the detection of ECa or Barrett’s Esophagus using DNA methylation-based biomarkers (Additional Table 1). Studies with a small cohort of patients [n ≤ 40 for each group (cases and controls)] were also excluded. Furthermore, only genes with a specificity higher than 98% were selected for testing. Of the remaining 5 genes, no specific primers were obtained to test ZNF345 and EPB41L3. Hence, promoter methylation levels of three genes selected [16, 17] - GPX3 (glutathione peroxidase 3), COL14A1 (collagen type XIV alpha 1 chain), and ZNF569 (zinc finger protein 569) - were tested in a series of ECa and normal esophageal tissues.
Patients and samples collection
A total of 124 formalin-fixed paraffin-embedded (FFPE) tissues samples from patients diagnosed with ECa between 2007 and 2017 at the Portuguese Oncology Institute of Porto (IPO-Porto) were included in this study (Table 1). Among the tumor samples, 88 were collected before any treatment previous surgery, and 36 were collected after neoadjuvant treatment (ChT and/or RT). Additionally, 56 FFPE tissues samples of normal esophagus from patients diagnosed with gastric carcinoma and without evidence of esophageal cancer were used as control. All samples were archived at the Department of Pathology of IPO-Porto. All the cases were revised by an experienced pathologist and classified according to the World Health Organization (WHO) classification of Tumors of the Digestive System (4th edition) and staged according to the 7th edition American Joint Committee on Cancer (AJCC) system [18, 19]. Relevant clinical data were collected from medical charts. For DNA extraction, a 4 μm section was cut from a representative tissue block and stained with hematoxylin-eosin. Tumor areas were delimited, enabling macrodissection in eight consecutive 8 μm sections. This study was approved by the institutional ethics committee of IPO Porto (CES 202/017).
Promoter methylation evaluation
DNA extraction from FFPE sections was performed using FFPE RNA/DNA Purification Plus Kit (Norgen Biotek, Thorold, Canada) following the manufacturer’s instructions. DNA concentrations and purity ratios were determined using the NanoDrop Lite spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and modified with sodium bisulfite, using the EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA, USA) according to manufacturer’s instructions. For quantitative methylation-specific PCR (QMSP), modified DNA was used as template. Primers to specifically amplify methylated bisulfite converted complementary sequences were used and are listed in Additional Table 2. QMSP reactions were carried out in LightCyler 480 II (Roche, Germany) using 2 μL of modified DNA and 5 μL Xpert Fast SYBR (2X) (GRiSP, Porto, Portugal). All samples were run in triplicate and melting curves were obtained for each case by gene. β-actin (ACTβ) was used to normalize for DNA input in each sample [20]. To ascertain PCR efficiency, and samples’ quantification, modified CpGenome™ Universal Methylated DNA (Merck Millipore, France) was used in each plate to generate a standard curve. The relative methylation for each gene was calculated by the ratio of mean quantity for the target gene and the mean quantity of ACTβ, multiplied by 1000 for easier tabulation.
Statistical analysis
All the comparisons were performed using non-parametric tests. Specifically, the Kruskal-Wallis test was used for comparisons among three or more groups, whereas the Mann-Whitney U test was used in comparisons between two groups.
To assess biomarker performance, Receiver Operator Characteristic (ROC) curves were constructed for each gene and the Area Under the Curve (AUC) was calculated. The highest value obtained by the ROC curve analysis [sensitivity + (1-specificity)] was established as cut-off to categorize samples as methylated or unmethylated, according to Youden’s J index method [21, 22]. Furthermore, specificity, sensitivity, accuracy, positive likelihood ratio (+LH), and negative likelihood ratio (−LH) were determined. For combination of biomarkers, cases were considered positive if at least one of the individual biomarkers was positive.
Spearman non-parametric correlation was used to assed the correlation methylation levels and age. Disease-specific and disease-free survival curves (Kaplan-Meier with log-rank test) were built for standard clinicopathological variables and categorized methylation status. Disease-specific survival curves and disease-free survival curves (Kaplan–Meier with log-rank test) were computed for standard clinicopathological variables and categorized methylation status.
Two-tailed P-values were derived from statistical tests, using a computer-assisted program (SPSS Version 25.0, Chicago, IL), and results were considered statistically significant at p < 0.05, with Bonferroni’s correction for multiple tests, when applicable (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; ns – non significant). Graphics were assembled using GraphPad 6 Prism (GraphPad Software, USA).
Results
Clinical and pathological data
The most relevant clinical and pathological data are depicted in Table 1. In addition to normal esophageal tissues, ECa cases were segregated into treatment-naïve (samples collected before any treatment) and post-neoadjuvant treatment groups. No significant differences were disclosed concerning age among the three groups of samples (p = 0.06).
Gene promoter methylation levels in naïve ECa tumors vs. normal esophagus samples
To assess performance for ECa detection, COL14A1 methylation (COL14A1me), GPX3 methylation (GPX3me), and ZNF569 methylation (ZNF569me) levels in naïve tumors (n = 88) were compared to normal esophagus samples (n = 56). ZNF569me levels significantly differed between cancerous and normal samples (p < 0.001, Fig. 1c), whereas no significant differences were found for COL14A1me and GPX3me levels (p = 0.382 and p = 0.094, respectively, Fig. 1a, b).

Boxplots with all points of COL14A1 (a), GPX3 (b) and ZNF569 (c) relative methylation levels in the normal esophagus (n = 56) and esophageal tumor tissues (n = 88). *** p < 0.001; n.s- non-significant
Using ROC curve analysis (Fig. 2), a cut-off of 55.17 was set for ZNF569me to assess biomarker performance. Thus, over 90% specificity and 69.3% sensitivity (Table 2) was disclosed, corresponding to an AUC of 0.8467 (Fig. 2).

Receiver Operating Characteristic Curve of the ZNF569 in Esophageal naïve tumors tissues
Association between promoter methylation levels and standard clinicopathologic features
COL14A1me, GPX3me, and ZNF569me levels were tested for associations with standard clinicopathological features in naïve ECa patients. All genes disclosed significant differences in promoter methylation levels according to histological subtype (Fig. 3). ESCC displayed higher COL14A1me and GPX3me levels than EA (p = 0.001 and p = 0.024, respectively), whereas EA displayed higher ZNF569me levels (p = 0.020). Additionally, COL14A1me levels were significantly higher in pT1 tumors compared to pT3 (p = 0.006) (Additional Fig. 1). However, no significant differences on methylation levels were found among different N stages (Additional Fig. 2).

Boxplots with all points of COL14A1 (a), GPX3 (b) and ZNF569 (c) relative methylation levels in Esophageal Squamous Cell Carcinoma (ESCC), n = 48 and Esophageal Adenocarcinoma (EA), n = 40 tissues. * p < 0.05; ** p < 0.01
Biomarker performance according to histological subtype
Because methylation levels differed between histological subtypes, samples were stratified according to this parameter for assessing histotype-specific biomarker performance. Concerning EA, COL14A1me and ZNF569me levels significantly differed from normal samples (p = 0.002 and p < 0.001, respectively). COL14A1me and ZNF569me levels identified EA with an AUC of 0.68 and 0.91, respectively (Fig. 4a), whereas the combination of both genes disclosed a sensitivity above 97% and 82.29 accuracy (Table 3). Furthermore, GPX3me and ZNF569me levels differed significantly between ESCC and normal samples (p = 0.009 and p < 0.001, respectively), individually discriminating this tumor type from controls with AUC of 0.65 and 0.79 (Fig. 4b), respectively. Accuracy of detection improved to 81.73 when the two genes were combined in a single panel (Table 3).

Receiver Operating Characteristic Curve of the two-gene panel (a) COL14A1 and ZNF569 in Esophageal Adenocarcinoma; (b) GPX3 and ZNF569 in Esophageal Squamous Cell Carcinoma
Assessment of promoter methylation in post-treatment samples
GPX3me and ZNF569me levels were significantly higher in residual EA samples after neoadjuvant treatment compared to normal samples (p < 0.001, for both). Nonetheless, COL14A1me levels in tumors after neoadjuvant treatment did not differ significantly from normal samples (p = 0.493) (Fig. 5). Concerning ESCC, only GPX3me levels remained significantly different between post-treatment tumor and normal samples (p = 0.001).

Boxplots with all points of COL14A1 (a), GPX3 (b) and ZNF569 (c) relative methylation levels in the normal esophagus, n = 56, Esophageal Adenocarcinoma (EA), n = 16 and Esophageal Squamous Cell Carcinoma (ESCC), n = 20 Post-Chemotherapy and/or Radiotherapy patient’s samples. * p < 0.05; *** p < 0.001
Survival analysis
Survival analysis was carried out in the treatment-naïve ECa cohort. For analysis, a maximum of 5 years follow-up was considered. During this period, 39 patients died from the disease (44.3%), three patients died with the disease (3.4%) and six patients died without evidence of cancer (6.8%). Among the remainder patients, 39 were alive without evidence of disease (44.3%) and one patient was alive with disease (1.2%).
No associations were depicted between COL14A1me, GPX3me, and ZNF569me levels and disease-specific or disease-free survival, whereas the pT stage, pN stage, and stage associated with both disease-specific and disease-free survival (p = 0.010, p = 0.002, p = 0.002 for disease-specific survival and p = 0.012, p = 0.001, p = 0.025 for disease-free survival).
Discussion
ECa remains a leading cause of cancer-related mortality globally [1] and in Portugal [23]. Most patients are diagnosed with locally advanced or metastatic disease, entailing poor 5-years survival rate (about 25 and 5%, respectively) [24]. Thus, new strategies for early detection of this malignancy are urgently needed. In this context, epigenetic alterations such as DNA methylation have emerged as promising biomarkers in several cancers, including ECa [15, 25].
Herein, we tested three gene promoters’ methylation as ECa DNA methylation-based biomarkers, following a literature review. We selected the genes ZNF569, GPX3, and COL14A1, all previously reported to harbor promoter methylation and suggested to have an oncossuppressive function. ZNF569 protein has been reported as a potential transcriptional repressor implicated in MAPK signaling pathway [26]. COL14A1 encodes for the alpha chain of type XIV collagen which interacts with decorin associated with cell growth and survival [27]. GPX3 is a glutathione peroxidase found to catalyze glutathione’s reduction of organic hydroperoxides and hydrogen peroxide and thereby protecting cells against oxidative damage [28, 29].
COL14A1 aberrant methylation has been reported in ESCC [16], as well as in renal cell carcinoma, sarcomas, and endometrial carcinoma [27, 30, 31], whereas hypomethylation has been shown in coronary artery disease [32]. GPX3 promoter methylation has been shown in ESCC and esophageal glandular lesions, including Barrett’s esophagus and EA [29, 33, 34]. In the same vein, ZNF569 promoter hypermethylation has been associated with glandular lesions, like Barrett’s esophagus [17] in comparison with the normal esophagus.
Interestingly, we showed that ZNF569me levels could discriminate between ECa and normal esophagus with high specificity, regardless of the histotype, extending those previous reports. Furthermore, ZNF569me was shown to play a tumor-suppressive role in head and neck squamous cell carcinoma [35] and a DNA-methylation based panel, which included ZNF569me, discriminated gastric adenocarcinoma from normal mucosa [36].
ESCC and EA displayed different cancer-specific methylation patterns. Accordingly, differential methylation patterns have been previously reported between ESCC and EA [37, 38]. Indeed, in treatment-naïve tumors, the three selected genes disclosed different methylation levels among ESCC and EA, variably comparing to normal esophageal tissue samples. In particular, available data support the value of identification specific ESCC methylation panels to enable early detection [16, 39, 40]. We found ZNF569 hypermethylated in both histological subtypes and, thus, this gene constitutes a promising biomarker for ECa detection, regardless of histological subtype. Moreover, we found COL14A1 promoter methylation levels slightly higher in ESCC when compared with normal esophagus samples, although not statistically significant. This can be partially explained by variations in the population (Asian vs. Caucasian), as previously attested for some genes [41], and the different nature of samples tested (plasma vs. FFPE). Notwithstanding, COL14A1 promoter methylation levels were significantly lower in EA compared to normal. To our knowledge, this is the first reported association between COL14A1 methylation levels and EA. Conversely, GPX3 promoter methylation levels did not differ between EA from normal tissues, although a few cases disclosed higher methylation levels (data not shown). Notwithstanding, different sample processing (fresh frozen tissues vs. FFPE) [29, 34], different methodologies to assess GPX3 methylation levels among studies (qMSP vs. MSP vs. methylation ligation-dependent macroarray vs. pyrosequencing), and the smaller size of some cohorts [29, 33] may explain some disparate results.
Overall, we propose two different methylation-based panels, both with high accuracy to early detect ECa according to histological subtype. The ESCC-panel displayed higher specificity (87.5%), whereas the EA-panel disclosed higher sensitivity (97.5%). In fact, the performance of both methylation-based panels compares well with that of other studies [38, 42, 43]. For EA detection, Moinova et al. reported a two-gene methylation panel, comprising CCNA1 and VIM, with higher specificity (91.7%), similar to TAC1 hypermethylation reported by Jin et al. [44, 45]. Additionally, for ESCC Li et al. and Wang et al. reported panels with higher performance in Asian populations [16, 46].
Currently, most ECa patients are treated with neoadjuvant therapy followed by surgery, if diagnosed with locally advanced disease [2]. The randomized CROSS trial showed that surgery has a major impact on ECa patients’ QoL. Features such as fatigue and physical performance are decreased even in long-term survivors. Those effects are similar in patients undergoing neoadjuvant treatment or surgery only, emphasizing the impact of surgery in QoL [7, 8]. Hence, biomarkers enabling the identification of patients complete response to neoadjuvant treatment (who might be spared surgery) and to early detect disease recurrence are needed [47] to improve QoL without risking the likelihood of cure. Thus, we evaluated methylation levels of candidate genes in 36 samples from non-complete responders after neoadjuvant treatment. In our series, ZNF569me levels only significantly differed in EA comparatively to the normal esophagus, whereas GPX3 promoter methylation levels were significantly higher both in ESCC and EA than in the normal esophagus. Because GPX3me has been associated with ChT resistance [28], GPX3me levels observed in EA after neoadjuvant treatment might be explained by selective pressure caused upon neoplastic cells, entailing adaptative alterations induced by treatment [48]. Several studies have associated DNA methylation with ChT or RT resistance [49,50,51,52]. However, most used samples before any treatment or in vitro studies with immortalized cell lines [51]. Thus, a direct comparison between our results and previously reported data should be made with caution. Nonetheless, the lack of information on methylation status before treatment, the small size of the pre-treated patient cohort along with the retrospective nature of our series, and the limited access to normal esophagus samples are major limitations of this study. Importantly, our series comprised ESCC and EA samples in similar proportions, contrarily to most of the previous studies which evaluated methylation status. Moreover, this study reported the potential of DNA methylation-based biomarkers for patients’ monitoring after neoadjuvant treatment.
Conclusions
In conclusion, we identified two gene panels that might detect ESCC and EA with good accuracy, which might prove useful for early disease detection among high-risk populations, as well as to detect residual disease after neoadjuvant treatment. As future perspectives, we intend to validate these panels in liquid biopsies, using plasma samples as a minimally invasive approach, not only for ECa early detection and diagnosis but also to identify patients with residual disease after neoadjuvant treatment, which are the most likely to benefit from surgery.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- -LH:
-
Negative likelihood ratio
- +LH:
-
Positive likelihood ratio
- ACTβ :
-
β-actin
- AJCC:
-
American Joint Committee on Cancer
- AUC:
-
Area Under the Curve
- ChT:
-
Chemotherapy
- COL14A1 :
-
Collagen Type XIV alpha 1 chain
- COL14A1me :
-
COL14A1 methylation
- EA:
-
Esophageal Adenocarcinomas
- ECa:
-
Esophageal cancer
- ESCC:
-
Esophageal Squamous Cell Carcinomas
- FFPE:
-
Formalin-fixed paraffin-embedded
- GPX3 :
-
Glutathione Peroxidase 3
- GPX3me :
-
GPX3 methylation
- IPO-Porto:
-
Portuguese Oncology Institute of Porto
- QMSP:
-
Quantitative methylation-specific PCR
- QoL:
-
Quality of Life
- ROC:
-
Receiver Operator Characteristic
- RT:
-
Radiotherapy
- WHO:
-
World Health Organization
- ZNF569 :
-
Zinc finger protein 569
- ZNF569me :
-
ZMF569 methylation
References
Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer J Int Du Cancer. 2019;144(8):1941–53.
Lordick F, Mariette C, Haustermans K, Obermannova R, Arnold D, Committee EG. Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v50–v7.
Abbas G, Krasna M. Overview of esophageal cancer. Ann Cardiothorac Surg. 2017;6(2):131–6.
Shah RD, Cassano AD, Neifeld JP. Neoadjuvant therapy for esophageal cancer. World journal of gastrointestinal oncology. 2014;6(10):403–6.
Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–89.
Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371(26):2499–509.
Noordman BJ, Verdam MGE, Lagarde SM, Hulshof M, van Hagen P, van Berge Henegouwen MI, et al. Effect of Neoadjuvant Chemoradiotherapy on health-related quality of life in esophageal or Junctional Cancer: results from the randomized CROSS trial. J Clin Oncol. 2018;36(3):268–75.
Noordman BJ, Verdam MGE, Lagarde SM, Shapiro J, Hulshof M, van Berge Henegouwen MI, et al. Impact of neoadjuvant chemoradiotherapy on health-related quality of life in long-term survivors of esophageal or junctional cancer: results from the randomized CROSS trial. Ann Oncol. 2018;29(2):445–51.
Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res. 1997;57(13):2619–22.
Klump B, Hsieh CJ, Holzmann K, Gregor M, Porschen R. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett's esophagus. Gastroenterology. 1998;115(6):1381–6.
Eads CA, Lord RV, Wickramasinghe K, Long TI, Kurumboor SK, Bernstein L, et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001;61(8):3410–8.
Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res. 2006;12(15):4515–22.
Kaz AM, Grady WM. Epigenetic biomarkers in esophageal cancer. Cancer Lett. 2014;342(2):193–9.
Kaz AM, Grady WM, Stachler MD, Bass AJ. Genetic and epigenetic alterations in Barrett's esophagus and esophageal adenocarcinoma. Gastroenterol Clin N Am. 2015;44(2):473–89.
Ma K, Cao B, Guo M. The detective, prognostic, and predictive value of DNA methylation in human esophageal squamous cell carcinoma. Clin Epigenetics. 2016;8:43.
Li X, Zhou F, Jiang C, Wang Y, Lu Y, Yang F, et al. Identification of a DNA methylome profile of esophageal squamous cell carcinoma and potential plasma epigenetic biomarkers for early diagnosis. PLoS One. 2014;9(7):e103162.
Chettouh H, Mowforth O, Galeano-Dalmau N, Bezawada N, Ross-Innes C, MacRae S, et al. Methylation panel is a diagnostic biomarker for Barrett's oesophagus in endoscopic biopsies and non-endoscopic cytology specimens. Gut. 2018;67(11):1942–9.
Bosnan F, Carneiro F, Hruban R, Theise N. WHO classification of tumours of the digestive system. Lyon: IARC; 2010.
Edge SB, Compton CC. The American joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471–4.
Martins AT, Monteiro P, Ramalho-Carvalho J, Costa VL, Dinis-Ribeiro M, Leal C, et al. High RASSF1A promoter methylation levels are predictive of poor prognosis in fine-needle aspirate washings of breast cancer lesions. Breast Cancer Res Treat. 2011;129(1):1–9.
Schisterman EF, Perkins NJ, Liu A, Bondell H. Optimal cut-point and its corresponding Youden Index to discriminate individuals using pooled blood samples. Epidemiology (Cambridge, Mass). 2005;16(1):73–81.
Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3(1):32–5.
Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, et al. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer; 2018. p. 2018.
Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M. SEER Cancer Statistics Review, 1975–2016. Bethesda, MD: National Cancer Institute; 2018. p. 2019.
Lin DC, Wang MR, Koeffler HP. Genomic and Epigenomic aberrations in esophageal squamous cell carcinoma and implications for patients. Gastroenterology. 2018;154(2):374–89.
Huang X, Yuan W, Huang W, Bai Y, Deng Y, Zhu C, et al. ZNF569, a novel KRAB-containing zinc finger protein, suppresses MAPK signaling pathway. Biochem Biophys Res Commun. 2006;346(3):621–8.
Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, et al. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 2010;29(14):2104–17.
Chen B, Rao X, House MG, Nephew KP, Cullen KJ, Guo Z. GPx3 promoter hypermethylation is a frequent event in human cancer and is associated with tumorigenesis and chemotherapy response. Cancer Lett. 2011;309(1):37–45.
Lee OJ, Schneider-Stock R, McChesney PA, Kuester D, Roessner A, Vieth M, et al. Hypermethylation and loss of expression of glutathione peroxidase-3 in Barrett's tumorigenesis. Neoplasia. 2005;7(9):854–61.
Chen YC, Tsao CM, Kuo CC, Yu MH, Lin YW, Yang CY, et al. Quantitative DNA methylation analysis of selected genes in endometrial carcinogenesis. Taiwan J Obstet Gynecol. 2015;54(5):572–9.
Renner M, Wolf T, Meyer H, Hartmann W, Penzel R, Ulrich A, et al. Integrative DNA methylation and gene expression analysis in high-grade soft tissue sarcomas. Genome Biol. 2013;14(12):r137.
Guay SP, Brisson D, Mathieu P, Bosse Y, Gaudet D, Bouchard L. A study in familial hypercholesterolemia suggests reduced methylomic plasticity in men with coronary artery disease. Epigenomics. 2015;7(1):17–34.
Guilleret I, Losi L, Chelbi ST, Fonda S, Bougel S, Saponaro S, et al. DNA methylation profiling of esophageal adenocarcinoma using methylation ligation-dependent macroarray (MLM). Biochem Biophys Res Commun. 2016;479(2):231–7.
Peng DF, Razvi M, Chen H, Washington K, Roessner A, Schneider-Stock R, et al. DNA hypermethylation regulates the expression of members of the mu-class glutathione S-transferases and glutathione peroxidases in Barrett's adenocarcinoma. Gut. 2009;58(1):5–15.
Liu X, Zhao X, Gou C. Identification of novel methylated DNA marker ZNF569 for head and neck squamous cell carcinoma. J Cancer. 2019;10(10):2250–60.
Anderson BW, Suh YS, Choi B, Lee HJ, Yab TC, Taylor WR, et al. Detection of gastric Cancer with novel methylated DNA markers: discovery, tissue validation, and pilot testing in plasma. Clin Cancer Res. 2018;24(22):5724–34.
Li D, Zhang L, Liu Y, Sun H, Onwuka JU, Zhao Z, et al. Specific DNA methylation markers in the diagnosis and prognosis of esophageal cancer. Aging (Albany NY). 2019;11(23):11640–58.
Gonzaga IM, Soares Lima SC, Nicolau MC, Nicolau-Neto P, da Costa NM, de Almeida ST, et al. TFF1 hypermethylation and decreased expression in esophageal squamous cell carcinoma and histologically normal tumor surrounding esophageal cells. Clin Epigenetics. 2017;9:130.
Tungekar A, Mandarthi S, Mandaviya PR, Gadekar VP, Tantry A, Kotian S, et al. ESCC ATLAS: a population wide compendium of biomarkers for esophageal squamous cell carcinoma. Sci Rep. 2018;8(1):12715.
He Y, Wang Y, Li P, Zhu S, Wang J, Zhang S. Identification of GPX3 epigenetically silenced by CpG methylation in human esophageal squamous cell carcinoma. Dig Dis Sci. 2011;56(3):681–8.
Chen S, Zhou K, Yang L, Ding G, Li H. Racial differences in esophageal squamous cell carcinoma: incidence and molecular features. Biomed Res Int. 2017;2017:1204082.
Pu W, Wang C, Chen S, Zhao D, Zhou Y, Ma Y, et al. Targeted bisulfite sequencing identified a panel of DNA methylation-based biomarkers for esophageal squamous cell carcinoma (ESCC). Clin Epigenetics. 2017;9:129.
Ross-Innes CS, Chettouh H, Achilleos A, Galeano-Dalmau N, Debiram-Beecham I, MacRae S, et al. Risk stratification of Barrett's oesophagus using a non-endoscopic sampling method coupled with a biomarker panel: a cohort study. Lancet Gastroenterol Hepatol. 2017;2(1):23–31.
Moinova HR, LaFramboise T, Lutterbaugh JD, Chandar AK, Dumot J, Faulx A, et al. Identifying DNA methylation biomarkers for non-endoscopic detection of Barrett's esophagus. Sci transl Med. 2018;10(424):eaao5848.
Jin Z, Olaru A, Yang J, Sato F, Cheng Y, Kan T, et al. Hypermethylation of tachykinin-1 is a potential biomarker in human esophageal cancer. Clin Cancer Res. 2007;13(21):6293–300.
Wang HQ, Yang CY, Wang SY, Wang T, Han JL, Wei K, et al. Cell-free plasma hypermethylated CASZ1, CDH13 and ING2 are promising biomarkers of esophageal cancer. J Biomed Res. 2018;32(5):424–33.
Noordman BJ, Spaander MCW, Valkema R, Wijnhoven BPL, van Berge Henegouwen MI, Shapiro J, et al. Detection of residual disease after neoadjuvant chemoradiotherapy for oesophageal cancer (preSANO): a prospective multicentre, diagnostic cohort study. Lancet Oncol. 2018;19(7):965–74.
Ramon YCS, Sese M, Capdevila C, Aasen T, De Mattos-Arruda L, Diaz-Cano SJ, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med (Berl). 2020;98(2):161–77.
Takahashi T, Yamahsita S, Matsuda Y, Kishino T, Nakajima T, Kushima R, et al. ZNF695 methylation predicts a response of esophageal squamous cell carcinoma to definitive chemoradiotherapy. J Cancer Res Clin Oncol. 2015;141(3):453–63.
Hamilton JP, Sato F, Greenwald BD, Suntharalingam M, Krasna MJ, Edelman MJ, et al. Promoter methylation and response to chemotherapy and radiation in esophageal cancer. Clin Gastroenterol Hepatol. 2006;4(6):701–8.
Kuo IY, Huang YL, Lin CY, Lin CH, Chang WL, Lai WW, et al. SOX17 overexpression sensitizes chemoradiation response in esophageal cancer by transcriptional down-regulation of DNA repair and damage response genes. J Biomed Sci. 2019;26(1):20.
Kurimoto K, Hayashi M, Guerrero-Preston R, Koike M, Kanda M, Hirabayashi S, et al. PAX5 gene as a novel methylation marker that predicts both clinical outcome and cisplatin sensitivity in esophageal squamous cell carcinoma. Epigenetics. 2017;12(10):865–74..
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from ESTIMA-NORTE-01-0145-740 FEDER-000027. SS was supported by a PhD fellowship IPO/ESTIMA-1 NORTE-01-0145- 740 FEDER-000027. VMG and NL received a post-doc fellowship from ESTIMA-NORTE-01-0145-740 FEDER-000027. CM-S was supported by a fellowship NORTE-01-0247-FEDER-033399.
Author information
Authors and Affiliations
Contributions
SS prepared samples for molecular analyses, including DNA extraction, bisulfite modification, performed qMSP assays, analyzed data and drafted the manuscript. CM-S and VMG assisted in samples preparation (DNA extraction, bisulfite modification, and qMSP). NL contributed in draft manuscript. DG collected clinical follow-up data and performed histopathological evaluation of FFPE stained by H&E. RG performed the FFPE sections of all cases. MF performed histopathological evaluation of FFPE stained by H&E. OS collected clinical follow-up data. RH and CJ designed and supervised the study and revised the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the institutional ethics committee of IPO Porto (CES 202/017).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Additional Table 1
– List of The Genes Considered for This Study. Additional Table 2 – Primers sequences and qMSP conditions for each gene studied in tissues samples. Additional Fig. 1 – Boxplots with all points of COL14A1 (a), GPX3 (b) and ZNF569 (c) relative methylation levels in the pT stage groups (pT1 n = 16, pT2 n = 14, pT3/pT4 n = 58). *** p < 0.001. Additional Fig. 2 – Boxplots with all points of COL14A1 (a), GPX3 (b) and ZNF569 (c) relative methylation levels in the pN stage groups (pN0 n = 39, pN1 n = 17 pN2 n = 21, pN3 n = 11).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Salta, S., Macedo-Silva, C., Miranda-Gonçalves, V. et al. A DNA methylation-based test for esophageal cancer detection. Biomark Res 8, 68 (2020). https://doi.org/10.1186/s40364-020-00248-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-020-00248-7