
Abstract
The development of neonatal chronic lung disease (nCLD), i.e., bronchopulmonary dysplasia (BPD) in preterm infants, significantly determines long-term outcome in this patient population. Risk factors include mechanical ventilation and oxygen toxicity impacting on the immature lung resulting in impaired alveolarization and vascularization. Disease development is characterized by inflammation, extracellular matrix remodeling, and apoptosis, closely intertwined with the dysregulation of growth factor signaling. This review focuses on the causes and consequences of altered signaling in central pathways like transforming growth factor (TGF), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF) driving these above indicated processes, i.e., inflammation, matrix remodeling, and vascular development. We emphasize the shared and distinct role of these pathways as well as their interconnection in disease initiation and progression, generating important knowledge for the development of future treatment strategies.
Similar content being viewed by others


Background
Neonatal chronic lung disease (nCLD), commonly known as bronchopulmonary dysplasia (BPD), is a frequent complication in very low birth weight preterm infants receiving mechanical ventilation with oxygen-rich gas (MV-O2) [1]. In this high-risk patient population, up to 30% of the infants born at less than 30 weeks of gestation (gestational age, GA) develop BPD [2]. It is associated with significant long-term pulmonary and neurologic sequelae persisting into adulthood [3, 4]. Clinically, BPD is defined by the need for supplemental oxygen or ventilator support at day 28 of life (mild BPD) or at 36 weeks gestational age (moderate and severe BPD) [5]. Both, shear stress as well as oxygen toxicity, are known to negatively impact development of the gas exchange area of lungs resulting in impaired structure and function of both vessels and alveoli, critically driven by an inflammatory response and significant extracellular matrix (ECM) remodeling [5]. Cause and consequence of these pathophysiologic processes is the dysregulation of central signaling pathways that play a critical role in both normal lung development as well as disease initiation and progression. In comparison to the understanding of a single pathway, knowledge about the interplay of these important regulators of disease pathophysiology is of significant importance for the development of future preventive and causative treatment strategies.
The review addresses important aspects of the crosstalk between key signaling pathways linking alveolar and vascular development with lung apoptosis and matrix remodeling [6]: part one of the review will focus on the platelet-derived growth factor (PDGF) as a critical driver of alveolar septation, whereas part two will address the role of the vascular endothelial growth factor (VEGF) as a key regulator in vascular development. Finally, a chapter reviewing the role of the transforming growth factor (TGF) β, known for its impact on apoptosis, matrix remodeling, and inflammatory processes in the developing lung, will connect important aspects discussed on behalf of the abovementioned signaling pathways in a disease-relevant context [7].
The role of PDGF signaling in (patho)-physiologic lung development
The saccular and alveolar phase of lung development is characterized by the process of secondary septation, critically driven by the migration of the PDGF receptor (PDGF-R) α expressing myofibroblast (MFB) towards the tip of the septal crest, and the deposition of elastin at this position providing the scaffold for the developing alveoli [8,9,10].
The important role of PDGF-Rα signaling in lung development is shown by studies performed on homozygous knockout mice for PDGF-A as well as PDGF-Rα. Very few mice pups from homozygous knockout of PDGF-A escape in utero death caused by distorted lung development, leading to an emphysematous-like phenotype in newborn mice [11]. Simultaneously, mice lacking the respective receptor only survive until early embryonic stages as a result of respiratory failure due to a simplified pulmonary structure with a decrease in secondary crests and a subsequent increase in alveolar size, thereby resembling the phenotype observed in neonatal rats treated with a PDGF-Rα antagonist [12, 13].
The failure in alveologenesis in PDGF-Rα knockout mice is assigned to the reduced migration of PDGF-Rα + alveolar smooth muscle progenitor cells, i.e., MFBs towards the secondary crest, confirming the important role of PDGF-Rα signaling in MFBs migration [14, 15]. Along these lines, Popova et al., demonstrate a reduced chemotaxis of pulmonary mesenchymal cells isolated from BPD patients with reduced mRNA levels for PDGF-Rα [16], that is in concert with studies performed on ventilated newborn mice and lamb connecting dysregulated PDGF-A and PDGF-Rα signaling to disease development [17,18,19,20]. In a three-dimensional study performed in lungs of hyperoxia-exposed mice, the dislocation of α-SMA positive MFBs corresponds with a disruption of the pulmonary elastin network [21]. As shown in patients with BPD and further evaluated by studies in newborn transgenic mice, the breakdown and pathologic remodeling of the elastic network characterizes alveolar branching arrest, increased airspace size, and simplification of the gas exchange area, all contributing to significant in long-term complications [10, 22,23,24,25,26].
In order to generate a deeper understanding of the interplay between early more generalized disease processes with cell-specific pathways and identify potential treatment targets, we will address the specific interaction of the pro-inflammatory and pro-apoptotic cytokine TGF-β and its interplay with the PDGF and VEGF pathway, linking the characteristic inflammatory response in BPD development to critical drivers of alveolarization and vascular development (see Chapter III TGF-β signalling enhancing pulmonary injury in neonates of this review).
Disturbed vascular growth factor signaling in the injured neonatal lung
Studies in premature rats undergoing postnatal hyperoxia exposure demonstrate a reduction in pulmonary VEGF-R2 expression, a receptor on endothelial cells mediating their survival and function [27]. Animal models of BPD using MV-O2 to trigger disease development lead to defective vasculature and alveolar structure accompanied by decreased VEGFA/VEGF-R2 signaling [28, 29]. According to the “vascular hypothesis of lung development,” endothelial cell signaling is a crucial pre-requisite for the formation of the alveolar structure [30]. In line with this, the use of a VEGF receptor inhibitor in animal models of BPD hinders both pulmonary angiogenesis and alveolar development, resulting in reduced lung weight [31,32,33]. In this process, VEGF holds an important role impacting on endothelial and epithelial cell signaling [34, 35]. Indicating the severely disturbed signaling process in BPD patients, studies in broncho-alveolar lavage fluid of BPD patients show significantly reduced VEGF levels [36], in line with genetic polymorphism in the VEGF gene associated with disease development [37]. Disturbed VEGF signaling is paralleled by increased levels of the anti-angiogenic proteins thrombospondin-1 and endoglin together with decreased expression levels of the pro-angiogenic factor Tie-2 [38, 39].
Retinopathy of prematurity (ROP), a condition in preterm infants characterized by comparable complications in vascular development, is associated with reduced levels of insulin-like growth factor (IGF)-1 that in turn abrogates VEGF-A expression through the MAPK pathway leading to endothelial cell apoptosis [40]. Coincidentally, low pulmonary IGF-1 expression can be considered as a driver of disrupted formation of the capillary bed in the developing lung undergoing postnatal injury in the same cohort of high-risk preterm infants [41].
Seemingly in contrast, pulmonary overexpression of VEGF-A in newborn mice leads to the development of a simplified lung structure with fewer and larger alveoli when the respective mice are exposed to hyperoxia [42]. These findings are in line with treatment studies using VEGF-A that failed to rescue the lung from postnatal injury but resulted in severe capillary leakage [43]. These findings further indicate the critical regulation of VEGF expression throughout lung development in concert with the complex crosstalk of other growth factors. The elevation of VEGF levels upon initiation of postnatal injury—as observed in some studies in BPD patients—may indicate a rescue mechanism underlining the essential role of VEGF-A in endothelial cell survival including mobilization of endothelial progenitor cells [44,45,46]. Another source for the (transitory) increase in pulmonary VEGF expression upon injury is monocytes/macrophages recruited to the injured lung [47].
With respect to its upstream regulation, hypoxia-inducible factor [9] plays an important role as it induces pulmonary VEGF expression [48]. Accordingly, the expression of a mutant HIF gene leads to the development of postnatal respiratory distress together with a simplified alveolar structure in the lungs of fetal mice. The outlined results point towards a role for the co-regulation of HIF and VEGF in BPD pathophysiology side by side with other potential regulators of VEGF signaling such as nuclear factor-ƙB, where abrogation of its expression induces apoptosis, simplifies alveolar structure, and reduces pulmonary capillary density in neonatal mice in concert with a significant reduction of VEGF-R2 expression [49].
Knowledge about reduced expression of VEGF-A and VEGF-R2 in ventilated neonatal mice in association with a decrease in PDGF-A expression first indicated a potential co-regulation of these pathways during disease development [19]. In our own studies, we therefore comprehensively investigate the specific interaction of these two signaling pathways and show that abrogation of PDGF-Rα signaling leads to reduced VEGF-A production by the MFB, thereby leading to endothelial cell apoptosis in ventilated newborn mice. Using transgenic mice in a pre-clinical model and detailed in vitro studies, we not only establish a causal relationship between the two pathways through the lung MFB but, at the same time, show that PDGF treatment is able to successfully restore VEGF signaling in the newborn mouse lung ([50] (in press). This first step towards the development of new treatment concepts has to be pursued further in order to address other co-regulated pathways in this context.
TGF-β signaling enhancing pulmonary injury in neonates
The TGF-β protein is secreted into the extracellular matrix (ECM) in its inactive form bound to latent TGF-β binding proteins (LTBPs) [51]. In turn, binding of LTPBs to other ECM proteins like fibrillin 1 and 5 facilitates elastogenesis in the developing lung [51]. Next to its secretion by inflammatory cells, the release of TGF-β is observed as a consequence of ECM remodeling [52, 53]. As a consequence, both, the phosphorylation of SMAD proteins as well as the upregulation of connective tissue growth factor (CTGF), are associated with the activation of the TGF-β pathway [54]. In addition to the important role of TGF-β for normal lung development, studies in different animal models of BPD confirm elevation of TGF-β expression levels and activation of its associated pathway as an important part of disease pathophysiology [24, 28, 55, 56]. Supporting the relevance of these findings from experimental studies, clinical studies show increased expression as well as activation of TGF-β, an inflammatory cytokine and growth factor in BALF obtained from ventilated preterm infants [57].
Increase in pulmonary TGFβ expression stems from the characteristic inflammatory response preceding most of the BPD-relevant pathopysiology [58]. The influx of inflammatory cells, i.e., neutrophils and monocytes, into the injured lung during disease development is well described in infants with BPD [59, 60]. Consequently, the recruited monocytes and macrophages are the most important source of TGF-β [61] together with a simultaneous increase in secreted cytokines such as interleukin (IL-)1β, IL-6, IL-8, tumor necrosis factor (TNF)-α, monocyte chemo-attractant proteins (MCP) 1 to 3, and the macrophage inflammatory proteins (MIP) 1a and 1b in preterm infants with evolving BPD [57, 62, 63]. Pro-inflammatory factors, in turn, provoke the development of pulmonary edema in ventilated preterm infants and lambs [64,65,66]. The pro-inflammatory response is typically associated with decreased levels of their anti-inflammatory counterparts, i.e., IL-10, IL-4, IL-12, and IL-13 or the IL-1 receptor antagonist [67,68,69,70].
Remodeling of the ECM not only leads to the release of TGF-β [36, 71, 72] but, at the same time, is a consequence of its increased expression and signaling. Both, BPD patients as well as animals with a BPD phenotype induced by postnatal hyperoxia, exhibit a TGF-β dependant upregulation of lysyl oxidases and transglutaminases—enzyme that aids in crosslinking of elastin, collagen, and fibronectin [73, 74]. The overexpression of TGF-β furthermore results in impaired branching morphogenesis, altered cellular composition, and ECM remodeling in neonatal mice [72]. In line with these findings, the use of a TGF-β neutralizing antibody rescues the alveolar and microvascular phenotype occurring in newborn mice exposed to hyperoxia [75].
A possible mechanism underlying the TGF-β-driven alveolar pathology is indicated by in vitro findings that show a reduction of PDGF expression by TGF β in adult pulmonary mesenchymal cells [76]. Heinzelmann et al. furthermore demonstrate a negative correlation between TGF-β stimulation and PDGF-Rα expression in adult human lung fibroblasts [77]. Translating these findings into the developing human lung, recent studies performed by Popova et al. display a downregulation in PDGF-Rα expression upon TGF-β treatment in lung mesenchymal-like cells (MSCs) obtained from BPD patients [16]. In support with the help of tailored in vitro assays employing primary mouse and human fibroblasts together with a unique mouse model of the disease, we confirm a causal relationship between reduced PDGF-Rα expression and increased TGF-β expression ([50] (in press). Not only does the increased TGF-β activation coincides with reduction in PDGF expression but also with a decrease in VEGF signaling in animal models employing MV-O2 to induce a BPD phenotype [22]. This finding is further supported by antibody blocking of TGF signaling leading to a VEGF-dependant improvement in microvascular structure and function [75]. Different effects attributed to TGF-β can play a causal role in this context: one, the induction of the CTGF by TGF-β as shown in airway smooth muscle cells facilitates the binding of VEGF to the ECM [78]; second, the bone morphogenic protein (BMP), part of the TGF-β superfamily, enhances secretion of the VEGF protein in murine proteoblastic KS483 cells [79]. This is in line with our data, showing a TGF-β-induced and PDGF-Rα-dependant regulation of VEGF expression in the injured neonatal lung ([50] (in press). TGF-α, a growth factor from the same cytokine family, is known for its role in cell proliferation, branching morphogenesis, and epithelial cell differentiation [80]. Accordingly, its overexpression in transgenic mice leads to pulmonary hypertension along with pathologic muscularization of small pulmonary arteries [81]. Likewise, FGF (fibroblast growth factor)-10 is expressed on mesenchymal cells and crucial for branching morphogenesis. The interaction of FGF-10 with PDGF and VEGF signaling and their role in BPD pathophysiology needs further investigation but holds promising value for the development of future treatment strategies [82, 83].
Summary and conclusion
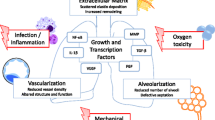
The multifactorial pattern of BPD requires a detailed understanding of different underlying signaling pathways, their critical crosstalk and the impact on disease pathophysiology. Figure 1 gives a schematic overview with respect to the interconnection of TGF activation, myofibroblast (PDGF) and endothelial cell (VEGF) signaling, and its impact on ECM remodeling eventually leading to defective alveolar and microvascular development. Deeper knowledge may reveal treatment options targeting different disease-relevant processes through one or two key processes.

A schematic model depicting important aspects of our current understanding with respect to the crosstalk between the PDGF, VEGF and TGF pathway in the developing lung. Chapter I (The role of PDGF signalling in (patho)- physiologic lung development) emphasizes the importance of PDGF-Rα expressing myofibroblasts (MFBs) in alveolarization that is hindered by mechanical ventilation and stretch causing abrogation in the receptor leading to reduced migration and disrupted elastin deposition. As a consequence of its reduced downstream signaling, a reduction in VEGF-A is observed accelerating endothelial cell (EC) apoptosis. This is the link to chapter II (Disturbed vascular growth factor signalling in the injured neonatal lung) of the review showing interaction of PDGF to disrupted VEGF, a pro-survival growth factor for ECs. Chapter III (TGF-β signalling enhancing pulmonary injury in neonates) of the review focuses on the aspect of upregulated TGF-β in injured lung that can be attributed to increased influx of monocytes and macrophages. The increased TGF-β expression and signaling interferes with PDGF-Rα and VEGF-A signaling hence affecting survival and function of MFBs and ECs.
References
Jobe AH (2011) The new bronchopulmonary dysplasia. Curr Opin Pediatr 23(2):167–172. doi:10.1097/MOP.0b013e3283423e6b
Hilgendorff A, Reiss I, Ehrhardt H, Eickelberg O, Alvira CM (2013) Chronic lung disease in the preterm infant: lessons learned from animal models. Am J Respir Cell Mol Biol. doi:10.1165/rcmb.2013-0014TR
Doyle, L. W., & Anderson, P. J. (2009). Long-term outcomes of bronchopulmonary dysplasia (Review). Semin Fetal Neonatal Med, 14(6), 391-395. doi:10.1016/j.siny.2009.08.004.
Ehrenkranz RA, Walsh MC, Vohr BR, Jobe AH, Wright LL, Fanaroff AA et al (2005) Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics 116(6):1353–1360. doi:10.1542/peds.2005-0249
Jobe, A. H., & Bancalari, E. (2001). Bronchopulmonary dysplasia (Congresses). Am J Respir Crit Care Med, 163(7), 1723-1729. http://www.ncbi.nlm.nih.gov/pubmed/11401896.
Mizikova, I., & Morty, R. E. (2015). The extracellular matrix in bronchopulmonary dysplasia: target and source (Review). Front Med (Lausanne), 2, 91. doi:10.3389/fmed.2015.00091.
Liwen Xu, Marlene Rabinovitch, B. (2006). Altered expression of key growth factors (TGF{alpha}, TGFß1, PDGF-A) and flawed formation of alveoli and elastin (Eln) in lungs of preterm (PT) lambs with chronic lung disease (CLD) FASEB J, 20 meeting abstract, A1442-A1443.
Mariani, T. J., Sandefur, S., & Pierce, R. A. (1997). Elastin in lung development (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review). Exp Lung Res, 23(2), 131-145. http://www.ncbi.nlm.nih.gov/pubmed/9088923.
Shifren, A., Durmowicz, A. G., Knutsen, R. H., Hirano, E., & Mecham, R. P. (2007). Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Am J Physiol Lung Cell Mol Physiol, 292(3), L778-L787. doi:10.1152/ajplung.00352.2006.
Wendel, D. P., Taylor, D. G., Albertine, K. H., Keating, M. T., & Li, D. Y. (2000). Impaired distal airway development in mice lacking elastin (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S). Am J Respir Cell Mol Biol, 23(3), 320-326. doi:10.1165/ajrcmb.23.3.3906.
Lindahl P, Karlsson L, Hellstrom M, Gebre-Medhin S, Willetts K, Heath JK et al (1997) Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124(20):3943–3953
Lau, M., Masood, A., Yi, M., Belcastro, R., Li, J., & Tanswell, A. K. (2011). Long-term failure of alveologenesis after an early short-term exposure to a PDGF-receptor antagonist (Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 300(4), L534-L547. doi:10.1152/ajplung.00262.2010.
Sun, T., Jayatilake, D., Afink, G. B., Ataliotis, P., Nister, M., Richardson, W. D., et al. (2000). A human YAC transgene rescues craniofacial and neural tube development in PDGFRalpha knockout mice and uncovers a role for PDGFRalpha in prenatal lung growth (Research Support, Non-U.S. Gov’t. Development, 127(21), 4519-4529. http://www.ncbi.nlm.nih.gov/pubmed/11023856.
Bland RD, Albertine KH, Pierce RA, Starcher BC, Carlton DP (2003) Impaired alveolar development and abnormal lung elastin in preterm lambs with chronic lung injury: potential benefits of retinol treatment. Biol Neonate 84(1):101–102 doi: 71012
Osornio-Vargas, A. R., Lindroos, P. M., Coin, P. G., Badgett, A., Hernandez-Rodriguez, N. A., & Bonner, J. C. (1996). Maximal PDGF-induced lung fibroblast chemotaxis requires PDGF receptor-alpha (Research Support, Non-U.S. Gov’t). Am J Phys, 271(1 Pt 1), L93-L99. http://www.ncbi.nlm.nih.gov/pubmed/8760137.
Popova, A. P., Bentley, J. K., Cui, T. X., Richardson, M. N., Linn, M. J., Lei, J., et al. (2014). Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia (Research Support, N.I.H., Extramural. Am J Physiol Lung Cell Mol Physiol, 307(3), L231-L239. doi:10.1152/ajplung.00342.2013.
Bland RD, Ertsey R, Mokres LM, Xu L, Jacobson BE, Jiang S et al (2008) Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice. Prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol 294(1):L3–14. doi:10.1152/ajplung.00362.2007
Bland, R. D., Ling, C. Y., Albertine, K. H., Carlton, D. P., MacRitchie, A. J., Day, R. W., et al. (2003). Pulmonary vascular dysfunction in preterm lambs with chronic lung disease (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.). Am J Physiol Lung Cell Mol Physiol, 285(1), L76-L85. doi:10.1152/ajplung.00395.2002
Bland, R. D., Mokres, L. M., Ertsey, R., Jacobson, B. E., Jiang, S., Rabinovitch, M., et al. (2007). Mechanical ventilation with 40% oxygen reduces pulmonary expression of genes that regulate lung development and impairs alveolar septation in newborn mice (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 293(5), L1099-L1110. doi:10.1152/ajplung.00217.2007
Hilgendorff A, e. a. (2011). Increased risk for lung injury in PDGF-Rα deficient newborn mice. Eur Respir J, 42 (suppl 57).
Branchfield, K., Li, R., Lungova, V., Verheyden, J. M., McCulley, D., & Sun, X. (2016). A three-dimensional study of alveologenesis in mouse lung (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Dev Biol, 409(2), 429-441. doi:10.1016/j.ydbio.2015.11.017
Bland, R. D., Xu, L., Ertsey, R., Rabinovitch, M., Albertine, K. H., Wynn, K. A., et al. (2007). Dysregulation of pulmonary elastin synthesis and assembly in preterm lambs with chronic lung disease (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 292(6), L1370-L1384. doi:10.1152/ajplung.00367.2006
Hilgendorff, A., Parai, K., Ertsey, R., Jain, N., Navarro, E. F., Peterson, J. L., et al. (2011). Inhibiting lung elastase activity enables lung growth in mechanically ventilated newborn mice (Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Am J Respir Crit Care Med, 184(5), 537-546. doi:10.1164/rccm.201012-2010OC.
Hilgendorff, A., Parai, K., Ertsey, R., Juliana Rey-Parra, G., Thebaud, B., Tamosiuniene, R., et al. (2012). Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 303(3), L215-L227. doi:10.1152/ajplung.00405.2011.
Pierce, R. A., Mariani, T. J., & Senior, R. M. (1995). Elastin in lung development and disease (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review). CIBA Found Symp, 192, 199-212; discussion 212-4. http://www.ncbi.nlm.nih.gov/pubmed/8575258.
Thibeault, D. W., Mabry, S. M., Ekekezie, II, & Truog, W. E. (2000). Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S). Pediatrics, 106(6), 1452-1459. http://www.ncbi.nlm.nih.gov/pubmed/11099603.
Wagenaar, G. T., ter Horst, S. A., van Gastelen, M. A., Leijser, L. M., Mauad, T., van der Velden, P. A., et al. (2004). Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress (Research Support, Non-U.S. Gov’t. Free Radic Biol Med, 36(6), 782-801. doi:10.1016/j.freeradbiomed.2003.12.007.
Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M et al (2010) Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298(1):L23–L35. https://doi.org/10.1152/ajplung.00251.2009
Rajatapiti P, van der Horst IW, de Rooij JD, Tran MG, Maxwell PH, Tibboel D et al (2008) Expression of hypoxia-inducible factors in normal human lung development. Pediatr Dev Pathol 11(3):193–199. doi:10.2350/07-04-0257.1.
Zhao, L., Wang, K., Ferrara, N., & Vu, T. H. (2005). Vascular endothelial growth factor co-ordinates proper development of lung epithelium and vasculature (Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S). Mech Dev, 122(7–8), 877-886. doi:10.1016/j.mod.2005.04.001.
Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF et al (2000) Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279(3):L600–L607 http://www.ncbi.nlm.nih.gov/pubmed/10956636
Prochilo T, Savelli G, Bertocchi P, Abeni C, Rota L, Rizzi A et al (2013) Targeting VEGF-VEGFR pathway by sunitinib in peripheral primitive neuroectodermal tumor, paraganglioma and epithelioid hemangioendothelioma: three case reports. Case Rep Oncol 6(1):90–97. doi:10.1159/000348429.
Tang, J. R., Seedorf, G., Balasubramaniam, V., Maxey, A., Markham, N., & Abman, S. H. (2007). Early inhaled nitric oxide treatment decreases apoptosis of endothelial cells in neonatal rat lungs after vascular endothelial growth factor inhibition (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Am J Physiol Lung Cell Mol Physiol, 293(5), L1271-L1280. doi:10.1152/ajplung.00224.2007.
Hoeben, A., Landuyt, B., Highley, M. S., Wildiers, H., Van Oosterom, A. T., & De Bruijn, E. A. (2004). Vascular endothelial growth factor and angiogenesis (Research Support, Non-U.S. Gov’t Review). Pharmacol Rev, 56(4), 549-580. doi:10.1124/pr.56.4.3.
Mammoto, T., Chen, J., Jiang, E., Jiang, A., Smith, L. E., Ingber, D. E., et al. (2012). LRP5 regulates development of lung microvessels and alveoli through the angiopoietin-Tie2 pathway (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). PLoS One, 7(7), e41596. doi:10.1371/journal.pone.0041596.
Been JV, Debeer A, van Iwaarden JF, Kloosterboer N, Passos VL, Naulaers G et al (2010) Early alterations of growth factor patterns in bronchoalveolar lavage fluid from preterm infants developing bronchopulmonary dysplasia. Pediatr Res 67(1):83–89. https://doi.org/10.1203/PDR.0b013e3181c13276
Fujioka, K., Shibata, A., Yokota, T., Koda, T., Nagasaka, M., Yagi, M., et al. (2014). Association of a vascular endothelial growth factor polymorphism with the development of bronchopulmonary dysplasia in Japanese premature newborns (Research Support, Non-U.S. Gov’t). Sci Rep, 4, 4459. doi:10.1038/srep04459.
De Paepe ME, Greco D, Mao Q (2010) Angiogenesis-related gene expression profiling in ventilated preterm human lungs. Exp Lung Res 36(7):399–410. https://doi.org/10.3109/01902141003714031
De Paepe, M. E., Patel, C., Tsai, A., Gundavarapu, S., & Mao, Q. (2008). Endoglin (CD105) up-regulation in pulmonary microvasculature of ventilated preterm infants (Research Support, N.I.H., Extramural). Am J Respir Crit Care Med, 178(2), 180-187. doi:10.1164/rccm.200608-1240OC
Hellstrom A, Perruzzi C, Ju M, Engstrom E, Hard AL, Liu JL et al (2001) Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci U S A 98(10):5804–5808. doi:10.1073/pnas.101113998
Lofqvist C, Hellgren G, Niklasson A, Engstrom E, Ley D, Hansen-Pupp I et al (2012) Low postnatal serum IGF-I levels are associated with bronchopulmonary dysplasia (BPD). Acta Paediatr 101(12):1211–1216. doi:10.1111/j.1651-2227.2012.02826.x
Syed, M. A., Choo-Wing, R., Homer, R. J., & Bhandari, V. (2016). Role of nitric oxide Isoforms in vascular and alveolar development and lung injury in vascular endothelial growth factor overexpressing neonatal mice lungs (Research Support, N.I.H., Extramural). PLoS One, 11(1), e0147588. doi:10.1371/journal.pone.0147588.
Akeson, A. L., Cameron, J. E., Le Cras, T. D., Whitsett, J. A., & Greenberg, J. M. (2005). Vascular endothelial growth factor-A induces prenatal neovascularization and alters bronchial development in mice (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.). Pediatr Res, 57(1), 82-88. doi:https://doi.org/10.1203/01.PDR.0000148070.89006.3F
Aicher, A., Heeschen, C., Mildner-Rihm, C., Urbich, C., Ihling, C., Technau-Ihling, K., et al. (2003). Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells (Research Support, Non-U.S. Gov’t). Nat Med, 9(11), 1370-1376. doi:10.1038/nm948
Matsumoto T, M. H. (2006). Signal transduction via vascular endothelial growth factor (VEGF) receptors and their roles in atherogenesis. J Atheroscler Thromb, 3(Jun 13), 130-135.
Meller, S., & Bhandari, V. (2012). VEGF levels in humans and animal models with RDS and BPD: temporal relationships (Research Support, N.I.H., Extramural Review). Exp Lung Res, 38(4), 192-203. doi:10.3109/01902148.2012.663454.
Lahm, T., Crisostomo, P. R., Markel, T. A., Wang, M., Lillemoe, K. D., & Meldrum, D. R. (2007). The critical role of vascular endothelial growth factor in pulmonary vascular remodeling after lung injury (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review). Shock, 28(1), 4-14. doi:10.1097/shk.0b013e31804d1998.
Huang, Y., Kempen, M. B., Munck, A. B., Swagemakers, S., Driegen, S., Mahavadi, P., et al. (2012). Hypoxia-inducible factor 2alpha plays a critical role in the formation of alveoli and surfactant (Research Support, Non-U.S. Gov’t). Am J Respir Cell Mol Biol, 46(2), 224-232. doi:10.1165/rcmb.2011-0024OC.
Iosef, C., Alastalo, T. P., Hou, Y., Chen, C., Adams, E. S., Lyu, S. C., et al. (2012). Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 302(10), L1023-L1036. doi:10.1152/ajplung.00230.2011.
Oak P, Pritzke T, Thiel I, Koschlig M, Mous DS, Windhorst A, Jain N, Eickelberg O, Foerster K, Schulze A, Goepel W, Reicherzer T, Ehrhardt H, Rottier RJ, Ahnert P, Gortner L, Desai TJ, Hilgendorff A (2017 (in Press) Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Molecular Medicine 9(10):1327–1462
Dabovic, B., Chen, Y., Choi, J., Davis, E. C., Sakai, L. Y., Todorovic, V., et al. (2011). Control of lung development by latent TGF-beta binding proteins (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). J Cell Physiol, 226(6), 1499-1509. doi:https://doi.org/10.1002/jcp.22479
Hinz, B. (2015). The extracellular matrix and transforming growth factor-beta1: tale of a strained relationship (Research Support, Non-U.S. Gov’t Review). Matrix Biol, 47, 54-65. doi:10.1016/j.matbio.2015.05.006.
ten Dijke, P., & Arthur, H. M. (2007). Extracellular control of TGFbeta signalling in vascular development and disease (Research Support, Non-U.S. Gov’t Review). Nat Rev Mol Cell Biol, 8(11), 857-869. doi:10.1038/nrm2262.
Parada, C., Li, J., Iwata, J., Suzuki, A., & Chai, Y. (2013). CTGF mediates Smad-dependent transforming growth factor beta signaling to regulate mesenchymal cell proliferation during palate development (Research Support, N.I.H., Extramural). Mol Cell Biol, 33(17), 3482-3493. doi:10.1128/MCB.00615-13.
Kompass, K. S., Deslee, G., Moore, C., McCurnin, D., & Pierce, R. A. (2010). Highly conserved transcriptional responses to mechanical ventilation of the lung (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Physiol Genomics, 42(3), 384-396. doi:10.1152/physiolgenomics.00117.2009.
Wallace, M. J., Probyn, M. E., Zahra, V. A., Crossley, K., Cole, T. J., Davis, P. G., et al. (2009). Early biomarkers and potential mediators of ventilation-induced lung injury in very preterm lambs (Comparative Study Research Support, Non-U.S. Gov’t. Respir Res, 10, 19. doi:10.1186/1465-9921-10-19.
Schultz C, Tautz J, Reiss I, Moller JC (2003) Prolonged mechanical ventilation induces pulmonary inflammation in preterm infants. Biol Neonate 84(1):64–66 doi:71446
Speer CP (2006) Pulmonary inflammation and bronchopulmonary dysplasia (review). J Perinatol. https://www.ncbi.nlm.nih.gov/pubmed/28923828
Ballabh P, Simm M, Kumari J, Krauss AN, Jain A, Califano C et al (2004) Neutrophil and monocyte adhesion molecules in bronchopulmonary dysplasia, and effects of corticosteroids. Arch Dis Child Fetal Neonatal Ed 89(1):F76–F83 http://www.ncbi.nlm.nih.gov/pubmed/14711863
Ogden, B. E., Murphy, S., Saunders, G. C., & Johnson, J. D. (1983). Lung lavage of newborns with respiratory distress syndrome. Prolonged neutrophil influx is associated with bronchopulmonary dysplasia (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.). Chest, 83(5 Suppl), 31S-33S. http://www.ncbi.nlm.nih.gov/pubmed/6601571.
Grotendorst, G. R., Smale, G., & Pencev, D. (1989). Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils (Comparative Study). J Cell Physiol, 140(2), 396-402. doi:10.1002/jcp.1041400226.
Groneck, P., Gotze-Speer, B., Oppermann, M., Eiffert, H., & Speer, C. P. (1994). Association of pulmonary inflammation and increased microvascular permeability during the development of bronchopulmonary dysplasia: a sequential analysis of inflammatory mediators in respiratory fluids of high-risk preterm neonates (Research Support, Non-U.S. Gov’t). Pediatrics, 93(5), 712-718. http://www.ncbi.nlm.nih.gov/pubmed/8165067.
Merritt TA, Deming DD, Boynton BR (2009) The ‘new’ bronchopulmonary dysplasia: challenges and commentary. Semin Fetal Neonatal Med 14(6):345–357. doi:10.1016/j.siny.2009.08.009
Carlton, D. P., Albertine, K. H., Cho, S. C., Lont, M., & Bland, R. D. (1997). Role of neutrophils in lung vascular injury and edema after premature birth in lambs (Research Support, U.S. Gov’t, P.H.S.). J Appl Physiol (1985), 83(4), 1307-1317. http://www.ncbi.nlm.nih.gov/pubmed/9338441
Jaarsma A, Braaksma MA, Geven WB, van Oeveren W, Oetomo SB (2004) Activation of the inflammatory reaction within minutes after birth in ventilated preterm lambs with neonatal respiratory distress syndrome. Biol Neonate 86:1–5
Kotecha, S., Mildner, R. J., Prince, L. R., Vyas, J. R., Currie, A. E., Lawson, R. A., et al. (2003). The role of neutrophil apoptosis in the resolution of acute lung injury in newborn infants (Clinical Trial Comparative Study Controlled Clinical Trial Research Support, Non-U.S. Gov’t). Thorax, 58(11), 961-967. http://www.ncbi.nlm.nih.gov/pubmed/14586049.
Baier RJ, Loggins J, Kruger TE (2003) Interleukin-4 and 13 concentrations in infants at risk to develop bronchopulmonary dysplasia. BMC Pediatr 3:8
Bhandari, V. (2014). Postnatal inflammation in the pathogenesis of bronchopulmonary dysplasia (Research Support, N.I.H., Extramural Review). Birth Defects Res A Clin Mol Teratol, 100(3), 189-201. doi:10.1002/bdra.23220
Bohrer, B., Silveira, R. C., Neto, E. C., & Procianoy, R. S. (2010). Mechanical ventilation of newborns infant changes in plasma pro- and anti-inflammatory cytokines (Research Support, Non-U.S. Gov’t). J Pediatr, 156(1), 16-19. doi:10.1016/j.jpeds.2009.07.027
Jónsson B, Tullus K, Brauner A, Lu Y, Noack G (1997) Early increase of TNFalpha and IL-6 in tracheobronchial aspirate fluid indicator of subsequent chronic lung disease in preterm infants. Arch Dis Child 77:F198–F201
Gauldie J, Galt T, Bonniaud P, Robbins C, Kelly M, Warburton D (2003) Transfer of the active form of transforming growth factor-beta 1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am J Pathol 163(6):2575–2584 http://www.ncbi.nlm.nih.gov/pubmed/14633629
Vicencio, A. G., Lee, C. G., Cho, S. J., Eickelberg, O., Chuu, Y., Haddad, G. G., et al. (2004). Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? (Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.). Am J Respir Cell Mol Biol, 31(6), 650-656. doi:10.1165/rcmb.2004-0092OC.
Kumarasamy, A., Schmitt, I., Nave, A. H., Reiss, I., van der Horst, I., Dony, E., et al. (2009). Lysyl oxidase activity is dysregulated during impaired alveolarization of mouse and human lungs (Research Support, Non-U.S. Gov’t). Am J Respir Crit Care Med, 180(12), 1239-1252. doi:10.1164/rccm.200902-0215OC.
Witsch, T. J., Niess, G., Sakkas, E., Likhoshvay, T., Becker, S., Herold, S., et al. (2014). Transglutaminase 2: a new player in bronchopulmonary dysplasia? (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). Eur Respir J, 44(1), 109-121. doi:10.1183/09031936.00075713.
Nakanishi, H., Sugiura, T., Streisand, J. B., Lonning, S. M., & Roberts, J. D., Jr. (2007). TGF-beta-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung (Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 293(1), L151-L161. doi:10.1152/ajplung.00389.2006.
Lindroos PM, Coin PG, Badgett A, Morgan DL, Bonner JC (1997) Alveolar macrophages stimulated with titanium dioxide, chrysotile asbestos, and residual oil fly ash upregulate the PDGF receptor-alpha on lung fibroblasts through an IL-1beta-dependent mechanism. Am J Respir Cell Mol Biol 16(3):283–292. doi:10.1165/ajrcmb.16.3.9070613
Heinzelmann K, Noskovicova N, Merl-Pham J, Preissler G, Winter H, Lindner M et al (2016) Surface proteome analysis identifies platelet derived growth factor receptor-alpha as a critical mediator of transforming growth factor-beta-induced collagen secretion. Int J Biochem Cell Biol 74:44–59. doi:10.1016/j.biocel.2016.02.013
Burgess, J. K., Ge, Q., Poniris, M. H., Boustany, S., Twigg, S. M., Black, J. L., et al. (2006). Connective tissue growth factor and vascular endothelial growth factor from airway smooth muscle interact with the extracellular matrix (Research Support, Non-U.S. Gov’t). Am J Physiol Lung Cell Mol Physiol, 290(1), L153-L161. doi:10.1152/ajplung.00287.2005
Deckers MM, van Bezooijen RL, van der Horst G, Hoogendam J, van Der Bent C, Papapoulos SE et al (2002) Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology 143(4):1545–1553. https://doi.org/10.1210/endo.143.4.8719
Ruocco S, Lallemand A, Tournier JM, Gaillard D (1996) Expression and localization of epidermal growth factor, transforming growth factor-alpha, and localization of their common receptor in fetal human lung development. Pediatr Res 39(3):448–455. doi:10.1203/00006450-199603000-00012.
Le Cras TD, Hardie WD, Fagan K, Whitsett JA, Korfhagen TR (2003) Disrupted pulmonary vascular development and pulmonary hypertension in transgenic mice overexpressing transforming growth factor-alpha. Am J Physiol Lung Cell Mol Physiol 285(5):L1046–L1054. doi:10.1152/ajplung.00045.2003
Benjamin JT, Smith RJ, Halloran BA, Day TJ, Kelly DR, Prince LS (2007) FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by Toll-like receptor activation. Am J Physiol Lung Cell Mol Physiol 292(2):L550–L558. doi:10.1152/ajplung.00329.2006
wa Maina C, Honkela A, Matarese F, Grote K, Stunnenberg HG, Reid G et al (2014) Inference of RNA polymerase II transcription dynamics from chromatin immunoprecipitation time course data. PLoS Comput Biol 10(5):e1003598. doi:10.1371/journal.pcbi.1003598
Acknowledgements
We sincerely thank the funding organizations German Center for Lung Research (DZL) for BMBF Grant 01KI1010C and BMBF Grant 01KI1010I and the Helmholtz Gemeinschaft and the Helmholtz Zentrum Muenchen, Germany, for Young Investigator Grant NWG VH-NG-829.
Availability of data and materials
Not applicable
Funding
This study was funded by the German Center for Lung Research (DZL), BMBF Grant 01KI1010C and BMBF Grant 01KI1010I, and Helmholtz Gemeinschaft and the Helmholtz Zentrum Muenchen, Germany, Young Investigator Grant NWG VH-NG-829.
Author information
Authors and Affiliations
Contributions
PO and AH contributed to the mini-review drafting and finalizing. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Oak, P., Hilgendorff, A. The BPD trio? Interaction of dysregulated PDGF, VEGF, and TGF signaling in neonatal chronic lung disease. Mol Cell Pediatr 4, 11 (2017). https://doi.org/10.1186/s40348-017-0076-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-017-0076-8