
Abstract
Chronic lung disease remains the major cause of morbidity and mortality in patients with cystic fibrosis (CF). Recent studies in young children with CF diagnosed by newborn screening identified neutrophil elastase (NE), a major product released from neutrophils in inflamed airways, as a key risk factor for the onset and early progression of CF lung disease. However, the understanding of how NE and potentially other proteases contribute to the complex in vivo pathogenesis of CF lung disease remains limited. In this review, we summarize recent progress in this area based on studies in βENaC-overexpressing (βENaC-Tg) mice featuring CF-like lung disease and novel protease-specific Förster resonance energy transfer (FRET) sensors for localization and quantification of protease activity in the lung. These studies demonstrated that NE is implicated in several key features of CF lung disease such as neutrophilic airway inflammation, mucus hypersecretion, and structural lung damage in vivo. Furthermore, these studies identified macrophage elastase (matrix metalloproteinase 12 (MMP12)) as an additional protease contributing to early lung damage in βENaC-Tg mice. Collectively, these results suggest that NE and MMP12 released from activated neutrophils and macrophages in mucus-obstructed airways play important pathogenetic roles and may serve as potential therapeutic targets to prevent and/or delay irreversible structural lung damage in patients with CF.
Similar content being viewed by others
Introduction
Cystic fibrosis (CF) is a complex disorder affecting multiple epithelial organs that is caused by over 2000 mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and remains the most common fatal genetic disease in Caucasian populations [13, 32]. Despite substantial improvements in clinical management, chronic lung disease remains the major cause of morbidity and mortality in patients with CF. Starting as a muco-obstructive lung disease with potentially reversible abnormalities such as airway mucus plugging, intermittent bacterial infection, and inflammation in the first months of life, CF lung diseases invariably progress towards a mucopurulent disorder characterized by chronic infection with specific pathogens such as Pseudomonas aeruginosa, non-resolving neutrophilic inflammation and irreversible structural lung damage, ultimately leading to respiratory failure [17, 32]. At the molecular and cellular levels, CF lung disease is caused by abnormal ion transport that disturbs the homeostasis of the thin liquid layer on airway surfaces producing a milieu that renders the airways susceptible for chronic infection and inflammation. CFTR functions as a cAMP-dependent anion (Cl− and bicarbonate) channel and regulator of the amiloride-sensitive epithelial Na+ channel ENaC and therefore plays a central role in the regulation of ion/fluid transport across airway epithelia [1, 9, 31, 51]. In CF, CFTR malfunction in the surface epithelium and submucosal glands renders airway surfaces dehydrated and slightly acidic [5, 33, 44]. These defects result in impaired mucus clearance and reduced bacterial killing by antimicrobial peptides thus setting the stage for airway mucus plugging and impaired host defenses that result in a vicious circle of chronic neutrophilic inflammation, infection, and progressive bronchiectasis in CF airways [36].
Because airway neutrophilia is associated with high levels of “free” NE activity leading to a protease-antiprotease imbalance in CF airways, NE has been implicated for a long time as a major player in the pathogenesis of structural lung damage in CF [4, 7, 40, 41, 46, 53]. This concept has been further substantiated by recent studies in infants and young children with CF who were diagnosed by newborn screening and followed longitudinally by annual chest computed tomography (CT) and bronchoalveolar lavage (BAL) [49, 50]. These studies demonstrated that CF lung disease starts in the first months of life, often in the absence of respiratory symptoms, and found a strong association between elevated NE activity in BAL fluid and the onset and progression of structural abnormalities including early bronchiectasis detected by chest CT [49, 50].
While these clinical association studies identified NE as a key risk factor, they do not provide mechanistic insights into how increased NE activity determines the progression of CF lung disease. In this context, it is noteworthy that a series of experimental studies demonstrated that NE has multiple functions that may be either disease promoting or protective in CF airways. Specifically, NE has been implicated in several key features of CF lung disease including airway inflammation, goblet cell metaplasia and mucus hypersecretion, and proteolytic damage of airway walls [41, 42, 45, 53–56]. Further, it was shown that increased NE activity can aggravate the basic CF ion transport defect via proteolytic degradation of CFTR and cleavage activation of ENaC [6, 27, 36]. On the other hand, NE can contribute to bacterial killing and may thus have important protective functions in host defense [3, 22]. Therefore, complementary studies in preclinical models of CF lung disease are needed to define the relative roles of these disease-promoting versus protective functions of NE and potentially other proteases in the in vivo pathogenesis of CF lung disease.
Review
NE is implicated in airway inflammation, mucus hypersecretion, and structural lung damage in mice with CF-like lung disease
For a systematic analysis of the diverse functions of NE in the complex in vivo pathogenesis, a recent study made use of the βENaC-Tg mouse as an established model of CF lung disease. The βENaC-Tg mouse features airway surface dehydration and reduced mucus clearance characteristic of CF and phenocopies key features of CF lung disease including early-onset airway mucus plugging, spontaneous bacterial infection, chronic inflammation, and structural lung damage [29, 30, 34, 35, 57, 61]. To determine the in vivo role of NE in CF-like lung disease, βENaC-Tg mice were crossed with NE-deficient (NE−/−) mice [3] and the impact of genetic deletion on these pulmonary disease phenotypes was determined. This classical candidate gene approach provided several novel insights into the pathogenesis of early CF-like lung disease that may support the development of novel therapies [15].
First, the cross of these mouse models demonstrated that genetic deletion of NE results in a substantial reduction of neutrophils in BAL from βENaC-Tg mice confirming an important role of this protease in neutrophilic airway inflammation [15]. In this context, as described in more detail below, we found high levels of active NE on the surface of BAL neutrophils from βENaC-Tg mice (Fig. 1). We therefore speculate that membrane-associated NE activity plays an important role in the transmigration of neutrophils from the blood to the airway lumen and that this mechanism is a critical component of the pro-inflammatory function of NE in CF-like lung disease [15].

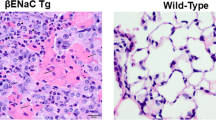
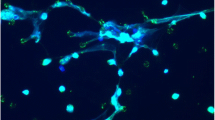
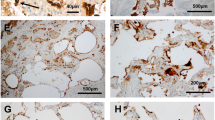
Neutrophil elastase (NE) and matrix metalloproteinase 12 (MMP12) activity is increased at the surface of bronchoalveolar neutrophils and macrophages and is associated with structural lung damage in βENaC-Tg mice. Protease activity was measured on the surface of neutrophils from bronchoalveolar lavage (BAL) using a lipidated FRET reporter for NE (NEmo-2) and representative ratio images calculated from donor and acceptor fluorescence are shown (left upper panels). NEmo-2 detects increased NE activity (green color) on neutrophils from βENaC-Tg compared to wild-type (WT) mice, and the specificity of the NEmo-2 FRET signal is confirmed by genetic deletion of NE (NE−/− and βENaC-Tg/NE−/− mice). Representative morphology of distal airspaces shows that increased NE activity on BAL neutrophils is associated with airspace enlargement and destruction in βENaC-Tg mice that is substantially reduced by genetic deletion of NE (left lower panels). Corresponding experiments using a lipidated FRET reporter for MMP12 (LaRee-1) show activity (green color) on macrophages from βENaC-Tg mice, but not from wild-type (WT) mice or mice that lack MMP12 (MMP12−/− and βENaC-Tg/MMP12−/− mice) (right upper panels). Representative morphology of lung sections from WT, βENaC-Tg, MMP12−/−, and βENaC-Tg/MMP12−/− mice demonstrates that increased MMP12 activity on BAL macrophages also contributes to structural lung damage (right lower panels). Reprinted from [15, 52] with permission from the American Thoracic Society
Next, these studies showed that NE is a potent trigger of goblet cell metaplasia and mucus hypersecretion in airways from βENaC-Tg mice. In fact, goblet cell metaplasia and increased expression of secreted mucins (Muc5ac and Muc5b) characteristic of βENaC-Tg mice [35] were completely abrogated in NE-deficient βENaC-Tg mice (βENaC-Tg/NE−/−) [15]. Surprisingly, even in the absence of mucus hypersecretion, βENaC-Tg/NE−/− mice were not protected from the development of severe airway mucus obstruction. However, measurements of the airway mucus concentration demonstrated that the mucus was dehydrated to similar levels in βENaC-Tg/NE−/− mice compared to βENaC-Tg littermates. These results suggest that airway surface dehydration may be sufficient to slow clearance of constitutively secreted mucus and trigger mucus plugging even in the absence of mucus hypersecretion [15].
In addition, these studies provided important information on the in vivo role of NE in antibacterial host defense. Mucociliary dysfunction in neonatal βENaC-Tg mice is associated with spontaneous bacterial infection dominated by species from the oropharyngeal flora [29]. In previous studies, lack of NE was shown to aggravate acute infection with P. aeruginosa in mice [3, 22]. On the other hand, high levels of NE activity were shown to cleave chemokine receptors on leukocytes compromising their ability to kill bacteria [20]. In this context, it was important to test the impact of genetic deletion of NE on the spontaneous airway infection of βENaC-Tg mice. These studies showed that lack of NE does not exacerbate bacterial infection in βENaC-Tg mice indicating that other innate and/or adaptive defense mechanisms [2] are sufficient to contain bacterial growth in the airways [15].
Finally, these studies demonstrated that NE is implicated in emphysema-like structural lung damage characteristic of βENaC-Tg mice [35, 57]. In contrast to patients with CF [50, 58], mice with chronic neutrophilic airway disease develop emphysema rather than bronchiectasis [35, 47, 57]. This species difference is probably related to anatomical differences including a substantially lower number of airway branching in the mouse compared to the human lung that may result in a faster spillover of damaging factors from the conducting airways to the distal airspaces in mice. Nevertheless, it was found that genetic deletion of NE leads to a significant reduction (~50 %) of distal airspace enlargement and alveolar destruction in βENaC-Tg mice (Fig. 1) [15]. Despite the species differences mentioned above, these results support the concept that NE plays a critical role in the in vivo pathogenesis of structural lung damage associated with neutrophilic airway inflammation.
To elucidate the localization of tissue damaging protease activity, highly sensitive FRET reporters were employed that can discriminate between free (NEmo-1) and membrane-bound (NEmo-2) NE activity in CF-like lung disease (Fig. 1) [16, 23]. Similar to infants and young children with CF, βENaC-Tg mice exhibit a moderate airway neutrophilia with 5–30 % of neutrophils in BAL fluid [30, 49, 61]. Using the NEmo FRET reporters, we found that NE activity is invariably increased on the surface of BAL neutrophils from βENaC-Tg mice compared to wild-type controls (Fig. 1), whereas no free NE activity was detected in cell-free BAL supernatant [15]. In addition, we found that the activity of purified NE is potently inhibited by BAL supernatant from βENaC-Tg mice indicating that NE secreted from activated neutrophils into the extracellular compartment is inhibited by a robust antiprotease shield [15]. In CF patients with advanced lung disease and higher neutrophil counts (>80 %), NEmo-2 also detected higher levels of NE activity on the surface of sputum neutrophils. Further, free NE activity was increased in sputum supernatant from patients with CF compared to healthy controls as expected from previous studies [15, 37, 46]. When viewed in combination, these results suggest (i) that free NE activity is inhibited as long as the antiprotease shield composed of NE inhibitors such as α1-antitrypsin and secretory leukocyte protease inhibitor (SLPI) is not overwhelmed [26, 42, 43] and (ii) that surface-bound NE activity may play a critical role in tissue damage, even in early CF lung disease with moderate airway neutrophilia [15].
MMP12 contributes to structural lung damage in mice with CF-like lung disease
Interestingly, quantitative phenotyping of the cross of βENaC-Tg mice with NE−/− mice also revealed substantial residual alveolar destruction in double-mutant βENaC-Tg/NE−/− mice indicating that increased NE activity only accounts for ~50 % of structural lung damage and that other factors contribute to emphysema formation in βENaC-Tg mice [15]. Spurred by this observation, whole-genome expression profiling of the lung tissues was used as a bottom-up approach to search for candidate genes and pathways responsible for residual emphysema formation in βENaC-Tg mice. This unbiased approach identified Mmp12, but no other candidates previously implicated in emphysema formation, as a strongly upregulated gene in the lungs from βENaC-Tg mice [52]. Matrix metalloproteinase 12 (MMP12) is a matrix metalloproteinase with elastolytic capacity [48] that is secreted by activated macrophages and has been implicated in emphysema pathogenesis in mouse models of COPD and asthma via several independent mechanisms including (i) degradation of the extracellular matrix, (ii) proteolytic inactivation of antiproteases such as α1-antitrypsin, and (iii) proteolytic activation of proinflammatory cytokines including TNFα [10, 21, 60]. Interestingly, MMP12 is also a signature gene of alternatively activated macrophages (AAM) and the majority of genes that were differentially upregulated in the lungs from βENaC-Tg mice (Alox15, Arg1, Chi3l3, Chi3l4, Mgl2, Retnla) belong to this AAM signature [18, 28]. These results suggest that the microenvironment of mucostatic CF-like airways triggers alternative macrophage activation, which in turn results in upregulation of MMP12 [52].
To validate the pathogenetic relevance of MMP12 in emphysema formation in βENaC-Tg mice, the temporal relationship between elevated MMP12 expression and structural lung damage was investigated by several independent approaches including (i) MMP12-specific FRET reporters to localize its activity in the lung [11], (ii) genetic deletion of MMP12, and (iii) pharmacological inhibition of MMP activity in βENaC-Tg mice [52]. These studies demonstrated a strong temporal association of increased Mmp12 expression with distal airspace enlargement and destruction. First, it was shown that these emphysema-like features are substantially (~50 %) reduced by deletion of MMP12 in βENaC-Tg mice (Fig. 1). Similar effects were observed when βENaC-Tg mice were treated with the MMP inhibitor GM 6001. However, in contrast with the findings in βENaC-Tg/NE−/− mice, neither genetic deletion nor pharmacological inhibition of MMP12 reduced airway inflammation or goblet cell metaplasia in βENaC-Tg mice indicating that MMP12 is not essential in the in vivo pathogenesis of CF-like airway diseases [52]. Similar to findings for NE on activated neutrophils, studies using FRET reporters capable of detecting cell membrane-bound (LaRee-1) and free (LaRee-5) MMP12 activity [11, 23] showed that the activity of this protease is significantly increased on the surface of BAL macrophages (Fig. 1), but not in BAL fluid from βENaC-Tg mice [52]. These results indicate that the zymogen form of MMP12 is activated at the macrophage surface and that secreted MMP12 is inhibited by antiproteases in the extracellular milieu [42]. Collectively, these studies identified MMP12 secreted by activated macrophages as an additional protease contributing to the in vivo pathogenesis of structural lung damage in CF-like lung disease. Further, these studies corroborate the concept that protease activity on the surface of activated inflammatory cells, via direct contact to the extracellular matrix, plays an important role in structural lung damage associated with chronic airway inflammation [52].
Following biological validation of a candidate gene in a mouse model, it remains critical to determine its respective role in human disease. Previous studies have implicated MMP12 in the pathogenesis of COPD and asthma. Specifically, it was shown that elevated levels of MMP12 in the sputum are associated with emphysema severity in COPD [8, 12, 38] and that a functional variant in the MMP12 promoter (rs2276109) [25, 59] is associated with a beneficial effect on lung function in children with asthma, as well as a reduced risk for adult smokers to develop COPD [24]. In comparison, current knowledge on the role of MMP12 in CF lung disease remains limited [14]. One study detected alternatively activated macrophages in BAL and demonstrated an inverse relationship with lung function in patients with CF; however, MMP12 levels were not determined [39]. More recently, the development of the LaRee FRET reporters enabled measurements of MMP12 activity in BAL samples from patients with CF [52]. Similar to the results obtained in βENaC-Tg mice (Fig. 1), these studies detected increased activity of MMP12 at the macrophage surface even in children with CF with early lung disease. The functional relevance of this finding is supported by a genetic association study that investigated the impact of SNP in MMP12 on lung function in a cohort of 442 patients with CF [52]. This study showed that the SNP in the MMP12 promoter (rs2276109), as well as a tightly linked SNP (rs737693), is positively associated with longitudinal lung function (FEV1 % predicted) in patients with CF. Taken together, these translational studies provide initial evidence that proteolytic activity of MMP12 secreted by macrophages that are activated on mucostatic airway surfaces may contribute to the pathogenesis of structural lung damage and lung function decline in patients with CF [52].
Conclusions
In summary, the cross of βENaC-Tg mice with NE−/− mice demonstrates that NE is implicated in the in vivo pathogenesis of several key features of CF-like lung disease including the modulation of neutrophilic airway inflammation, induction of goblet cell metaplasia and mucin hypersecretion, and structural lung damage [15]. Further, whole-genome expression profiling as an unbiased bottom-up approach led to the identification of MMP12 released from activated macrophages as an important contributor to tissue damage in CF-like lung disease [52]. Importantly, genetic deletion of NE and MMP12 did not exacerbate spontaneous airway infection in βENaC-Tg mice. These data support that these proteases are promising targets for novel anti-inflammatory and tissue-protective therapies in CF. Interestingly, localization of protease activity with sensitive FRET reporters showed that the activities of both NE and MMP12 are invariably increased on the surface of activated neutrophils and macrophages, respectively, even under conditions when “free” activity of these secreted proteases is absorbed by an intact antiprotease shield [15, 52]. These results suggest that membrane-bound protease activity may play a critical role in airway damage and that NE and MMP12 may have to be inhibited at the surface of inflammatory cells to achieve maximal therapeutic effects. In this context, translational studies in clinical specimens (BAL and sputum) from patients with CF indicate that lipidated FRET reporters designed to measure protease activity at the surface of inflammatory cells, such as NEmo-2 and LaRee-1 [11, 16], may be sensitive novel tools to identify CF patients with the greatest risk to develop severe lung damage. However, additional studies in a larger number of patients that address the relationship with clinical indices of lung disease severity, such as lung function and imaging endpoints, will be required to determine the value of membrane-bound protease activity as an inflammation biomarker in CF and potentially other neutrophilic airway diseases. Of note, studies in βENaC-Tg mice also demonstrated that genetic or pharmacological inhibition of NE and MMP12 does not prevent dehydration-induced airway mucus plugging in vivo indicating that additional rehydration therapies such as osmolytes [19] or modulators of epithelial ion channels including CFTR, ENaC, or alternative Cl− channels [33] may be required for effective treatment of airway mucus plugging in CF.
References
Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ (1991) Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253:202–5
Bals R, Weiner DJ, Wilson JM (1999) The innate immune system in cystic fibrosis lung disease. J Clin Invest 103(3):303–7
Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD (1998) Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med 4(5):615–8
Birrer P, McElvaney NG, Rudeberg A, Sommer CW, Liechti-Gallati S, Kraemer R, Hubbard R, Crystal RG (1994) Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med 150(1):207–13
Boucher RC (2007) Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58:157–70
Caldwell RA, Boucher RC, Stutts MJ (2005) Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol 288(5):L813–L819
Cantin AM, Hartl D, Konstan MW, Chmiel JF (2015) Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros 14(4):419–30
Chaudhuri R, McSharry C, Brady J, Donnelly I, Grierson C, McGuinness S, Jolly L, Weir CJ, Messow CM, Spears M, Miele G, Nocka K, Crowther D, Thompson J, Brannigan M, Lafferty J, Sproule M, Macnee W, Connell M, Murchison JT, Shepherd MC, Feuerstein G, Miller DK, Thomson NC (2012) Sputum matrix metalloproteinase-12 in patients with chronic obstructive pulmonary disease and asthma: relationship to disease severity. J Allergy Clin Immunol 129(3):655–63
Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, Muallem S (2001) Aberrant CFTR-dependent HCO3 − transport in mutations associated with cystic fibrosis. Nature 410(6824):94–7
Churg A, Wright JL (2005) Proteases and emphysema. Curr Opin Pulm Med 11(2):153–9
Cobos-Correa A, Trojanek JB, Diemer S, Mall MA, Schultz C (2009) Membrane-bound FRET probe visualizes MMP12 activity in pulmonary inflammation. Nat Chem Biol 5(9):628–30
Demedts IK, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos GF, Pauwels RA, Brusselle GG (2006) Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax 61(3):196–201
Elborn JS (2016) Cystic fibrosis. Lancet 16:10–6736
Gaggar A, Hector A, Bratcher PE, Mall MA, Griese M, Hartl D (2011) The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur Respir J 38(3):721–7
Gehrig S, Duerr J, Weitnauer M, Wagner CJ, Graeber SY, Schatterny J, Hirtz S, Belaaouaj A, Dalpke AH, Schultz C, Mall MA (2014) Lack of neutrophil elastase reduces inflammation, mucus hypersecretion, and emphysema, but not mucus obstruction, in mice with cystic fibrosis-like lung disease. Am J Respir Crit Care Med 189(9):1082–92
Gehrig S, Mall MA, Schultz C (2012) Spatially resolved monitoring of neutrophil elastase activity with ratiometric fluorescent reporters. Angew Chem Int Ed Engl 51(25):6258–61
Gibson RL, Burns JL, Ramsey BW (2003) Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168(8):918–51
Gordon S, Martinez FO (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32(5):593–604
Graeber SY, Zhou-Suckow Z, Schatterny J, Hirtz S, Boucher RC, Mall MA (2013) Hypertonic saline is effective in the prevention and treatment of mucus obstruction, but not airway inflammation, in mice with chronic obstructive lung disease. Am J Respir Cell Mol Biol 49(3):410–7
Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, Krauss-Etschmann S, Koller B, Reinhardt D, Roscher AA, Roos D, Griese M (2007) Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med 13(12):1423–30
Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD (1997) Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277(5334):2002–4
Hirche TO, Benabid R, Deslee G, Gangloff S, Achilefu S, Guenounou M, Lebargy F, Hancock RE, Belaaouaj A (2008) Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J Immunol 181(7):4945–54
Hu HY, Gehrig S, Reither G, Subramanian D, Mall MA, Plettenburg O, Schultz C (2014) FRET-based and other fluorescent proteinase probes. Biotechnol J 9(2):266–81
Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J, Stidley C, Melen E, Soderhall C, Hallberg J, Kull I, Kere J, Svartengren M, Pershagen G, Wickman M, Lange C, Demeo DL, Hersh CP, Klanderman BJ, Raby BA, Sparrow D, Shapiro SD, Silverman EK, Litonjua AA, Weiss ST, Celedon JC (2009) MMP12, lung function, and COPD in high-risk populations. N Engl J Med 361(27):2599–608
Jormsjo S, Ye S, Moritz J, Walter DH, Dimmeler S, Zeiher AM, Henney A, Hamsten A, Eriksson P (2000) Allele-specific regulation of matrix metalloproteinase-12 gene activity is associated with coronary artery luminal dimensions in diabetic patients with manifest coronary artery disease. Circ Res 86(9):998–1003
Korkmaz B, Poutrain P, Hazouard E, de Monte M, Attucci S, Gauthier FL (2005) Competition between elastase and related proteases from human neutrophil for binding to alpha1-protease inhibitor. Am J Respir Cell Mol Biol 32(6):553–9
Le Gars M, Descamps D, Roussel D, Saussereau E, Guillot L, Ruffin M, Tabary O, Hong SS, Boulanger P, Paulais M, Malleret L, Belaaouaj A, Edelman A, Huerre M, Chignard M, Sallenave JM (2013) Neutrophil elastase degrades cystic fibrosis transmembrane conductance regulator via calpains and disables channel function in vitro and in vivo. Am J Respir Crit Care Med 187(2):170–9
Lewis CC, Aronow B, Hutton J, Santeliz J, Dienger K, Herman N, Finkelman FD, Wills-Karp M (2009) Unique and overlapping gene expression patterns driven by IL-4 and IL-13 in the mouse lung. J Allergy Clin Immunol 123(4):795–804
Livraghi-Butrico A, Kelly EJ, Klem ER, Dang H, Wolfgang MC, Boucher RC, Randell SH, O'Neal WK (2012) Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial infection and inflammation. Mucosal Immunol 5(4):397–408
Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC (2004) Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10(5):487–93
Mall M, Hipper A, Greger R, Kunzelmann K (1996) Wild type but not DF508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. FEBS Lett 381:47–52
Mall MA, Boucher RC (2014) Pathophysiology of cystic fibrosis lung disease. In: Mall MA, Elborn JS (eds) Cystic fibrosis. European Respiratory Society, Sheffield, pp 1–13
Mall MA, Galietta LJ (2015) Targeting ion channels in cystic fibrosis. J Cyst Fibros 14(5):561–70
Mall MA, Graeber SY, Stahl M, Zhou-Suckow Z (2014) Early cystic fibrosis lung disease: role of airway surface dehydration and lessons from preventive rehydration therapies in mice. Int J Biochem Cell Biol 52:174–9
Mall MA, Harkema JR, Trojanek JB, Treis D, Livraghi A, Schubert S, Zhou Z, Kreda SM, Tilley SL, Hudson EJ, O'Neal WK, Boucher RC (2008) Development of chronic bronchitis and emphysema in b-epithelial Na+ channel-overexpressing mice. Am J Respir Crit Care Med 177(7):730–42
Mall MA, Hartl D (2014) CFTR: cystic fibrosis and beyond. Eur Respir J 44(4):1042–54
Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, Sagel SD, Ramsey BW (2007) Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med 175(8):822–8
Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, Planquois JM, Delaval P, Lagente V (2005) Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res 54(1):31–6
Murphy BS, Bush HM, Sundareshan V, Davis C, Hagadone J, Cory TJ, Hoy H, Hayes D Jr, Anstead MI, Feola DJ (2010) Characterization of macrophage activation states in patients with cystic fibrosis. J Cyst Fibros 9(5):314–22
Nakamura H, Yoshimura K, McElvaney NG, Crystal RG (1992) Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J Clin Invest 89(5):1478–84
Nichols DP, Chmiel JF (2015) Inflammation and its genesis in cystic fibrosis. Pediatr Pulmonol 50(Suppl 40):S39–56. doi:10.1002/ppul.23242.:S39-S56
Owen CA (2008) Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 3(2):253–68
Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ (1995) Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 131(3):775–89
Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J (2012) Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487(7405):109–13
Pham CT (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6(7):541–50
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET (2012) Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 186(9):857–65
Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A (2003) Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 163(6):2329–35
Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD (1996) Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A 93(9):3942–6
Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, Stick SM, Robinson PJ, Robertson CF, Ranganathan SC (2009) Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med 180(2):146–52
Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, Murray CP, Stick SM (2013) Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 368(21):1963–70
Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC (1995) CFTR as a cAMP-dependent regulator of sodium channels. Science Vol 269:847–50
Trojanek JB, Cobos-Correa A, Diemer S, Kormann M, Schubert SC, Zhou-Suckow Z, Agrawal R, Duerr J, Wagner CJ, Schatterny J, Hirtz S, Sommerburg O, Hartl D, Schultz C, Mall MA (2014) Airway mucus obstruction triggers macrophage activation and matrix metalloproteinase 12-dependent emphysema. Am J Respir Cell Mol Biol 51(5):709–20
Twigg MS, Brockbank S, Lowry P, FitzGerald SP, Taggart C, Weldon S (2015) The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators Inflamm 2015:293053. doi:10.1155/2015/293053, Epub 2015 Jun 21
Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, Ho SB, Foster WM (2004) Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol 287(6):L1293–L1302
Voynow JA, Fischer BM, Zheng S (2008) Proteases and cystic fibrosis. Int J Biochem Cell Biol 40(6–7):1238–45
Voynow JA, Young LR, Wang Y, Horger T, Rose MC, Fischer BM (1999) Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am J Physiol 276(5 Pt 1):L835–L843
Wielpütz MO, Eichinger M, Zhou Z, Leotta K, Hirtz S, Bartling SH, Semmler W, Kauczor HU, Puderbach M, Mall MA (2011) In vivo monitoring of cystic fibrosis-like lung disease in mice by volumetric computed tomography. Eur Respir J 38(5):1060–70
Wielputz MO, Puderbach M, Kopp-Schneider A, Stahl M, Fritzsching E, Sommerburg O, Ley S, Sumkauskaite M, Biederer J, Kauczor HU, Eichinger M, Mall MA (2014) Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med 189(8):956–65. doi:10.1164/rccm.201309-1659OC
Wu L, Tanimoto A, Murata Y, Sasaguri T, Fan J, Sasaguri Y, Watanabe T (2003) Matrix metalloproteinase-12 gene expression in human vascular smooth muscle cells. Genes Cells 8(3):225–34
Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr, Chapman HA Jr, Shapiro SD, Elias JA (2000) Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 106(9):1081–93
Zhou Z, Duerr J, Johannesson B, Schubert SC, Treis D, Harm M, Graeber SY, Dalpke A, Schultz C, Mall MA (2011) The βENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J Cyst Fibros 10(Suppl 2):S172–S182
Acknowledgements
This review is based on a presentation at the Lung Symposium held during the 2015 Annual Meeting of the German Society of Pediatrics and Adolescent Medicine (DGKJ) in Munich, Germany. We thank our colleagues who contributed to the work cited in this review.
Funding
This work was supported in part by the German Federal Ministry of Education and Research (82DZL00401 and 82DZL004A1 to MAM and 82DZL00403 and 82DZL004A3 to CS) and the European Commission (Sixth and Seventh Framework Programme Projects No. 603038 CFMatters to MAM and LSHG-CT-2003-503259 Molecular Imaging and Health-2009-1.2-5-241919 LIVIMODE to CS).
Authors’ contributions
CJW and MAM wrote the manuscript. CS edited the manuscript and contributed to the discussion. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wagner, C.J., Schultz, C. & Mall, M.A. Neutrophil elastase and matrix metalloproteinase 12 in cystic fibrosis lung disease. Mol Cell Pediatr 3, 25 (2016). https://doi.org/10.1186/s40348-016-0053-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-016-0053-7