
Abstract
Malaria is a major cause of morbidity and mortality in many African countries and parts of Asia and South America. Novel approaches to combating the disease have emerged in recent years and several drug candidates are now being tested clinically. However, it is long before these novel drugs can hit the market, especially due to a scarcity of safety and efficacy data.
To reduce the malaria burden, the Medicines for Malaria Venture (MMV) was established in 1999 to develop novel medicines through industry and academic partners’ collaboration. However, no reviews were focused following various preclinical and clinical studies published since the MMV initiation (2000) to till date.
We identify promising approaches in the global portfolio of antimalarial medicines, and highlight challenges and patient specific concerns of these novel molecules. We discuss different clinical studies focusing on the evaluation of novel drugs against malaria in different human trials over the past five years.
The drugs KAE609 and DDD107498 are still being evaluated in Phase I trials and preclinical developmental studies. Both the safety and efficacy of novel compounds such as KAF156 and DSM265 need to be assessed further, especially for use in pregnant women. Synthetic non-artemisinin ozonides such as OZ277 raised concerns in terms of its insufficient efficacy against high parasitic loads. Aminoquinoline-based scaffolds such as ferroquine are promising but should be combined with good partner drugs for enhanced efficacy. AQ-13 induced electrocardiac events, which led to prolonged QTc intervals. Tafenoquine, the only new anti-relapse scaffold for patients with a glucose-6-phosphate dehydrogenase deficiency, has raised significant concerns due to its hemolytic activity. Other compounds, including methylene blue (potential transmission blocker) and fosmidomycin (DXP reductoisomerase inhibitor), are available but cannot be used in children.
At this stage, we are unable to identify a single magic bullet against malaria. Future studies should focus on effective single-dose molecules that can act against all stages of malaria in order to prevent transmission. Newer medicines have also raised concerns in terms of efficacy and safety. Overall, more evidence is needed to effectively reduce the current malaria burden. Treatment strategies that target the blood stage with transmission-blocking properties are needed to prevent future drug resistance.
Similar content being viewed by others

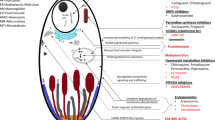
Multilingual abstract
Please see Additional file 1 for translations of the abstract into the five offical working languages of the United Nations.
Background
Malaria is an infectious disease caused by the protozoa of the genus Plasmodium, transmitted through the bite of the female Anopheles mosquito. It is a major public health problem in many endemic countries including Sub-Saharan Africa (SSA); in 2015, an estimated 438 000 malaria deaths were reported globally [1]. Human pathogenic Plasmodium species include Plasmodium falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi. Sporozoites are injected into the skin through a mosquito bite, invading hepatocytes and causing liver infection. The merozoites released from the liver rapidly infect the erythrocytes during the erythrocytic stage. Multiple rounds of the erythrocytic stage produce larger numbers of parasites that invade the blood, consequently causing clinical illness. Erythrocytic parasites develop into sexual gametocytes and are transmitted to humans through a mosquito bite.
The Plasmodium-infected female Anopheles mosquito is the deadliest known disease vector causing as many deaths as deaths from HIV/AIDS and tuberculosis [2]. In 2008, the World Health Organization (WHO) initiated the Global Malaria Action Plan (GMAP) to reduce rates of malaria incidence and mortality by at least ten-fold by 2030. Despite numerous advances over the past decade, new drugs are urgently needed. To reduce the malaria burden in developing countries, the not-for-profit organization Medicines for Malaria Venture (MMV) was established in 1999. Its main goal is to initiate collaborations with industry and academic partners in order to develop novel approaches to combat malaria [3]. This has led to the design and discovery of new medicines for human malaria cases. Between 2000 and 2015, malaria incidence rates decreased globally by 37 % and mortality rates decreased by 60 %. Most malaria cases (89 %) and deaths (91 %) from malaria globally were reported in SSA [4]. The emergence of resistance to traditional therapies including chloroquine, primaquine, quinine, and mefloquine has also revealed novel antimalarial targets. In the past decades, a consortium of researchers from academia and industry was created to develop new remedies focusing on chemotypes [5]. Consequently, the discovery of artemisinin-based combination therapies (ACTs) by Chinese scientists has tremendously benefitted hundreds of thousands of patients. However, resistance to antimalarial drugs continues to pose a major threat to malaria eradication [6]. Furthermore, recent advances in the development of species-specific malaria vaccines have emerged as the most prominent approach to eradicating malaria.
24 malarial vaccines are currently being tested in 99 clinical sites in Africa and 30 in Southeast Asia [7]. The most advanced recombinant protein-based malaria vaccine is Mosquirix™ (RTS, S), a combination of 25 % fusion protein RTS and 75 % wild-type hepatitis B surface antigen (HBsAg). The vaccine is effective against P. falciparum malaria and was developed by GlaxoSmithKline (GSK), the PATH Malaria Vaccine Initiative, and other partners. In a late-stage Phase III trial, Mosquirix™ showed poor efficacy with only 27 % protection against severe malaria in infants [8]. The European Medicines Agency approved the use of Mosquirix™ in young African children in July 2015, although final consent from the WHO is still needed. While these vaccines may provide partial protection, their widespread use in Africa may be affected by high costs, as well as compliance and feasibility challenges.
Challenges for the eradication of malaria
Since 2000, malaria infection rates have been reduced by over 50 %, but complete eradication is still a challenge [9]. To completely wipe out malaria, multifaceted strategies are essential. One approach is single exposure radical cure and prophylaxis (SERCaP), proposed as a global agenda in 2007 for identifying ideal drugs to treat malaria [10]. International guidelines have recommended the use of ACTs as a first-line treatment on a three-day course schedule, as ACTs, particularly artesunate, can rapidly reduce the parasitic load by at least 10 000-fold within 48 h of the parasitic life cycle, resulting in >95 % clearance of initial infection. However, subtherapeutic doses and non-adherence are additional barriers promoting the emergence of resistant malarial strains and contributing to treatment failure. Medicines with longer durations of drug action and higher minimum inhibitory concentrations (MICs) in the plasma for at least one week can suppress the parasitic load and offer post-exposure and post-treatment protection. Chemoprotectants are an emerging class of drugs. Sulfadoxine-pyrimethamine is a chemoprotectant combination drug that was widely used among patients until the emergence of resistance last decade [11, 12]. Novel chemoprotective agents acting against the schizont stage of the malaria parasite are needed to prevent merozoite production from blood cells.
In this study, we reviewed various preclinical and clinical studies published during the period directly following the establishment of the Medicines for Malaria Venture (2000) and 2015.
We discuss different preclinical and clinical studies focusing on the evaluation of novel drugs against malaria in different human trials over the past five years registered in the clinicaltrials.gov database [13] (see Table 1). We also address additional approaches to treating malaria with a special focus on safety.
Methods
To summarize the existing evidence related to the portfolio of novel antimalarial drugs, we conducted a systematic review using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Statement [14]. We searched publicly available sources such as PubMed, Web of Science, clinicaltrials.gov, and drug company websites. Original and unoriginal peer-reviewed articles published between 2000 (after the establishment of MMV) and 2015 were retrieved. We included preclinical studies and all clinical trial phases. If trial results were unavailable, we referenced the study either by its clinicaltrial.gov identifier or using the company press release newsletters.
Two of the authors of this paper (ASB and AAE) checked all potentially relevant studies and reached a consensus on all items. One of the authors (ASB) screened the titles and abstracts. Two authors (ASB and AAE) selected the studies for inclusion after reviewing full-text articles. The following keywords were entered into the search field to search for titles, abstracts, and index terms: novel* AND antimalarial* AND preclinical* AND clinical trials* AND plasmodium falciparum* AND plasmodium vivax* AND malaria* AND medicine for malaria venture*. Data were updated in January 2016.
Extracted data were based on information reported in or calculated from the included studies. Authors were not contacted for additional information regarding the drug or trial information. The following information was retrieved from clinicaltrials.gov therapeutic agent, class of drug, site of action, clinical phase, control group, study population, and therapeutic doses. We only examined information on antimalarial drug efficacy and safety.
Pharmacological approaches
Phenotypic molecules for malaria
KAE609
Advances in automation and phenotypic assay screening techniques have aided the discovery of innovative compounds effective against both asexual and sexual stages of P. falciparum. Spiroindolone KAE609 (cipargamin), a potential Na+-ATPase 4 ion channel (PfATP4) inhibitor, was discovered by scientists from the Swiss Tropical and Public Health Institute and developed by the Novartis Institute of Tropical Diseases in Singapore. KAE609 originated from the high-throughput phenotypic screening of nearly 12 000 natural compounds evaluated for their activity against erythrocytic stages of P. falciparum [15]. Encouraging results were reported in a Phase I human trial with elevated MICs in the plasma for several days, and total efficacy doses of 300 mg (single) and 150 mg (multiple) for three days [16, 17]. Interestingly, KAE609 is seven times more potent than artesunate and 40 times more potent than 4-aminoquinolines [18]. Results from a recent Phase II clinical trial conducted among Thai patients indicated a clearance half-life of 0.90 h for P. falciparum and 0.95 h for P. vivax. Furthermore, the mean terminal half-life for the elimination of KAE609 was 20.8 h, supporting a once-daily oral dosing regimen [18]. The promising profile of KAE609 will be evaluated further in upcoming early phase trials. In vitro, KAE609 showed active against artemisinin-resistant K 13 mutant parasite and prevents recrudescence of dihydeoartemisinin (DHA)- arrested ring at minimal concentration (50 nM) [19]. Thus can as a broad range antimalarial and in treatment of multidrug-resistant P.facliparum malaria.
DDD107498
Advances in natural-product-based screening techniques have improved our understanding of medicinal chemistry via chemoinformatics. A high-throughput screening of more than 4 700 compounds resulted in a promising scaffold, which led to the discovery of DDD107498, a novel phenotypic molecule that specifically acts against liver-stage P. falciparum malaria. This molecule was developed at the University of Dundee, UK by a research consortium [20]. The DDD107498 compound is a 2,6-disubstituted quinoline-4-carboxamide scaffold effective against the liver (schizont formation) stage. In vitro assays against different P. falciparum laboratory strains, such as artemisinin-resistant strains, chloroquine-, amodiaquine- and mefloquine-resistant strains, revealed a low micromolar range against the parasite. In addition, the compound impaired the growth of other strains including P. berghei and P. yoelii during their schizont formation stage. DDD107498 may be effective against multidrug resistant Plasmodium strains (Dd2 and 7G8). Remarkably, the ex vivo efficacy of DDD107498 has been shown to be higher than artesunate against P. falciparum (median EC50 = 0.81 nM [range 0.29–3.29 nM]) and P. vivax (median EC50 = 0.51 nM [range 0.25–1.39 nM]) [20]. DDD107498 has shown excellent oral bioavailability and a longer plasma half-life, which is preferable for single-dose treatment in vitro. These results suggest that DDD107498 can achieve complete parasitic clearance in the blood stage by rapid killing for more than 48 h. DDD107498 is currently in the developmental stage and needs to be tested for approval in human clinical trials.
KAF156
KAF156 (also known as imidazolopiperazine), a promising chemoprevention molecule, is a cyclic amine resistance locus inhibitor (PfCARL) developed by the Novartis research consortium [21]. In vitro KAF156 is active against uncomplicated P. falciparum and P. vivax strains in the liver, asexual erythrocytic, and transmission stages. One recently published paper reported a KAF156 Phase II proof-of-concept trial [22] conducted among Vietnamese and Thai monoinfection patients, who were treated with 400 mg/day for three days and a single 800 mg dose. No efficacy data are yet available on KAF156 and no predictions can be made about its potential as a future antimalarial drug. Adverse events reported with higher doses of KAF156 include sinus bradycardia, thrombocytopenia, and hypokalemia. Further studies are needed to evaluate the molecule’s side effects.
DSM265
DSM265, a dihydroorotate dehydrogenase (DHODH) inhibitor acting against the liver (schizont formation) stage, is proving to be promising as a one-dose (400 mg) malaria cure in a Phase I trial in healthy volunteers, with an encouraging safety profile. DSM265 is currently in the clinical developmental stage (Phase II) in Peru (NCT02123290). Its activity against uncomplicated P. falciparum and P. vivax parasites is being assessed in adult patients using a single dose treatment (400 mg) [23]. However, no clinical data are yet available to confirm DSM265 as a potential antimalarial armory. Although DSM265 showed robust results in Phase I trials, further studies are needed to predict its safety for use in pregnant women.
In most countries in SSA, malaria in pregnancy contributes to significant maternal and perinatal mortality. It is not recommended to use ACTs during the first trimester due to side effects observed in preclinical models [24]. Currently, sulfadoxine-pyrimethamine is used in pregnant women as an intermittent preventive treatment to reduce infections and improve pregnancy outcomes. Several optional antibacterials and antifolate combinations have emerged including azithromycin-chloroquine, mefloquine, and dihydroartemisinin-piperaquine. Antibacterial combinations potentially reduce the risk of sexually transmitted diseases to mothers and newborns [25]. Studies registered in clinicaltrials.gov on co-trimoxazole prophylaxis for prevention of malaria in pregnancy (NCT01053325) and co-infection with malaria and HIV in women (NCT00970879) were completed in 2013, but results have not yet been published. Moreover, mefloquine has shown significant benefits but may cause nausea and neuropsychiatric side effects [26]. To fulfill the GMAP portfolio, determining the safety of novel chemoprotective molecules in pregnancy should be considered a priority in clinical investigations.
Other compounds under development
Several molecules are currently being tested in preclinical models. Examples include SJ557733, developed in collaboration between St. Jude Children’s Research Hospital, TN, USA and Rutgers University, NJ, USA [27], and PA21A092 developed at Drexel University, PA, USA [28]. Both molecules target the PfATP4 of multiple Plasmodium species at different stages of infection. Another similar phenotypic molecule known as MMV390048, developed by researchers at the University of Cape Town in South Africa, targets lipid phosphatidyl inositol 4-kinase (PfPI4K) [29]. The MMV390048 research group has completed a Phase I trial on healthy African volunteers for the first time, but results have not yet been published (registered in clinicaltrials.gov; NCT02230579). Although additional novel phenotypic molecules are currently being clinically tested against malaria (see Table 2), more studies are needed to elucidate their clinical effectiveness and safety. Genetic polymorphism in pfcrt is associated with chloroquine resistance. Additional polymorphisms (dhfr and dhps) for sulfadoxine-pyrimethamine and polymorphism of P. falciparum multidrug resistance protein 1 (pfmdr1) are associated with resistance to chloroquine, mefloquine, quinine, and artemisinin [30]. Novel loci such as encoding the mu chain of the adopter protein 2 (ap2-mu), P. falciparum ap2-mu (Pfap2-mu) homologue [27], gene mutations encoding pfmdr1, and sarco-endoplasmic reticulum calcium ATPase6 (PfSERCA) [31] may be associated with antimalarial resistance. Emerging evidence shows that pfmdr 1, pfcrt, and pf3d7-1343700 Kelch propeller (K13-propeller) mutations are potential markers indicating that P. falciparum is developing resistance to artemisinin and its derivatives [32, 33].
Synthetic medicinal arsenals
OZ277 and OZ439
Quinine, first used in Europe in the 17th century, chloroquine [34], and 4-aminoquinoline scaffolds are some of the semi-synthetic drugs that have shown good antimalarial activity over the years. Fixed-dose combinations of artemisinin derivatives are currently considered to be the gold-standard malaria treatment. Synthetic artemisinin-like endoperoxides and their derivatives (artesunate, artemether, and dihydroartemisinin) have been proven to be more effective than chloroquine. OZ277 (arterolane), a novel non-artemisinin ozonide compound, has been developed by Ranbaxy Laboratories in collaboration with MMV in 2004. The clinical activity of OZ277 in a Phase II dose-finding trial for uncomplicated P. falciparum malaria was shown to be not as effective as artemisinin. This was indicated by the reduced parasitic clearance on day 28 after seven days (60–70 %) compared to artesunate dose–response (95 %) [34]. Thus, increasing the dose does not necessarily decrease parasitic recrudescence. Following a Phase III trial in 2013, a fixed-dose combination of OZ277 (arterolane) (150 mg) and long-acting piperaquine (750 mg) (Synriam™) was tested for treating P. falciparum malaria in India and received mark approval from the Drug Controller General of India. It was subsequently put on the market in seven African countries [35]. Due to a suboptimal half-life and reduced stability in low-level parasitemia (1 % at 45 % haematocrit), OZ277 failed to show efficacy against high parasitic loads [36]. The results of these trials confirmed the safety of the compounds but not the efficacy, even when doses were increased in the presence of a high level of infected erythrocytes. Drug partners may be needed to increase efficacy. These weaknesses have led MMV to develop a potential next generation synthetic endoperoxide ozonide, OZ439 (artefenomel), which has a longer half-life (30 h) and a MIC of more than one week, after a single dose. OZ439 is the first highly active ozonide against Plasmodium [36].
Different doses of artefenomel (200–1 200 mg) were tested in a Phase IIA exploratory, open-label trial and revealed promising safety and efficacy profiles among Southeast Asian adults with uncomplicated P. falciparum and P. vivax malaria. Due to the reduced elimination half-life of 46–62 h, a single dose of OZ439 alone or in combination with piperaquine can eliminate 98.0 % of P. falciparum and 99.6 % of P. vivax within 36 h. Artefenomel has demonstrated a higher parasitic clearance within the first 24 h in P. vivax patients as compared to P. falciparum patients (30–36 h). However, gametocyte clearance was 100 % in patients who were administered 1 200 mg of artefenomel within 48 h [36]. OZ439 is now being evaluated with piperaquine in Phase IIB combination trials.
One of the major concerns about the use of OZ compounds is that they have a similar endoperoxide structure to artemisinin, indicating possible treatment failures. Previous data suggest that artemisinin derivatives are associated with the risk of spontaneous abortions in early pregnancies [37], but recent clinical evidence confirmed the safety of ACTs against P. falciparum and P. vivax in the first trimester, with no risk of spontaneous abortions or major congenital malformations [38]. Similarly, preclinical studies have shown that OZ compounds are also safe for embryos and fetuses [39]. No clinical data are yet available to prove the safety of using these compounds in pregnancy and more tests are therefore needed for their evaluation.
Other compounds
Two interesting endoperoxides from artesunate derivatives including artemisone (BAY 44–9585) and tetraxoane (TDD E209), are examples of other synthetic candidates currently under development. Artemisone is a semi-synthetic second-generation artemisinin derivative developed in collaboration between Bayer HealthCare Pharmaceuticals in Germany and the Hong Kong University of Science and Technology. Results of preclinical studies are highly promising as compared to other novel artemisinins. Artemisone is more effective than artesunate against P. falciparum and multidrug resistant strains [40, 41]. Dose-escalating Phase I trials on healthy volunteers have shown that artemisone is a rapidly effective treatment as it achieves peak plasma concentrations within 30 min following oral administration [41]. A Phase II interventional study testing artemisone for treating uncomplicated P. falciparum malaria planned for Western Cambodia (NCT00936767) has been withdrawn for unknown reasons. Some studies reported neurological and auditory side effects such as ataxia and slurred speech [42, 43] due to ACTs. However, no strong evidence exists to confirm neurological side effects. Furthermore, the activity of artemisone has shown a GM IC50s correlation with pfmdr1 Y184F mutations, which potentially reduces sensitivity to artemisinin-resistant strains and contributes to emerging ACT resistance [44]. Recent genome-wide association studies revealed that artemisone does not interact with Y1915 and has no effect on P.falciparum phosphatidylinositol-3 kinase (PfPI3K) [45].
Aminoquinoline scaffolds
Ferroquine
Ferroquine is an ameliorated blood-schizonticidal 4-aminoquinoline developed by Sanofi-Aventis. Along with OZ439, it is a more effective parasite-killing compound against Plasmodium strains when compared to artesunate. Several preclinical studies have shown its benefits, particularly for treating patients infected with chloroquine-, amodiaquine-, and mefloquine-resistant malaria strains [46–48]. The greatest advantage of using ferroquine is its 30-h half-life, which is highly superior to that of other artemisinin derivatives. Two ferroquine Phase II trials were recently registered in the clinicaltrials.gov database (NCT02497612 and NCT00988507) focusing on P. falciparum and P. vivax malaria at the multicenter level. One study has been completed (NCT00988507) but no results are yet available. Most recently, a ferroquine-artesunate dose-ranging Phase II trial on P. falciparum-infected adults and children in eight African hospitals was conducted [49]. The research findings were astonishing: 97 % polymerase chain reaction (PCR)-confirmed cure rates (95 % CI: 90–100) after treatment with 2 mg/kg ferroquine combined with 4 mg/kg artesunate. However, the cure rate was reduced (79 %; 95%CI: 68–88) when ferroquine monotherapy 4 mg/kg/day for 3-days regimen was used. Furthermore, exacerbated malaria symptoms were observed in 14 % of the individuals in the treatment cohort.
AQ-13
Another 4-aminoquinoline derivative called AQ-13 (Ro47-0543), a similarly structured chloroquine with a modified propyl side chain from the aminoquine panel, was developed in collaboration between Tulane University and Louisiana State University both located in LA, USA. Preclinical studies have indicated increased efficacy of AQ-13 when compared to other derivatives [50]. Phase I first-in-human safety and efficacy studies have shown results similar to those observed with chloroquine. Adverse events include electrocardiac changes, especially prolonged QTc intervals, which are commonly encountered with many quinolones [51]. AQ-13 did not present any advantages over other aminoquinolines, and further observation of this compound has currently been halted.
Tafenoquine
The majority of clinical trials focus on malaria caused by P. falciparum, while fewer studies evaluate treatments against P. vivax and P. ovale malaria. A deficiency of glucose-6-phosphate dehydrogenase (G6PD) is a hereditary enzyme defect condition that causes episodic hemolysis. Patients with a G6PD deficiency are common in malaria-endemic countries and are at high risk of hemolysis due to treatment with antimalarial drugs (primaquine, chloroquine, quinine, and sulfamethoxazole). These patients are generally not included in trials due genotypic variations. For these individuals, tafenoquine (WR 238605) is a good alternative drug. It is an 8-aminoquinoline derivative and has a similar mode of action to primaquine against hypnozoites, gametocytes, and liver stages [52]. Tafenoquine is more potent during blood stages due its longer half-life (14 days) as compared to primaquine. Nevertheless, slower parasitic clearance was observed with tafenoquine monotherapy. Therefore, combing tafenoquine with other partner drugs may ideally benefit G6PD-deficient patients. So far, chloroquine combined with primaquine has been used for the radical cure of P. vivax malaria. Tafenoquine with chloroquine was tested in studies against P. vivax malaria. In a Phase IIB dose-ranging trial, different doses of tafenoquine alone (50, 100, 300, or 600 mg) or in combination with 15 mg primaquine for 14 days were tested, with a fixed dose of chloroquine for three days. A single dose of tafenoquine (300 mg) co-administered with chloroquine was shown to prevent relapse in 89.2 % (95 % CI: 77–95) of people as compared to chloroquine alone (51.7 %; 95 % CI: 36–69) during the first six months of follow-up [53]. Recent results of a Phase IIB dose-ranging trial (DETECTIVE study) conducted on monoinfected P. vivax patients for radical cure showed that single-dose tafenoquine (300 mg) combined to chloroquine is more efficacious in preventing relapses as compared to chloroquine alone, with a similar safety profile. Based on these observations, GSK and MMV announced two new Phase III studies: 1) a DETECTIVE study (TAF112582) to evaluate the efficacy, safety, and tolerability of tafenoquine co-administered with chloroquine as a radical cure for P. vivax malaria (blood-stage antimalarial treatment); and 2) a GATHER study (TAF 116546) to assess the incidence of hemolysis and the efficacy and safety of tafenoquine over primaquine [54].
Biomolecular approaches
Methylene blue
A century ago, the German scientist Paul Ehrlich discovered the antiplasmodial activity of methylene blue [55]. The chemotherapeutic use of synthetic methylene blue in treating methemoglobinemia and cancer-induced neurotoxicity was tested in 1995 [56]. Additional experiments were conducted using methylene blue and its analogs against P. falciparum isolates [57]. Methylene blue combined with chloroquine has been shown to prevent hemolysis in G6PD-deficient adult patients. Other studies assessed the use of different doses of methylene blue with chloroquine for three days and showed 90 % recovery rates in patients with uncomplicated P. falciparum malaria. Although results were promising, adverse effects were reported including vomiting, as well as the discoloration of urine, mucous surfaces, and teeth [58]. Drug resistance to chloroquine has also emerged globally [59]. In 2006, methylene blue was evaluated in combination with artesunate but showed poor cure rates despite rapid parasitic clearance [60]. In 2011, treatment with artesunate-amodiaquine-methylene blue was studied in children aged between six and 50 months with uncomplicated P. falciparum malaria. This combination showed poor efficacy (71 %) when compared to the control group (artesunate-amodiaquine; 85 %) [61]. However, after comparing a fixed dose 15 mg/kg of methylene blue co-administered with artesunate or amodiaquine versus artesunate-amodiaquine for three days, decreased gametocytes (from 100 to 36 %) were reported within seven days of treatment. Interestingly, the pronounced effect on gametocyte clearance indicates that methylene blue is a new promising drug component to reduce P. falciparum transmission. A Phase I trial testing the combination of methylene blue with primaquine is currently registered in the clinicaltrials.gov database (NCT01668433), but results are not yet available.
Antibiotics
Fosmidomycin
Isoprenoids are derived from the mevalonate pathway in humans, an essential metabolic pathway for parasite synthesis. Jomaa Pharma GmbH developed a synthetic antibiotic agent called fosmidomycin derived from Streptomyces lavendulae bacterial isolates. This compound inhibits the non-mevalonate pathway (also known as the DOXP pathway), essential for the synthesis of parasite isoprenoids [62]. Fosmidomycin has a half-life of only two hours and acts rapidly upon oral administration. Additional trials to evaluate the efficacy of different doses of fosmidomycin monotherapy undertaken for more than four days are required. One study indicated complete parasitic clearance on day seven following administration of fosmidomycin (1 200 mg four times a day) in adult patients with uncomplicated P. falciparum malaria. On day 28, recrudescence was observed in seven out of nine patients, indicating monotherapy failure [63]. Fosmidomycin co-administered with clindamycin has been proven to be effective in adults and older children with acute uncomplicated P. falciparum malaria. Poor efficacy was observed due to poor immunity in children aged between one an>d two years [64]. Two additional short half-life combinations (fosmidomycin with artesunate) were evaluated in 50 children aged between six and 12 years. Five different fosmidomycin-artesunate regimens achieved complete cure rates within three days of administration, and no resistant alleles were detected after seven and 28 days [65]. However, no evidence of prolonged protection by this combination was provided. A Phase IIA open-label efficacy trial focusing on fosmidomycin (450 mg capsule; twice daily) and piperaquine (320 mg; once daily) for treating patients with uncomplicated P. falciparum malaria, aged between one and 60 years and with a body weight between 5 and 90 kg, is currently registered in the clinicaltrials.gov database (NCT02198807). Overall, studies indicated that fosmidomycin is only effective for short-term treatment. Studies on finding a potential partner drug to prove the efficiency of fosmidomycin urgently need to be conducted.
Conclusions
In this review, we summarized the different approaches tested over the years to control the malaria pandemic, and possibly reduce global malaria incidence and mortality by 90 % before 2030. Novel chemotherapeutic approaches have emerged over the past five years, with promising results. Nevertheless, the efficacy and safety of these drugs need to be studied further. These novel antimalarial approaches are multifaceted, thus there is an urgent need for effective single-dose molecules to act during the liver and blood stages of malaria. Effective compounds should be developed before global emergence of resistance to artemisinin derivatives and 4-aminoquinoline. There is currently no low-dose primaquine regimen for pediatric use. Novel blood-stage compounds such as DDD107498 and tafenoquine should focus on blocking parasite transmission in children and adolescents, and pregnant women. Molecules such as ferroquine should be combined with a potential partner drug to enhance efficacy. Additional challenges in preventing the relapse of malaria episodes include hemolysis in patients with a G6PD deficiency, treatment for drug-resistant strains, pediatric dosing, serious drug-drug interactions, transmission blocking, radical cure, and relapse prevention. Potentially targeting mitochondrial electron-transport chain of P.falciparum and protein inhibition in blood- and liver-stage parasites could be ideal for future drug development.
Abbreviations
- ACT:
-
Artemisinin-based combination therapy
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GMAP:
-
Global Malaria Action Plan
- GSK:
-
GlaxoSmithKline
- MIC:
-
Minimum inhibitory concentration
- MMV:
-
Medicines for malaria venture
- PfATP4:
-
Na+-ATPase 4 ion channel
- SSA:
-
Sub-Saharan Africa
- WHO:
-
World Health Organization
References
World Health Organization. 10 facts on malaria. Available at: http://www.who.int/features/factfiles/malaria/en.html. Accessed 9 Feb. 2016.
Nosten F, Brasseur P. Combination therapy for malaria. Drugs. 2002;62(9):1315–29.
Medicines for Malaria venture. Available information: http://www.mmv.org/about-us/our-history.html. Accessed 9 Feb. 2016.
World Health Organization: Malaria fact sheet. Available at http://www.who.int/mediacentre/factsheets/fs094/en.html. Accessed 28 Jan. 2016
Biamonte MA, Wanner J, Le Roch KG. Recent advances in malaria drug discovery. Bioorg Med Chem Lett. 2013;23:2829–43.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Taming T, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67.
Clinical trials.gov. Available at https://clinicaltrials.gov/ct2/results?term=Phase+3+malaria+vaccine&Search=Search.html. Accessed 10 June 2015
RTS,S Clinical Trials Partnership. Efficacy and safety of the RTS,S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS Med. 2014;11:e1001685.
Alonso PL, Brown G, Arevalo-Herrera M, Binka F, Chitnis C, Collins F, et al. A research agenda to underpin malaria eradication. PLoS Med. 2011;8:e1000406.
Ashley EA, Dhorda RM, Fairhurst C, Amaratunga P, Lim S, Suon S, et al. Spreading of artemisinin resistance in Plasmodium falciparum malaria. N Eng J Med. 2014;371(5):411–23.
Vinayak S, Alam MT, Mixaon-Hayden T, McCollum AM, Sem R, Shah NK, et al. Origin and evolution of sulfadoxine resistant Plasmodium falciparum. PLoS Pathog. 2010;6(3):e1000830.
McCollum AM, Mueller K, Villegas L, Udhyakumar V, Escalante AA. Common origin and fixation of Plasmodium falciparum and dhps mutations associated with sulphadoxine-pyramethamine resistance in a low-transmission area in South America. Antimicrob Agents Chemother. 2007;51(6):2085–91.
Clinical trials.gov. Available at https://clinicaltrials.gov/ct2/results?term=novel+antimalarial+drug&Search=Search.html Accessed 22 June 2015.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097.
Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329(5996):1175–80.
Leong FJ, Li R, Jain JP, Lefevre G, Magnusson B, Diagana TT, et al. A first-in-human randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study of novel antimalarial spiroindolone KAE609 (cipargamin), to assess the safety, tolerability and pharmacokinetics in healthy adult volunteers. Antimicrob Agents Chemother. 2014;58:6209–14.
Stein DS, Jain JP, Kangas M, Lefevre G, Machineni S, Griffin P, et al. Open-label, parallel-group study in healthy volunteers to determine in the safety.in healthy volunteers to determine the drug-drug interactions potential between KAE609 (Cipargamin) and piperaquine. Antimicrob Agents Chemother. 2015;59(6):3493–500.
Nicholus JW, Sasithon P, Phyo AP, Reuangweerayut R, Nosten F, Podjanee J, et al. Spiroindolone KAE609 for falciparum and vivax malaria. N Eng J Med. 2014;371:403–10.
Chavchich M, Van Breda K, Rowcliffe K, Diagana TT, Edstein MD. The spiroindolone KAE609 does not induce dormant ring stage in Plasmodium falciparum parasites. Antimicrob Agents Chemother. 2016;60(9):5167–74.
Baragana B, Hallyburton I, Lee MC, Norcross NR, Raffaella G, Otto TD, et al. A novel multiple-stage antimalarial agent than inhibits protein synthesis. Nature. 2015;522:315–20.
Vaidya AB, Morrisey JM, Zhang Z, Das S, Daly TM, Otto TD, et al. Pyrazoleamide compounds are potent antimalarials that target Na + homeostasis in intraerythrocytic Plasmodium falciparum. Nature Commun. 2014;5:5521.
Novartis Pharmaceuticals. Efficacy, safety, tolerability and pharmacokinetics of KAF156 in adult patients with acute, uncomplicated Plasmodium falciparum or vivax malaria mono-infection. In:ClinicalTrials.gov. Bethesda (MD). [Cited 2015 Nov 19] Available from: http://clinicaltrial.gov/show/NCT01753323.
Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Translat Med. 2015;7(269):296ra111.
Beckman DA, Youreneff M, Butt MT. Neurotoxicity assessment of artemether in juvenile rats. Birth Defects Res B Dev Reprod Toxicol. 2013;98(2):183–99.
Luntamo M, Rantala AM, Meshnick SR, Cheung YB, Kulmala T, Maleta K, et al. The effect of monthly sulfadoxine-pyrimethamine, alone or with azithromycin, on PCR-diagnosed malaria at delivery: a randomized controlled trial. PLoS One. 2012;7(7):e41123.
Tansley R, Lotharius J, Priestley A, Bull F, Duparc S, Mohrle J. A randomized, double-blind, placebo-controlled study to investigate the safety, tolerability, and pharmacokinetics of single enantiomer (+)-mefloquine compared with racemic mefloquine in healthy persons. Am J Trop Med Hyg. 2010;83:1195–201.
Jimenez-Diaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre ME, et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci U S A. 2014;111:E5455–62.
McNamara CW, Lee MC, Lim CS, Lim SH, Roland K, Nagle A, et al. “Targeting Plasmodium PI(4) K to eliminate malaria. Nature. 2013;504(7479):248–53.
Younis Y, Douelle F, Feng TS, Gonzales Cabrera D, Le Manach C, Nichinda AT, et al. 3,5-Diaryl-2-aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. J Med Chem. 2012;55(7):3479–87.
Ariey F, Watkowski C, Amaratunga J, Beghain AC, Langois N, Khim S, et al. A molecular-marker of artesmisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Henriques G, van Schalkwyk DA, Burrow R, Warhurst DC, Thompson E, Baker DA, et al. The Mu subunit of Plasmodium falciparum clathrin-associated adaptor protein 2 modulates in vitro parasite response to artesmisinin and quinine. Antimicrob Agents Chemother. 2015;59(5):2540–7.
Li J, Chen J, Xie D, Monte-Nguba SM, Eyi JU, Matesa RA, et al. High prevalence of pfmdr1 N86Y and Y184F mutations in Plasmodium falciparum isolates from Bioko Island, Equatorial Guinea. Pathog Glob Health. 2014;108:339–43.
St Laurent B, Miller B, Burton TA, Amaratunga C, Men S, Sovannaroth S, et al. Artemisinin-resistant Plasmodium falciparum clinical isolates can infect diverse mosquitoes vectors of Southeast Asia and Africa. Nat Commun. 2015;6:8614.
Valecha N, Krudsood S, Tangpukdee N, Mohanty S, Sharma SK, Tyagi PK, et al. Arterolane maleate plus piperaquine phosphate for treatment of uncomplicated Plasmodium falciparum malaria: a comparative, multicenter, randomized clinical trial. Clin Infect Dis. 2012;55(5):663–71. Patil CY, Katara SS, Baig MS, Doifode SM. Fixed dose combination of arterolane and piperaquine: A newer prospect in antimalarial therapy. Ann Med Health Sci Res 2014;4(4):466–471.
Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, et al. Indentification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430(7002):900–4.
Phyo AP, Jittamala P, Nosten FH, Pukrittayakamee S, Imwong M, White NJ, et al. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: a open-lable phase 2 trial. Lancet Infect Dis. 2016;16:61–9.
Ritter S, Laffan SB, Clark RL. Artesunate-induced depletion of embryonic erythroblasts precedes embryolethality and teratogenicity in vivo. Birth Defects Res B Dev Reprod Toxicol. 2006;77:413–29.
Moore KA, Simpson JA, Paw MK, Pimanpanarak MP, Wiladphaingern J, Rijken MJ, et al. Safety of artemisinins in first trimester of prospectively followed pregnancies: an observational study. Lancet Infect Dis. 2016. doi:10.1016/S1473-3099(15)00547-2.
Wells TNC, van Huijsduijnen RH, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015;14(6):424–42.
Vivas L, Rattray L, Stewart LB, Robinson BL, Fugmann B, Haynes RK, et al. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J Antimicrob Chemother. 2007;59(4):658–65.
Nagelschmitz J, Voith B, Wensing G, Roemer A, Fugmann B, Haynes RK, et al. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone. Antimicrob Agents Chemother. 2008;52(9):3085–91.
Medhi B, Patyar S, Rao RS, Bayrav DSP, Praksh A. Pharmacokinetic and toxicological profile of artemisinin compounds: an update. Pharmacol. 2009;84(6):323–32.
Carrasquilla G, Baron C, Monsell EM, Cousin M, Walter V, Lefevre G, et al. Randomized, prospective, three-arm study to confirm the auditory safety and efficacy of artemether-lumefantrine in Colombian patients with uncomplicated Plasmodium falciparum malaria. Am J Trop Med Hyg. 2012;86(1):75–83.
Lanteri CA, Chaorattanakawee S, Lon C, Saunders DL, Rutvisuttinunt W, Yingyuen K, et al. Ex Vivo activity of endoperoxide antimalarials, including artemisone and arteolane, against multidrug-resistant Plasmodium falciparum isolates from Cambodia. Antimicrob Agents Chemother. 2014;58(10):5831–40.
Mbengye A, Bhattacharjee S, Pandhakar T, Liu H, Estiu G, Stahelin RV, et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature. 2015;520(7549):628–30.
Mombo-Ngoma G, Sapan C, Dal-Bianco MP, Missinou MA, Matsiegui PB, Ospina Salazar CL, et al. Phase I randomized dose-ascending placebo-controlled trials of ferroquine—a candidate anti-malarial drug-in adults with asymptomatic Plasmodium falciparum infection. Malar J. 2011;10:53.
Marfurt K, Chalfein F, Prayoga P, Wabiser F, Kenangalem E, Piera KA, et al. Ex vivo drug susceptibility of ferroquine against chloroquine-resistant isolates of Plasmodium falciparum and P.vivax. Antimicrob Agents Chemother. 2011;55(9):4461–4.
Marfurt K, Chalfein F, Prayoga P, Wabiser F, Wirjanata G, Sebayang B, et al. Comparative ex vivo activity of novel endoperoxides in multidrug-resistant Plasmodium falciparum and P.vivax. Antimicrob Agents Chemother. 2012;56(10):5258–63.
Held J, Supan C, Salazar CL, Tinto H, Bonkian LN, Nahum A. Ferroquine and artesunate in African adults and children with Plasmodium falciparum malaria: a phase 2, multicentre, randomized, double-blind, dose-ranging, non-inferiority study. Lancet Infect Dis. 2015;15(12):1409–19.
Ramanathan-Girish S, Catz P, Creek MR, Wu B, Thomas D, et al. Pharmacokinetics of the antimalarial drug, AQ-13, in rats and cynomolgous macaques. Int J Toxicol. 2004;23:179–89.
Mzayek F, Deng H, Mather JF, Wasilevich EC, Liu H, Hadi CM, et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin Trials. 2007;2(1):e6.
Marcsisin SR, Sousa JC, Reichard GA, Caridha D, Zeng Q, Careaganarja J, et al. Tafenoquine and NPC-1161B requires CYP2D metabolism for anti-malarial activity: implications for the 8-aminoquinoline class of anti-malarial compound. Malaria J. 2014;13(2):10–1186.
Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, Krudsood S, Gupta SK, Kochar SK. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet. 2014;383(9922):1049–58.
GSK and MMV announce start of Phase III programme of tafenoquine for Plasmodium vivax malaria. Available at: http://us.gsk.com/en-us/media/press-release/2014/gsk-and-mmv-announce-start-of-phase-iii-programme-of-tafenoquine-for-plasmodium-vivax-malaria.html. Accessed: 11 Jul 2015.
Ehrlich P, Guttmann P. Ueber die Wirkung des Methylenblaubei Malaria. BerlKlinWochenschr. 1891;28:953–6.
Atamna H, Krugliak M, Shalmiev G, Deharo E, Pescarmona G, Ginsburg H. Mode of antimalarial effect of methylene blue and some of its analogues on Plasmodium falciparum in culture and their inhibition of P. vinckeipetteri and P. yoeliinigeriensis in vivo. Biochem Pharmacol. 1996;51(5):693–700.
Bountogo M, Zoungrana A, Coulibaly B, Klose C, Mansmann U, Mockenhaupt FP, et al. Efficacy of methylene blue monotherapy in semi-immune adults with uncomplicated falciparum malaria: a controlled trial in Burkina Faso. Trop Med Int Health. 2010;15(6):713–7.
Rengelshausen J, Burhenne J, Frohlich M, Tayrouz Y, Singh SK, Riedel KD, et al. Pharmacokinetic interaction of chloroquine and methylene blue combination against malaria. Eur J Clin Pharmacol. 2004;60(10):709–15.
Takala-Harrison S, Laufer MK. Antimalarial drug resistance in Africa: key lessons for the future. Ann N Y Acad Sci. 2015;1342:62–7.
Zoungrana A, Coulibaly B, Sie A, Walter-Sack I, Mockenhaupt FP, Kouyate B, et al. Safety and efficacy of methylene blue combined with artesunate or amodiaquine for uncomplicated falciparum malaria: a randomized controlled trial from Burkina Faso. PLoS One. 2008;3(2):e1630.
Coulibaly B, Pritsch M, Bountogo M, Meissner PE, Nebie E, Klose C, et al. Efficacy and safety of triple combination therapy with artesunate-amodiaquine-methylene blue for falciparum malaria in children: a randomised controlled trial in Burkina Faso. J Infect Dis. 2015;211(5):689–97.
Oyakhirome S, Issifou S, Pongratz P, BorondiF RM, Kun JF, et al. Molecular basis of fosmidomycin’s action on the human malaria parasite Plasmodium falciparum. Sci Res. 2011;1:1–8.
Lee B, Ruangweerayut R, Wiesner J, Missinou MA, Schindler A, Baranek T, et al. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob Agents Chemother. 2003;47(2):735–8.
Ruangweerayut R, Looareesuwan S, Hutchinson D, Chauemung A, Banmairuroi V, Na-Bangchang K. Assessment of the pharmacokinetics and dynamics of two combination regimens of fosmidomycin clindamycin in patients with acute uncomplicated falciparum malaria. Malar J. 2008;7:225.
Borrmann S, Adegnika AA, Moussavou F, Oyakhirome S, Esser G, Matseiegui P, et al. Short-course regimens of artesunate-fosmidomycin in treatment of uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2005;49(9):3749–54.
Abbas S, Jain V, Bhartam PV. Origin of the specificity of inhibitor P218 towards wild-type and mutant PfDHFR: a molecular dynamics analysis. J Biomol Struct Dyn. 2014;17:1–16.
Corteron JM, Marco M, Esquivias J, Deng X, White KL, White J, et al. Structure-guided lead optimization of triazolopyrimidine-ring substituents identified potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitor with clinical candidate potential. J Med Chem. 2011;54(15):5540–61.
Kumar S, Kumari R, Pandey R. New insight-guided approaches to detect, cure, prevent and eliminate malaria. Protoplasma. 2015;252(2):717–53.
Kuhen KL, Chatterjee AK, Rottmann M, Gagaring K, Borboa R, Buenviaje J, et al. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58(9):5060–7.
Miley GP, Pou S, Winter R, Nilsen A, Li Y, Kelly JX, et al. ELQ-300 prodrug for enhanced development and single dose cure of malaria. Antimicrob Agents Chemother. 2015. doi:10.1128/ACC.01183-15.
Antoine T, Fisher N, Amewu R, O’Neill PM, Ward SA, Biagini GA. Rapid kill of malaria parasites by artemisin and semi-synthetic endoperoxides involves ROS-dependent polorization of the membrane potential. J Antimicrob Chemother. 2014;69(4):1005–16.
Cassera MB, Hazleton KZ, Merino EF, Obaldia 3rd N, Ho MC, Murkin AS, et al. Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in primate animal model. PLoS One.2011;6(11):e26916.
Dutta AK, Avery BA, Wyandt CM. Development and validation of a stability-indicating reversed-phase high performance method for NPC1161C, a novel 8-aminoquinoline anti-malarial drug. J Chromatogr A. 2006;1110(1–2):35–45.
Sumanadasa SD, Goodman CD, Lucke AJ, Skinner-Adams T, Sahama I, Haque A, et al. Antimalarial activity of the anticancer histone deacetylase inhibitor SB939. Antimicrob Agents Chemother. 2012;56(7):3849–56.
Somanadhan B, Kotturi SR, Yan Leong C, Glover RP, Huang Y, Flotow H, et al. Isolation and synthesis of falcitidin, a novel myxobacterial-derived acylretrapeptide with activity against the malaria target falcipain-2. J Antibiot (Tokyo). 2013;66(5):259–64.
Bueno JM, Herreros E, Angulo-Barturer I, Ferrer S, Fiandor JM, Gamo FJ, et al. Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochromebc1. Future Med Chem. 2012;4(18):2311–23.
Trenholme K, Marek L, Duffy S, Pradel G, Fisher G, Hansen FK, et al. Lysine acetylation in sexual stage malarial parasites is a target for antimalarial small molecules. Antimicrob Agents Chemother. 2014;58(7):3666–78.
Jaudzems K, Tars K, Maurops G, Ivdra N, Otikovs M, Leitans J, et al. Plasmepsin inhibitory activity and structure-guided optimization of a potent hydroxyethylamine-based antimalarial hit. ACS Med Chem Lett. 2014;5(4):373–7.
Working together to fight malaria with new medicines: Development of an active Toll-like receptor (TLR) 9 antagonist, Access to medicines: Eisai. Available at http://www.eisai.com/company/atm/activities/04.html Accessed on 12 Feb 2016
Barker Jr RH, Urgaonkar S, Mazitschek R, Celatka C, Skerlj R, Cortese JF, et al. Aminoindoles, a novel scaffold with potent activity against Plasmodium falciparum. Antimicrob Agents Chemother. 2011;55:2612–22.
Booker ML, Bastos CM, Kramer ML, Barker Jr RH, Skerlj R, Sidhu AB, et al. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J Biol Chem. 2010;285:33054–64.
O’Neill PM, Amewu RK, Nixon GL, Bousejra ElGarah F, Mungthin M, Chadwick J, et al. Identification of a 1,2,4,5-tetraoxane antimalarial drug-development candidate (RKA 182) with superior properties to the semisynthetic artemisinins. Angew Chem Int Ed Engl. 2010;49:5693–97.
Acknowledgements
We thank Dr. Damiana Chiavolini for editing the paper.
Funding
None.
Authors’ contributions
ASB, AAE and AS conceived and designed the study, and drafted the paper. AAE analysed the literature and reviewed the manuscript. All authors read and approved the final paper.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Multilingual asbtracts in the five official working languages of the United Nations. (PDF 561 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bhagavathula, A.S., Elnour, A.A. & Shehab, A. Alternatives to currently used antimalarial drugs: in search of a magic bullet. Infect Dis Poverty 5, 103 (2016). https://doi.org/10.1186/s40249-016-0196-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40249-016-0196-8