
Abstract
Background
Coronaviruses (CoV) are a large family of viruses that are common in humans and many animal species. Animal coronaviruses rarely infect humans with the exceptions of the Middle East respiratory syndrome (MERS-CoV), the severe acute respiratory syndrome corona virus (SARS-CoV), and now SARS-CoV-2, which is the cause of the ongoing pandemic of coronavirus disease 2019 (COVID-19). Several studies suggested that genetic variants in the ACE2 gene may influence the host susceptibility or resistance to SARS-CoV-2 infection according to the functional role of ACE2 in human pathophysiology. However, many of these studies have been conducted in silico based on epidemiological and population data. We therefore investigated the occurrence of ACE2 variants in a cohort of 131 Italian unrelated individuals clinically diagnosed with COVID-19 and in an Italian control population, to evaluate a possible allelic association with COVID-19, by direct DNA analysis.
Methods
As a pilot study, we analyzed, by whole-exome sequencing, genetic variants of ACE2 gene in 131 DNA samples of COVID-19 patients hospitalized at Tor Vergata University Hospital and at Bambino Gesù Children’s Hospital, Rome. We used a large control group consisting of 1000 individuals (500 males and 500 females).
Results
We identified three different germline variants: one intronic c.439+4G>A and two missense c.1888G>C p.(Asp630His) and c.2158A>G p.(Asn720Asp) in a total of 131 patients with a similar frequency in male and female. Thus far, only the c.1888G>C p.(Asp630His) variant shows a statistically different frequency compared to the ethnically matched populations. Therefore, further studies are needed in larger cohorts, since it was found only in one heterozygous COVID-19 patient.
Conclusions
Our results suggest that there is no strong evidence, in our cohort, of consistent association of ACE2 variants with COVID-19 severity. We might speculate that rare susceptibility/resistant alleles could be located in the non-coding regions of the ACE2 gene, known to play a role in regulation of the gene activity.
Similar content being viewed by others


Introduction
Since the end of last year, in December 2019, Chinese authorities have reported several cases of pneumonia in Wuhan City, Hubei Province of China [1]. A novel betacoronavirus was identified as the causative agent of the viral acute respiratory human distress [2, 3]. Afterwards, the disease was named “Coronavirus Disease 2019 (COVID-19)” by the World Health Organization (WHO) [4].
Coronaviruses (CoV) are a large family of viruses that are common in humans and other animal species, including bats [5], camels, cattle, and cats. Animal coronaviruses rarely infect humans and then spread between subjects with the exceptions of the Middle East respiratory syndrome (MERS-CoV), the severe acute respiratory syndrome coronavirus (SARS-CoV), and now SARS-CoV-2, which is the cause of the ongoing pandemic [6]. A critical step for a viral infection is receptor recognition and binding to the host-cell surface. The angiotensin-converting enzyme 2 (ACE2) has been identified as a functional receptor for SARS-CoV-2, allowing host-cell entry [7]. SARS-CoV-2 uses an extensively glycosylated spike (S) protein that protrudes from the viral envelope and mediates the binding to ACE2 [5], the carboxypeptidase that catalyzes the hydrolysis of angiotensin II to angiotensin (1-7) [8]. The S protein is a 1273 amino acid (aa) long-structural glycoprotein located on the outer envelope of the virus. It has two functional subunits: an N-terminal S1 subunit and a shorter C-terminal S2 subunit. ACE2 is a single-pass type I membrane protein (805 aa) and it contains an N-terminal peptidase M2 domain and a C-terminal collectrin domain. The binding affinity of the ACE2 receptor-binding domain (RBD) to the C-terminal domain of S1 subunit of the SARS-CoV-2 S protein is 10- to 20-fold higher than that of SARS-CoV, which may contribute to the higher infectivity and transmissibility of SARS-CoV-2 [9,10,11]. The high variation in clinical severity observed among patients may be suggestive of a critical role of the inter-individual variability in the host genetic background. It is consequently conceivable that the distribution of ACE2 determines the virus cell and tissue tropism and pathogenesis [12]. Investigating the molecular virus-receptor interactions is a crucial step through the understanding of viral pathogenesis and host susceptibility. Clinical conditions include pulmonary and extrapulmonary manifestations. Despite a standardized molecular mechanism of infection, SARS-CoV-2 shows highly variable clinical presentations, amenable to a variety of factors that range from viral strain to the host genetic background [12]. Indeed, several studies inferred that genetic variants in ACE2 gene may influence the individual susceptibility or resistance to SARS-CoV-2 according to the functional role of ACE2 in human pathophysiology [12]. It is possible that the affinity of binding of SARS-CoV-2 to ACE2 could be modulated by genetic variants within the RBD and/or other ACE2 domains. As a matter of fact, genetic variants affecting the receptor might influence infection rates and severity of the disease. ACE2 functional variants might enhance or reduce the binding affinity for the RBD by altering the residues accessibility or protein conformation. Seeking for variants that show a correlation with the disease severity and investigating their influence on the viral replicative cycle is a first step to unveil the reasons behind the broad range of disease outcomes. Furthermore, it might provide insights for the development of antiviral therapies.
In this study, we, therefore, investigated the occurrence of ACE2 variants in a cohort of 131 Italian SARS-CoV-2-positive patients, extracting data on ACE2 variants by direct DNA analysis. We also verified the existence of an association of ACE2 variants with severity of the disease.
Materials and methods
Clinical study
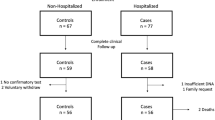
For our study, we enrolled a total of 131 subjects with COVID-19. More than half were hospitalized at Tor Vergata University Hospital (n = 89, 68%) and the remaining at Bambino Gesù Children’s Hospital of Rome (n = 42, 32%). One hundred fourteen patients (87%) showed clinical symptoms of COVID-19. All were diagnosed with COVID-19 after positive results of naso-oropharyngeal swabs. They were admitted to the relevant wards for appropriate care and checks, while the asymptomatic subjects (n = 17, 13%) returned home or were kept under brief observation for few days.
Most of the enrolled subjects were male (81/131; 62%). Age ranged between 6 and 92 years old (median age ± SD 57 ± 19.7). Fifty subjects were female (38%), with their age ranging from 2 to 93 years old (median age ± SD 55 ± 22.9). 13 were children (median age ± SD 11 ± 4.2), classified as asymptomatic or with mild disease severity. None of the patients showed symptoms of Kawasaki-like syndrome [13].
We clustered all the patients in four disease severity group as stated by their hospitalization outcomes:
-
Asymptomatic: absence of clinical symptoms (n = 17, 13%; median age ± SD 39 ± 16.2 years old);
-
Mild: presence of few symptoms, but not requiring ventilation, except for cases of respiratory support via Venturi Mask (VMK) (n = 16, 12%; median age ± SD 48 ± 23.3 years old);
-
Moderate: showing respiratory impairment, requiring non-invasive ventilation and CPAP (continuous positive airway pressure) or BiPAP (bilevel positive airway pressure) cycles (n = 43, 33%; median age ± SD 61 ± 14.7 years old);
-
Severe: defined as respiratory failure, requiring invasive ventilation and intensive care unit (ICU) admission (n = 55, 42%; median age ± SD 65 ± 17.6 years old).
Venous blood samples from patients and control individuals (1000 Italian subjects, 500 males, and 500 females) were collected for the Whole Exome Sequencing (WES).
Our investigations received approval by the local ethics committee at Tor Vergata University Hospital (protocol no. 50/20). The study was conducted in agreement with the principles of the Declaration of Helsinki. Informed written consent was obtained from each patient.
Whole exome sequencing and data preprocessing
Library preparation and whole exome capture were performed by using the Twist Human Core Exome Kit (Twist Bioscience) according to the manufacture's protocol and sequenced on the Illumina NovaSeq 6000 platform. The BaseSpace pipeline (Illumina) and the TGex software (LifeMap Sciences) were used for the variant calling and annotating variants, respectively. Sequencing data were aligned to the hg19 human reference genome. Based on the guidelines of the American College of Medical Genetics and Genomics (ACMG), a minimum depth coverage of 30X was considered suitable for analysis. Variants were examined for coverage and Qscore (minimum threshold of 30), and visualized by the Integrative Genome Viewer (IGV). For this study, we analyzed only data on the ACE2 candidate gene.
Statistical analysis
Differences in alleles frequencies between groups were evaluated by the Pearson χ2 test or by Fisher’s exact test, as requested according to the numbers of samples in the compared groups. P values less than 0.05 were considered statistically significant. Since we considered only ACE2 gene, with a “candidate gene” approach, we did not perform corrections for multiple comparison normally used for exome sequencing data analyses of thousands of genes. The Hardy-Weinberg equilibrium was evaluated, where possible, by the Pearson χ2 test.
Results
We identified three different germline variants, one intronic c.439+4G>A (rs2285666) and two missense c.1888G>C p.(Asp630His) (rs140312271) and c.2158A>G p.(Asn720Asp) (rs41303171), in a total of 30 patients (14 females and 16 males). Seven out of 30 were asymptomatic (23%; median age ± SD 42 ± 19.4 years old); 3 out of 30 were mild (10%; median age ± SD 15 ± 32.7 years old); 6 out of 30 were moderate (20%; median age ± SD 66 ± 19.1 years old); and 14 out of 30 were severe (median age ± SD 70.5 ± 10.6 years old). Four out of 30 passed away (1 male and 3 female; median age ± SD 74 ± 11.9 years old). The frequency of the three identified variants are similar between male and female patients suggesting also there is no gender effect underlying the frequency distribution of ACE2 variants (Table 1). GnomAD database analysis revealed that these identified ACE2 variants are reported with a cumulative frequency of 0.2289 in ethnically matched populations (EUR). The cumulative frequency of these variants in our examined Italian cohort is 0.2353 and is not statistically different (Table 1). A significant difference was detected only for the c.1888G>C p.(Asp630His) even if this result is to be confirmed in a larger cohort since it was found only in a heterozygous female (p = 0.0088) (Table 1). The allelic frequency of this variant in GnomAD for the EUR reference population is 0.0000368 confirming that this is a very rare allele. This variant was not found in our Italian control population. In order to predict the functional impact of this variant on the protein, we used several tools (PolyPhen2, Mutation Taster, SIFT) and two ensemble score (MetaLR_pred, MetaSVM_pred.). The in silico analysis gave conflicting computational verdicts because of 3 benign predictions vs. 2 pathogenic predictions. The sequence alignment of the ACE2 protein with its orthologous proteins shows that the wild type residue is not highly conserved in species implying an irrelevant functional or structural role of this residue in the ACE2 protein. However, this variant deserves further investigation in a larger COVID-19 cohorts as well as functional studies. Concerning the other two variants, the recurrent c.439+4A>G (rs2285666) intronic variant has been previously reported by Strafella et al. [14] and by Asselta et al. [15] in two different Italian cohorts representative of the country’s population. The variant is located in the intron 3 in a splice site region of the gene. However, using Human Splicing Finder (HSF) no significant splicing alterations were suggested. The missense variant c.2158A>G p.(Asn720Asp) was found in two patients, one female in heterozygous state and one male, with a frequency in line with our Italian control population and with the frequency reported for the European non-Finnish population in the GnomAD database. This variant is located in the C-terminal domain, which is not involved in the SARS-CoV-2 S protein interaction. The in silico analysis to predict the potential impact of this variant on the protein sequence gave benign computational verdict because of 4 benign predictions vs. 1 pathogenic prediction. We tested the hypothesis if these variants were associated with COVID-19 severity. We analyzed the SNP rs2285666 (for which we observed more variant alleles) comparing both asymptomatic vs mild-moderate-severe and severe vs moderate-mild asymptomatic, but we did not find any kind of significant difference (data not shown). For the other two SNPs, allelic variants were observed only in asymptomatic subjects in heterozygous status. However, the small number of patients in each subgroup considered does not allow us to make definitive conclusions.
Discussion
Several in silico data suggested that the ACE2 variants in structural part of the protein could have an impact on the pathogen binding dynamics or increase the quantitative expression of ACE2 [7, 10,11,12]. ACE2 receptor binds the SARS-CoV-2 spike protein at least 10 times more tightly than SARS-CoV-1 [16]. This might explain some of the differences between the two viruses in the way they infect people and cause disease. For this reason, we wanted to analyze in detail the genetic variability of ACE2 in our population, among the most affected by the COVID-19 pandemic. Until now, most available published studies were carried out on an epidemiological basis of population allele frequencies deposited in the various available databases. We first tested the hypothesis on the existence of an enrichment of coding-region variants in ACE2 gene, able to affect the binding dynamics of SARS-CoV-2 to the receptor. Despite the number of patients systematically analyzed in this series, it is small for definitive conclusions, we believe that there are no enrichments of rare functional alleles in ACE2 capable of influencing the binding capacity of the receptor. We identified in a single COVID-19 patient a variant (p.Asp630His), very rare in European population and not detected in our Italian control population. Similarly, we did not observe significant differences by stratifying patients according to the clinical phenotype. In fact, 28 out of 30 patients (93%), with a asymptomatic, mild, moderate, and severe outcomes, presented c.439+4G>A variant; 2 asymptomatic out 30 patients (6%) presented c.2158A>G, p.Asn720Asp variant; only one asymptomatic woman (1%) presented c.1888G>C, p.Asp630His variant. However, these preliminary results should be verified in a much larger cohort for a more accurate risk assessment.
Our results confirm and extend the knowledge that ACE2 is a gene with a low allelic frequency of missense variants as expected on the basis of GnomAD population data. In fact, we provide evidence that the rate of amino acid changes at the binding region with SARS-CoV-2 and at the protein cleavage sites is very low. This suggests that these regions have been under evolutionary pressure, probably for the essential catalytic role of ACE2 as transmembrane carboxypeptidase. It is possible that rare susceptibility alleles are located in the non-coding regions of the gene, involved in the regulation of ACE2 gene activity. Also, a recent GWA study on a high number of patients did not show evidence of association with ACE2 variability [17]. Mutant alleles in non-coding DNA can cause alterations in expression levels or timing. These variations concern enhancers, promoters, insulators, and silencers or regions that provide instructions for producing functional RNA molecules, such as transfer RNA, miRNAs, or long non-coding RNA [18]. By inspecting the human genetic variants pool available at https://www.ncbi.nlm.nih.gov/snp/, ∼ 16,493 SNPs were extracted after filtering for the non-coding regions of ACE2. We are aware that the totality of these variants has no functional meaning. However, some of these may influence the expression of the receptor in a tissue-dependent way. It is therefore of interest to explore the existence of ACE2 susceptibility alleles to SARS-CoV-2 in these regulatory regions. Interestingly very recently, Bunyavanich et al. [19] showed age-dependent expression of ACE2 gene in nasal epithelium, highlighting that the different levels of ACE2 expression may be the reason for a lower incidence of COVID-19 in children. Several studies have shown that ACE2 gene undergoes the action of at least four miRNAs: miR-200c, let-7b, miR-1246, and miR-125b [20,21,22,23]. Polymorphisms within genes coding for these miRNAs could be of great help with regards to investigations on the regulation of ACE2 gene expression and the possible significance of variations in further more in depth studies.
Conclusions
Our study suggests that there is no strong evidence, in our cohort, of consistent association of ACE2 genomic variants with COVID-19 susceptibility or clinical phenotype. However, we cannot rule out a type II error considered to be a relatively small size of the samples tested. Despite this, we might speculate that rare susceptibility alleles could be located in the non-coding regions of the ACE2 gene, known to have a role in regulating gene activity. It should be therefore interesting to explore the existence of ACE2 susceptibility alleles to SARS-CoV-2 in the regulatory regions of the gene.
References
Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–3. https://doi.org/10.1016/S0140-6736(20)30185-9.
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Thian JH, Pei YY, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–9. https://doi.org/10.1038/s41586-020-2008-3.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, et al. A novel coronavirus from patients with pneumonia in China. N Engl J Med. 2020;382:727–33. https://doi.org/10.1056/NEJMoa2001017.
Sohrabi C, Alsafi Z, O'Neill N, Khan M, Kerwan A, Al-Jabir A, et al. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int J Surg. 2020;76:71–6. https://doi.org/10.1016/j.ijsu.2020.02.034.
Zhou P, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;426:270–3. https://doi.org/10.1038/s41586-020-2012-7.
de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–34. https://doi.org/10.1038/nrmicro.2016.81.
Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562–9. https://doi.org/10.1038/s41564-020-0688-y.
Santos RAS, Sampaio WO, Alzamora AC, et al. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol Rev. 2018;98(1):505–53. https://doi.org/10.1152/physrev.00023.2016.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–3. https://doi.org/10.1126/science.abb2507.
Yan R, Zhang Y, Li Y, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–8. https://doi.org/10.1126/science.abb2762.
Wang Q, Zhang Y, Wu L, et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 2020; 181(4): 894-904.e9. doi: 10.1016/j.cell.2020.03.045.
Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. 2020 2020;10.1002/jmv.25832. doi: 10.1002/jmv.25832.
Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020. https://doi.org/10.1016/S0140-6736(20)31103-X.
Strafella C., Caputo V., Termine A., Barati S., Gambardella S., Borgiani P., Caltagirone C., Novelli G., Giardina E., Cascella R. Analysis of ACE2 genetic variability among populations highlights a possible link with COVID-19-related neurological complications. Genes (Basel). 2020;11(7):741. doi: 10.3390/genes11070741. PMID: 32635188; PMCID: PMC7397291.
Asselta R., Paraboschi EM., Mantovani A., Duga S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging (Albany NY). 2020; 12(11): 10087-10098. doi: 10.18632/aging.103415.
Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M. Jr, Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020:eabd4251. doi: 10.1126/science.abd4251. Epub ahead of print. PMID: 32694201.
Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, Fernández J, Prati D, Baselli G, Asselta R, Grimsrud MM, Milani C, et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. NEJM. 2020. https://doi.org/10.1056/NEJMoa2020283.
Gloss BS, Dinger ME. Realizing the significance of noncoding functionality in clinical genomics. Exp Mol Med. 2018;50(8):97. https://doi.org/10.1038/s12276-018-0087-0.
Bunyavanich S, Do A, Vicencio A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA. 2020. https://doi.org/10.1001/jama.2020.8707.
Liu Q, Du J, Yu X, Xu J, Huang F, Li X, et al. miRNA-200c-3p is crucial in acute respiratory distress syndrome. Cell Discov. 2017; 3: 17021. doi: 10.1038/celldisc.2017.21.
Zhang R, Su H, Ma X, Xu X, Liang L, Ma G, et al. MiRNA let-7b promotes the development of hypoxic pulmonary hypertension by targeting ACE2. Am J Physiol Lung Cell Mol Physiol. 2019;316(3):L547–L57. https://doi.org/10.1152/ajplung.00387.2018.
Fang Y, Gao F, Hao J, Liu Z. microRNA-1246 mediates lipopolysaccharide-induced pulmonary endothelial cell apoptosis and acute lung injury by targeting angiotensin-converting enzyme 2. Am J Transl Res. 2017;9(3):1287–96.
Huang YF, Zhang Y, Liu CX, Huang J, Ding GH. microRNA-125b contributes to high glucose-induced reactive oxygen species generation and apoptosis in HK-2 renal tubular epithelial cells by targeting angiotensin-converting enzyme 2. Eur Rev Med Pharmacol Sci. 2016;20(19):4055–62.
Acknowledgements
We thank Dr. Paolo Gravina, Dr. Laura Liberatoscioli, and Valentina Lanari for their technical support. This work is part of the GEFACOVID2.0 Consortium. We also appreciate discussions with and the assistance of Juergen Reichardt.
Author information
Authors and Affiliations
Contributions
DC, MRD, carried out the molecular genetic studies; PB and MB, performed the statistical analysis; VLC, PR, SZ, FL, AC, and MR, contributed to the interpretation of the results and provided clinical phenotyping; AN, MB, MA, SG and GN, conceived the study and participated in its design and coordination. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Biological samples enrolled in the study were collected according to the ethical procedures of the GEFACOVID2.0 research program promoted by the University of Rome Tor Vergata. This program will ensure that its work is carried out with the highest regard for ethical issues and with respect to the rights, integrity, and privacy of patients. All consent, material/information storage, and distribution procedures have been approved by the local Ethics Committees (CEI PTV protocol no. 50/20). SARS-CoV-2-positive patients who will be offered participation in a research study will sign an informed consent prepared ad hoc and which will provide detailed information on the type of test, the implications of the genetic results and the possible psychosocial implications. As regards the participation of children in the research, consent and authorization are expected to be signed by the parents in accordance with the rules laid down by the Ethics Committee of the Bambino Gesù Hospital in Rome (http://www.ospedalebambinogesu.it/en/home).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Novelli, A., Biancolella, M., Borgiani, P. et al. Analysis of ACE2 genetic variants in 131 Italian SARS-CoV-2-positive patients. Hum Genomics 14, 29 (2020). https://doi.org/10.1186/s40246-020-00279-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-020-00279-z