
Abstract
Transcription factors (TFs) consisting of zinc fingers combined with BTB (for broad-complex, tram-track, and bric-a-brac) domain (ZBTB) are a highly conserved protein family that comprises a multifunctional and heterogeneous group of TFs, mainly modulating cell developmental events and cell fate. LRF/ZBTB7A, in particular, is reported to be implicated in a wide variety of physiological and cancer-related cell events. These physiological processes include regulation of erythrocyte maturation, B/T cell differentiation, adipogenesis, and thymic insulin expression affecting consequently insulin self-tolerance. In cancer, LRF/ZBTB7A has been reported to act either as oncogenic or as oncosuppressive factor by affecting specific cell processes (proliferation, apoptosis, invasion, migration, metastasis, etc) in opposed ways, depending on cancer type and molecular interactions. The molecular mechanisms via which LRF/ZBTB7A is known to exert either physiological or cancer-related cellular effects include chromatin organization and remodeling, regulation of the Notch signaling axis, cellular response to DNA damage stimulus, epigenetic-dependent regulation of transcription, regulation of the expression and activity of NF-κB and p53, and regulation of aerobic glycolysis and oxidative phosphorylation (Warburg effect). It is a pleiotropic TF, and thus, alterations to its expression status become detrimental for cell survival. This review summarizes its implication in different cellular activities and the commonly invoked molecular mechanisms triggered by LRF/ZBTB7A’s orchestrated action.
Similar content being viewed by others


Introduction
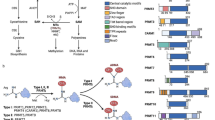
Zinc finger and BTB domain containing 7A (ZBTB7A) appears in the literature with several synonyms: POK erythroid myeloid ontogenic factor or POZ and Krüppel-erythroid myeloid ontogenic factor (Pokemon), factor binding IST protein-1 (FBI-1), HIV-1 inducer of short transcripts binding protein, TTF-I-interacting peptide 21, DKFZp547O146, etc. The lymphoma/leukemia-related factor (LRF) preferentially refers to the protein product encoded by the ZBTB7A gene.
Characteristic domains of the LRF/ZBTB7A transcription factor are the four C-terminal Krüppel-type zinc fingers with a sequence-specific DNA-binding capacity and the N-terminal (broad-complex, tram-track, and bric-a-brac) BTB domain, capable for the formation of homo- or hetero-dimers. These features provide LRF/ZBTB7A with collaboration abilities and highly contextual activities towards cellular function, such as transcription co-repressor activity, protein and histone acetyltransferase binding, proximal promoter sequence-specific DNA-binding, and DNA-binding with consecutive attraction and modulation of other TFs activities.
LRF/ZBTB7A exerts its action within the nucleus compartment; herein, cell responses may differ in different tissues and local cell microenvironments. Cell processes disturbed by the aberrant expression of LRF/ZBTB7A TF are discussed in the present manuscript in an effort to give prominence to its both widespread gene regulatory effects and components of cellular life-cycle affected. Its basic ability to cooperate and attract various complexes to the broader “targeted” regulated genome area determines not only its pleiotropic and sometimes conflicting action, but also its potential to act on a case-by-case basis, depending on the epigenetic profile of the genome, due to its preferred binding sites within rich CG regions [1,2,3]. In humans, it has been extensively implicated in cell fate and cell differentiation abnormalities, highly significant for malignant manifestations and disease outcome. Besides its potent implications to the hematopoietic tissue, lymphoid development, and adipogenesis, involvement in specific downstream intracellular pathways, cell cycle, apoptosis, and glycolysis is presented, along with a fine tuning on the insulin’s thymic expression regulation, highlighting the universal role of this TF towards genome regulation.
The LRF/ZBTB7A repertoire during erythroid development
Hematopoietic stem cells (HSCs) residing in specified compartments of the bone marrow, called “niche,” coordinate the replenishment of all types of blood cells through a series of lineage restriction steps, leading to pluripotency depletion and increasing commitment to a single pathway (lineage commitment). Differentiation of HSCs to mature erythrocytes involves the transition to common myeloid progenitors (CMP), early erythroid progenitors (burst-forming unit-erythroid, BFU-E), late erythroid progenitors (colony-forming unit-erythroid, CFU-E), and the morphologically recognizable erythroid precursors (proerythroblasts, basophilic erythroblasts, polychromatophilic erythroblasts, and orthochromatic erythroblasts) that undergo terminal maturation to erythrocytes by extruding their nuclei and progressively losing cytoplasmic organelles [4]. During effective erythropoiesis LRF/ZBTB7A, activated by the erythroid-specific transcription factor GATA1, directly binds to the promoter of the pro-apoptotic factor BCL2 like 11 (BIM), inhibits its transcription and suppresses cell apoptosis, ensuring the final production of erythroid cells [5]. Furthermore, KLF1 (Krüpel-like factor 1) cooperates with GATA1 to upregulate LRF/ZBTB7A expression in both human and mouse erythroid cell lines [6]. By its turn, LRF/ZBTB7A binds at GATA1 occupancy sites of a number of direct GATA1 target genes and recruits the chromatin repressive complex Polycomb Repressive Complex 2 (PRC2), directing epigenetic gene silencing [7] (Fig. 1).

LRF/ZBTB7A regulates hematopoiesis and lymphoid development. LRF/ZBTB7A contribution in specific stages of cell differentiation is indicated. Abbreviations: HSC, hematopoietic stem cell; CMP, common myeloid progenitors; BFU-E, burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid; ProE, proerythroblasts; BasoE, basophilic erythroblasts; PolyE, polychromatophilic erythroblasts; OrthoE, orthochromatic erythroblasts; RBC, matured erythrocytes; CLP, common lymphoid progenitors; ProB, progenitor B cell; PreB, precursor B cell; DN T cell, double negative or CD4-/8- negative; DP T cell, double positive or CD4+/8+ positive; CD4+ single positive expressing Thpok (T-helper inducing POZ/Krüppel-like factor); CD8+ single positive expressing the Runx3; FOB, follicular B cells; MZB, marginal zone B cells; GCΒ, germinal center B cells
Masuda and coworkers [8] demonstrated that LRF/ZBTB7A depletion reactivates the embryonic/fetal hemoglobin expression in adult mice and the γ-globin expression in human erythroblasts, due to permissive changes at the local chromatin conformation surrounding the γ-globin genes. LRF/ZBTB7A acts as a γ-globin repressor, during hemoglobin transversion from the fetal (HbF) to adult (HbA), independently from the master transcription factor B cell lymphoma/leukemia 11A (BCL11A), via the recruitment of the Nucleosome Remodeling and Deacetylase (NuRD) chromatin remodeling complex. In support to this mechanism, Zbtb7a-/- mice embryos exhibit early lethal anemia, while conditional inactivation of Lrf/Zbtb7a in adult mice blocks the terminal erythroid differentiation and leads to macrocytic anemia [5].
Reactivation of HbF expression, with regard to the use of pharmacological factors, has been applied to the clinic for the treatment of β-hemoglobinopathies, especially for the homozygous Sickle Cell Disease (SCD) patients and double heterozygotes with β-thalassemia and SCD (β-thal/SCD). Hydroxyurea (HU) or hydroxycarbamide is the major therapeutic agent used for the management of these patients, approved by the Food and Drug Administration (FDA) in severely affected adults, since 1998. Of the key parameters of HU action in patients suffering from SCD and β-thal/SCD is the successful reproduction of HbF, which improves their pathological phenotype resulting in reduced or eliminated transfusion needs, though with controversial results, because of the moderate and heterogeneous HbF expression levels produced and the lack of specificity [9]. Recent evidence supports the suppressive role of LFR/ZBTB7A to the reinduction of HbF, among the non-responders group of SCD and β-thal/SCD patients to HU, suggested to depend on an epigenetic mechanism and mediated by the HU driven hypomethylation of its 5’ CpG island and the consequent elevated expression of LRF/ZBTB7A. Recognition binding sites for the LRF/ZBTB7A are apparent at CpG islands settled at the 5’ areas of several “modifier genes” of the β-globin genes’ cluster, such as the erythroid-specific transcription factors KLF1 and GATA2, the regulators of hematopoiesis and erythropoiesis MYB, SIN3A, and BCL11A, members of the MAPK signalling pathway (MAP3K5), and the ZBTB7A gene itself, implying an auto- and inter-regulatory role for this TF [2] within specific gene networks.
Furthermore, LRF/ZBTB7A has been demonstrated to directly bind the γ-globin genes’ promoter and known recognition sequences responsible for the Hereditary Persistence of Fetal Hemoglobin (HPFH) syndrome. HPFH is a benign condition which is characterized by high HbF expression throughout adulthood, and regarding SCD and β-thal/SCD patients, it plays a pivotal role in ameliorating the severity of their symptoms. HPFH-associated mutations ordinarily disrupt the LRF/ZBTB7A binding sites and thus raise the γ-globin gene expression, constituting an interesting therapeutic goal aiming to reactivate the developmentally silenced γ-globin gene expression [10].
Lymphoid lineage fate decisions directed by LRF/ZBTB7A
LRF/ZBTB7A has been reported to regulate B versus T lymphocyte fate decisions by interfering with the Notch signaling pathway [11]. B cell development involves the sequential transition from the common lymphoid progenitors (CLP) to pro-B cell (progenitor B cell), pre-B cell (precursor B cell), immature naïve B cell, transitional naïve B cell, and ultimately mature B cell. The immature B cells normally migrate from the bone marrow to the secondary lymphoid organs in the body’s periphery, thereby reinforcing the ongoing maturation process [12]. During fetal lymphopoiesis in mice embryos, homozygous Zbtb7a deletion affects the early B cell development and leads to a significant reduction of the Pro-B stage cells, whereas HSC and CLP cell populations remain intact. However, in adult mice, the inducible inactivation of Lrf/Zbtb7a results in the drastic reduction of circulating B220+ cells and in the extrathymic double-positive (CD4/8) T cell development, limited in the bone marrow, at the expense of B cell development [13]. Furthermore, LRF/ZBTB7A prevents HSCs from premature differentiation towards T cell and supports adult HSC homeostasis by suppressing the Notch-mediated signals [14].
T cell differentiation process in the thymus is subcategorized into discrete stages based on the expression of the co-receptor molecules CD4 and CD8. The earliest thymocytes, which are CD4-/8- negative (double negative, DN) differentiate into CD4+/8+ positive (double positive, DP) and further mature in CD4+ single positive expressing Thpok (T-helper inducing POZ/Krüppel-like factor) encoded by the Zbtb7b gene or in CD8+ single positive cells expressing the Runx3 TF [15]. Inactivation of both Thpok/Zbtb7b and Lrf/Zbtb7a in mice, revealed that Thpok is required for the intrathymic T regulatory (Treg) differentiation, and both TFs redundantly promote CD4+ T cell lineage maintenance [16] and Treg-mediated immune homeostasis. Thpok/Zbtb7b and Lrf/Zbtb7a deletion in Treg cells leads to a lethal inflammatory disease similar to that of Scurfy mice which carry a missense mutation in the Foxp3 gene and therefore lack functional CD4+Foxp3+ Treg cells. Thpok/Zbtb7b and Lrf/Zbtb7a support Foxp3-directed gene expression in Tregs, specifically through activation of IL-2-dependent genes [17, 18].
Activation of the conserved Notch signaling pathway is essential for the T cell differentiation, but is also critical for distinct cell fate decisions, in the secondary lymphoid organs, during the transition of the long-lived mature B cell pool towards the follicular B cells (FOB) versus the marginal zone B cells (MZB). Lrf opposes Notch function under normal conditions as defined in Lrf/Zbtb7a conditional knockout mice, which showed excessive MZB differentiation against FOB, though inactivation of the Delta-like 4 (DLL4), component of the Notch axis, rescued the aberrant lymphoid differentiation and the balance between MZB and FOB development was restored [19].
The concurrent elevated expression levels of LRF/ZBTB7A and BCL6 (ZBTB27), another member of the ZBTB family, have been reported to regulate the formation of Germinal Centers B (GCΒ) cells. GCs are formed by rapidly proliferating B cells after activation of naïve B cells through interaction with helper T cells and antigen-presenting cells and appear as a distinct histologic structure found in secondary lymphoid organs [20, 21]. Defected GC formation was obtained in conditional inactivated Lrf/Zbtb7a adult mice, which interestingly was not rescued by Notch loss, suggesting that Lrf is required for the maintenance and function of GCB cells rather than commitment to this stage. Downregulation of the tumor suppressor gene p19Arf was confirmed to be responsible for the impaired proliferation and increased apoptosis seen in Lrf/Zbtb7a-deficient GCB-cells in mice [19].
Collectively, LRF/ZBTB7A plays a vital role to lineage cell fate decisions within the hematopoietic compartment [22, 23] illustrated in Fig. 1.
LRF/ZBTB7A function in hematological cancers and solid tumors
Direct and indirect involvement of LRF/ZBTB7A in glycolysis
To meet the demands of a highly proliferative state and survival in various unfavorable microenvironments, tumors undergo fundamental alterations in their metabolism regarding carbohydrates, lipids, and glutamine [24] herein presenting, dependence on glycolytic ATP as the major energetic pathway and elevated de novo lipid synthesis to provide building blocks for membrane biosynthesis [25]. The most prominent aspect of malignant metabolic transformation is the glycolytic phenotype or the Warburg effect, whereby cancer cells exhibit high glycolytic activity under aerobic conditions [26, 27] and favor glycolysis, although it yields lower amounts of ATP than mitochondrial oxidative phosphorylation (OXPHOS) [28]. The pentose phosphate pathway (PPP) acts as an auxiliary secondary pathway to produce both NADPH and ribose-5-phosphate (R5P) for biosynthetic reactions and nucleic acids synthesis and it is enhanced by the increased aerobic glycolysis rate [28].
Current advances in molecular biology and cancer genetics provide evidence for mechanistic links between dysfunction of oncogenic proteins or tumor suppressors and hyperactive glycolysis in cancer [29]. KRAS and MYC proto-oncogenes, protein kinase B (AKT), epidermal growth factor receptor (EGFR), BCR-ABL fusion gene, and the receptor tyrosine kinase (ALK) promote independently glycolysis via upregulation of various glycolytic enzymes [30,31,32] or intermediates including glucose transporters I and III (GLUT1, GLUT3) [33, 34]. In addition, many oncoproteins activate the hypoxia inducible factor (HIF) via hypoxia-independent mechanisms or a pseudohypoxic state to enhance tumor glycolysis [30], while several glycolytic enzymes, including hexokinase 2 (HK2), phosphofructokinase 2 (PFK2), pyruvate kinase M2 (PKM2), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase (PDK), have been identified as HIF-targeted genes [35]. Contrariwise, the tumor suppressor p53 promotes mitochondrial respiration through multiple mechanisms and ultimately dampens aerobic glycolysis mainly by inducing the cytochrome c oxidase (SCO2) and the TP53-induced glycolysis and apoptosis regulator (TIGAR), respectively [36]. The LRF/ZBTB7A’s silencing has been shown to induce p53 expression and phosphorylation [37, 38] indicating that LRF/ZBTB7A loss-of-function indirectly inhibits aerobic glycolysis and enhances OXPHOS, compromising thus the Warburg effect in cancer cells. However, Liu et al. [34] demonstrated that LRF/ZBTB7A mediates the transcriptional repression of glycolytic genes, including GLUT3, phosphofructokinase (PFKP), and PKM, indicating a second, opposing to the first, direct nuclear upregulating effect of LRF/ZBTB7A’s silencing on glycolysis. Whether and which mechanism predominates in the cellular transformation extended in a tissue- or cell-specific phenomenon, remains to be elucidated. Additionally, it has been shown that p53 inhibits the diversion of glycolytic intermediates into the PPP by binding and inhibiting glucose-6-phosphate dehydrogenase (G6PDH) [36]. To this end, LRF/ZBTB7A’s silencing is expected to further compromise PPP indirectly via p53-dependent inhibition of G6PDH (Fig. 2).

LRF/ZBTB7A’s silencing compromises Warburg effect and induces apoptosis in cancer cells. The inhibition of aerobic glycolysis and concomitant activation of oxidative phosphorylation (OXPHOS) is promoted by the LRF/ZBTB7A’s silencing and the subsequent activation of the expression and phosphorylation of p53, leading thus to compromised Warburg effect in cancer cells. Furthermore, LRF/ZBTB7A’s silencing induces apoptosis in cancer cell lines by mediating both known apoptotic pathways (intrinsic and extrinsic) as well as their cross-talk. That is, upon LRF/ZBTB7A’s silencing and the subsequent activation of p53, the pro-apoptotic Bcl-2 family proteins as well as AIF are activated and promote cytochrome c release from mitochondria and subsequent activation of caspase-9 and caspase-3 (intrinsic apoptotic pathway). Meanwhile, LRF/ZBTB7A’s silencing promotes the expression of the Fas receptor (death receptor), leading to the activation of the downstream caspase-10 and caspase-8 and leading thus to activation of the extrinsic apoptotic pathway. In addition, caspace-8 further enhances the activation of Bcl2 family members, as well as the activation of caspase-9 and caspase-3 supporting the hypothesis that LRF/ZBTB7A, besides affecting both apoptotic pathways, potentially mediates the cross-talk between the intrinsic and extrinsic apoptotic pathways in cancer cells (in red color are cellular processes inhibited and in green color are cellular processes activated by LRF/ZBTB7A silencing). Abbreviations: AIF, apoptosis inducing factor; FasR, Fas receptor; G6PDH, glucose-6-phosphate dehydrogenase; OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; SCO2, cytochrome c oxidase 2; TIGAR, TP53-induced glycolysis and apoptosis regulator
LRF/ZBTB7A as an oncogene
LRF/ZBTB7A has been characterized as an oncogene in many human cancers. It has been shown to be overexpressed in most of the human non-Hodgkin lymphoma cases (NHL) and commonly in conjunction with BCL6 (ZBTB27) [38]. Furthermore, frequent mutations of the LRF/ZBTB7A zinc finger coding sequences are identified in acute myeloid leukemia (AML) with translocation t(8;21), which links to a co-operational mechanism of action with the RUNX1/RUNX1T1 fusion gene towards leukemogenesis [39].
However, LRF/ZBTB7A association with cancer progression goes far beyond hematological cancers. After the initial report associating LRF/ZBTB7A with T cell lymphoblastic lymphoma/leukemia through the suppression of the Arf tumor suppressor (p19Arf) gene and the consequent decrease of p53 activity [38], accumulating evidence further show that LRF/ZBTB7A is overexpressed in several other human cancers, including non-small cell lung cancer (NSCLC) [40,41,42,43], hepatocellular carcinoma [44,45,46,47,48], prostate [49, 50], ovarian [51], breast [52,53,54] and gastric cancers [55], glioma [56], sarcomas [57, 58], colorectal cancer [59,60,61,62], and renal carcinoma [63]. Further elucidation of these oncogenic functions revealed that LRF/ZBTB7A can influence cancer cell survival and proliferation, apoptosis, invasion and migration/metastasis, traits comprising some of the key biological capabilities required for the multistep development of human cancer, also presented as “hallmarks of cancer” [64]. LRF/ZBTB7A expression also positively correlates with many clinicopathologic parameters of human cancers like tumor size, histological grade, and overall patient survival, findings rendering this molecule a potential biomarker for human cancers, as well as an attractive therapeutic target.
In order to exert these functions, LRF/ZBTB7A interacts with or regulates many different binding partners, forming a complex network of downstream pathways. These data highlighted not only the diversity of its roles but also their context-dependency. Cell cycle-related genes are frequent targets of LRF/ZBTB7A [48, 56, 57, 59, 65,66,67], offering a mechanistic explanation of the factor’s ability to affect cell proliferation. In human hepatocellular carcinoma cell lines, LRF/ZBTB7A regulates cell cycle progression by either suppressing or promoting the expression of key cell cycle regulators like cyclin-dependent kinase inhibitors 2B, 1B, and 1A (p15, p27, and p21 genes, respectively), tumor protein p53 (TP53), CYCLIN D1, CYCLIN D3, and cyclin-dependent kinases CDK4 and CDK6. Commonly, LRF/ZBTB7A has been shown to promote progression through the S phase of the cell cycle in colorectal, osteosarcoma, and cervical cancer cells by transcriptionally repressing p21 [59, 68], whereas its depletion induces G1 cell cycle arrest through increased expression of p53 and p21 in chondrosarcoma and lung cancer cells [57, 59, 69]. Additional cell cycle-related transcriptional targets of LRF/ZBTB7A include the well-known tumor suppressor retinoblastoma (RB) gene, and by negatively regulating its transcription, LRF/ZBTB7A can alleviate the cell-cycle arrest effect of RB [70].
Suggested underlying mechanisms for these functions, at least in hepatocellular carcinoma, involve the phosphoinositide-3-kinase-protein kinase B/Akt (PI3K/Akt) and RAS/RAF/MEK/ERK pathways [65, 66]. The PI3K/Akt pathway is activated in response to extracellular stimuli through the high-affinity cell surface receptor tyrosine kinases and has multiple downstream targets regulating among others cell growth and survival, cell cycle progression and proliferation, apoptosis, and glycogen synthesis [71, 72]. Along this pathway, LRF/ZBTB7A was found to inhibit phosphatase and tensin homolog (PTEN), one of its negative regulators, thereby enabling an incessant signaling within cells with aberrant consequences [66].
The RAS/RAF/MEK/ERK pathway involves a cascade of events starting with an extracellular mitogen and usually resulting in the regulation of cell proliferation and division, through the consecutive activation of proto-oncogenes KRAS and c-RAF (GTP-ase and serine/threonine kinase respectively), mitogen-activated protein kinases, and extracellular signal-regulated kinases, also known as mitogen-activated protein kinases (MAPK), which can eventually activate the expression of transcription factors responsible for cell proliferation. Accumulating evidence supports the idea that blockade of one pathway enhances the other via key components participating in both pathways and implying potential cross-talk mechanisms between them [71, 73]. As both pathways are deregulated in many human cancers [71], the key participants of these signaling cascades, including LRF/ZBTB7A, represent attractive targets for therapeutic interventions.
Closely related to the effects on cell cycle progression are the LRF/ZBTB7A’s implications in regulating senescence and apoptosis [37, 57, 74]. LRF/ZBTB7A deletion can urge cells to attain senescence through upregulation of p21 and p53 [57]. Zbtb7a-knockout mouse embryonic fibroblasts also exhibit premature growth arrest and subsequent senescence caused by upregulated p19Arf and Trp53 [38]. On the other hand, overexpression of LRF/ZBTB7A can rescue this phenotype and maintain cells in a proliferative state, by enhancing E2F-dependent transcription and upregulation of CYCLIN E [74].
In hepatocellullar carcinoma cells, silencing of LRF/ZBTB7A increased p53 expression and initiated caspase-dependent apoptosis via death receptor- and mitochondria-mediated pathways [37], whereas in breast cancer, LRF/ZBTB7A anti-apoptotic function involved Survivin, a negative regulator of apoptosis [52], discussed in a separate section of this review.
LRF/ZBTB7A overexpression is also associated with enhanced cancer cell invasiveness and metastasis. Potential mechanisms involve the LRF/ZBTB7A-mediated transcriptional activation of myocyte enhancer factor 2D (MEF2D), an oncogene favoring the invasion of hepatocellular carcinoma cells [46, 47], and independently, the activation in ovarian cancer of membrane type 1-matrix metalloproteinase (MT1-MMP), a molecule playing a vital role in the dissociation of the extracellular matrix and the consequent dissemination of tumor cells [51].
Other targets of LRF/ZBTB7A-mediated regulation of oncogenesis include Striatin 4 (STRN4), a protein implicated in the progression of various human cancers [50, 75] and protein C-ets-1 (ETS-1), which is overexpressed in several cancers and implicated in cell proliferation, apoptosis, invasion, and migration [61]. Additionally, in breast cancer cells, LRF/ZBTB7A has been proposed to participate in transforming growth factor beta (TGF-β) signaling, a critical pathway for tumor progression [76], by binding to specificity protein 1 (SP1) and downregulating Smad4 expression [77]. A repressive effect on the TGF-β pathway was also attributed to the recruitment of HDAC to the Smad4 complex mediated by the POZ domain of LRF/ZBTB7A [78]. LRF/ZBTB7A has also been described as a target of key tumorigenic cascades like epidermal growth factor (EGF), SP1, endothelial PAS domain-containing protein 1 (EPAS-1), and TGF-β pathways [79,80,81,82]. Furthermore, LRF/ZBTB7A seems to be a frequent target of various microRNAs (miRNAs) during carcinogenesis, as miR21, miR100, miR125, miR137, miR520e, and miR663 are indicated as modulators of the LRF/ZBTB7A expression levels in many different cancers [44, 46, 55, 58, 63, 69, 83,84,85]. Finally, gene amplification was suggested as a potential mechanism driving overexpression of LRF/ZBTB7A in non-small-cell lung carcinoma, accompanied by transcriptional and post-translational aberrations [43].
It is therefore plausible that, at least in some human cancers, LRF/ZBTB7A represents one of the incipient traits rendering cancer cells tumorigenic and ultimately malignant, thus favoring cancer progression.
LRF/ZBTB7A as a tumor suppressor
LRF/ZBTB7A plays an even more multifaceted role in carcinogenesis. A number of studies have indicated that LRF/ZBTB7A exerts also tissue- and context-dependent oncosuppressive functions. LRF/ZBTB7A’s expression was found to be repressed by the heterochromatin protein 1γ (HP1γ), which localizes to both the heterochromatic and euchromatic regions within the cell nucleus and is known to be involved in gene expression regulation [86]. Supporting evidence shows that LRF/ZBTB7A’s depletion restores the proliferation and migration defects caused by HP1γ upregulation in lung adenocarcinoma cells, linked to a poor prognosis in patients. Furthermore, HP1γ-induced downregulation of the LRF/ZBTB7A favors the expression of several tumor-promoting factors, such as AXL receptor tyrosine kinase (AXL), plasmacytoma variant translocation 1 (PVT1), and ETS Like-1 protein (ELK1) [87].
Lrf/Zbtb7a was also shown to interact with and antagonize the transcriptional activity of Sry-related HMG box 9 (Sox9), which drives expression of several genes contributing to increased proliferation and oncogenesis, and that loss of Lrf/Zbtb7a bypasses cellular senescence and promotes invasion in Pten-null prostate cancers [88, 89]. Interestingly, this finding was shown to have clinical implications, as analysis of human prostate cancer samples showed that LRF/ZBTB7A expression can stratify patients’ response to androgen-deprivation therapy [22, 90, 91]. Through SOX9, LRF/ZBTB7A also affects the commitment of adult mesenchymal stem cells towards undifferentiated sarcomas [92], whereas in breast cancer, LRF/ZBTB7A regulates estrogen receptor’s alpha (ERα) expression, one of the major markers used to determine course of treatment, and predicts a favorable/unfavorable outcome for patients treated with endocrine therapies [93].
In vitro and in vivo studies with hepatocellular carcinoma cells revealed another regulatory pathway of LRF/ZBTB7A expression, mediated by miR106, and showed that LRF/ZBTB7A overexpression, caused by miR106 inhibition, abrogates cell growth [83]. Further oncosuppressive functions of LRF/ZBTB7A include S cell cycle arrest, promotion of apoptosis and repression of migration in gastric cancer [94], and suppression of metastasis through transcriptional repression of melanoma cell adhesion molecule (MCAM) in melanoma [95]. An additional interesting mechanism related to the LRF/ZBTB7A’s oncosuppressive functions involves the transcriptional repression of key oncogenic glycolytic genes like glucose transporters 1 and 3 (GLUT1, GLUT3), phosphofructokinase (PFKP), and pyruvate kinase muscle isozyme (PKM), thereby inhibiting cancer metabolism [33, 34].
Cooperation of LRF/ZBTB7A with NF-κB
Another significant interaction mediating LRF/ZBTB7A functions in cancer is through binding and promoting the signaling of nuclear factor (NF)-κB, a transcription factor with well-known properties in regulating many aspects of cancer, inflammation, and immune responses [96]. NF-κB signaling can be activated in both immune and non-immune tissues by various extracellular signals and positively or negatively modulate the cell proliferation and apoptosis [97]. NF-κB signaling comprises a canonical pathway, involving the subunits p50/p65, the activating kinase IKKβ and the inhibitor IκBα, and a non-canonical pathway, involving the subunits RelB/p52, the activating kinase IKKα and the inhibitor IκBβ. Activation of NFκB is transiently induced in response to various stressful stimuli like infections, DNA damage or pro-inflammatory cytokines, and pathogen- and damage-associated molecular pattern (PAMPs and DAMPs) [96]; however, in cancerous tissues, NF-κB signaling can be constitutively active [96]. LRF/ZBTB7A was found to impact the transcription of p65 and IκBα [3, 98], i.e., members of the canonical pathway of NF-κB signaling and has been proposed to positively affect transcription of NF-κB target genes by increasing nuclear localization of the p65 subunit of NF-κB [99], as well as by facilitating the accession of NF-κB to its target genes [3].
Additional pathways of LRF/ZBTB7A functions, implicated in cancer
Interestingly, accumulating evidence reveals a fascinating plurality of LRF/ZBTB7A roles, in a transcription-independent manner of function. In a recent study, LRF/ZBTB7A was described to hold a key role in maintaining genome integrity by regulating the non-homologous end joining pathway of double-strand break DNA repair [100]. Along the same line, LRF/ZBTB7A was also found to participate in alternative splicing modulation [101]. Another unexpected function of the ZBTB7A gene was recently reported, showing that the locus can actually produce two different types of RNA, a linear one and a non-coding circular one, which can nonetheless contribute to cancer progression independently from and possibly antagonistically to the linear counterpart, further reinforcing LRF/ZBTB7A tumorigenic implications [102]. Strikingly, LRF/ZBTB7A was also found to define the tumor microenvironment and the influx of tumor infiltrating immune cells in prostate cancer, and in particular the attraction of polymorphonuclear cells [103]. Finally, the chromosomal region 19p13.3, encompassing the LRF/ZBTB7A, is frequently lost in human cancers, as presented in the Cancer Genome Atlas database (PMID: 22901813), while somatic loss-of-function zinc finger mutations of LRF/ZBTB7A present another mechanism promoting cancer progression [104].
Collectively, the data presented so far suggest that LRF/ZBTB7A is a pivotal factor regulating many different aspects of cancer progression (Fig. 2 and Table 1).
LRF/ZBTB7A silencing facilitates apoptosis
Recent studies show that LRF/ZBTB7A is capable of promoting apoptosis via the p53 pathway [37, 68]. To this end, LRF/ZBTB7A acts as a master administrator of cellular transformation and proliferation and its silencing potently induces the p53 pathway and the two subsequent apoptotic signaling pathways: (1) the mitochondria-mediated (intrinsic) and (2) the death receptor-mediated (extrinsic) pathway, thought to be distinct until recently [37].
The intrinsic, mitochondrial apoptotic pathway is regulated by the Bcl-2 family of proteins that administrate the release of cytochrome c from the mitochondria [105]. Bcl-2 pro-apoptotic (Bax, Bak, Bad, Bid, Bik, and Bim) protein family promotes the release of cytochrome c from the mitochondria, which initiates the apoptotic cascade by activating caspase-9, followed by the cleavage and activation of downstream effector proteases, such as caspase-3 [106]. Once activated, caspase-3 cleaves the Poly (ADP-ribose) polymerase-1 (PARP-1) into p89 and p24 fragments, promoting DNA fragmentation and eventually triggering cell apoptosis [107]. Elevated expression of p53, pro-apoptotic Bcl-2 family proteins and corresponding changes in other apoptosis-related factors, including apoptosis inducing factor (AIF) expression levels and cytochrome c release from mitochondria, other apoptosis-related factors derived from the ZBTB7A-knockdown HepG2 cell line support the proposed intrinsic mechanism [37]. Besides, the extrinsic apoptotic pathway is mediated by death receptors. Fas ligand interacts with the Fas receptor, causing caspase-8 and caspase-10 activation, which subsequently cleave directly and activate downstream effector proteases, such as caspase-3, causing cell apoptosis [108,109,110,111]. Upon LRF/ZBTB7A’s silencing the expression of the Fas receptor is increased, leading to the activation of the downstream caspase-10 and caspase-8, as were evidently upregulated in the ZBTB7A-knockdown HepG2 cell line compared to the controls [37].
In addition, caspase-8 and caspase–10 may cleave the Bcl-2 family member Bid into truncated Bid (tBid) and thus resulting in disruption and release of cytochrome c [112, 113] (Fig. 2). It is therefore speculated that LRF/ZBTB7A potentially mediates the cross-communication between the intrinsic and extrinsic apoptotic pathways, notably in cancer cells.
LRF/ZBTB7A expression levels during adipogenesis
Ιn vitro differentiation of preadipocytes is a step-forward process. That is, upon reaching confluence, preadipocytes become contact-inhibited and proliferation stops at the G1/S phase boundary. Following hormonal induction, preadipocytes complete two cycles of cell division known as mitotic clonal expansion (days 1–2), and finally, after a second growth arrest (days 3–4), they undergo terminal differentiation (days 4–10) resulting in the expression of genes defining the adipocyte phenotype [114]. The molecular mechanisms regulating the transition between cellular proliferation and differentiation of preadipocytes remain in part elusive.
Ιn human adipose tissue, LRF/ZBTB7A mRNA is normally expressed, though the highest levels are found in samples from morbidly obese subjects as displayed in oligonucleotide microarray experiments, originally designed to identify novel rosiglitazone [oral antidiabetic drug that activates gamma isoform of peroxisome proliferator-activated receptor (PPARγ)] targeted genes in early human preadipocyte differentiation [115, 116]. Consistently, in similar experiments, using murine 3T3-L1 preadipocytes over-expressing Lrf/Zbtb7a, it was evident that PPARγ and aP2 mRNAs were both significantly upregulated [117], and this induction was related to the early phase of differentiation process (days 2-4), urging terminal cell differentiation towards adipogenesis. Furthermore, murine cell lines constitutively expressing Lrf/Zbtb7a showed evidence for accelerated adipogenesis with earlier induction of differentiation markers and enhanced lipid accumulation, suggesting that this TF contributes significantly in the differentiation process due to downregulation of E2F-4 [117], a transcriptional factor known to inhibit PPARγ expression [118]. Since the LRF/ZBTB7A protein levels peak at the end of mitotic clonal expansion, it has been hypothesized that this TF facilitates the cells’ terminal differentiation during adipogenesis. Indeed, 3T3-L1 cells stably overexpressing Lrf/Zbtb7a showed reduced DNA synthesis and reduced expression of cyclin A, cyclin-dependent kinase 2, and p107, proteins known to be involved in the regulation of mitotic clonal expansion. In addition, Lrf/Zbtb7a reduced the transcriptional activity of the cyclin A promoter [117]. Similarly, human LRF/ZBTB7A reduces the activity of cyclin A promoter indirectly by inhibiting Sp1 while represses the activity of E2F-4 promoter directly [119] (Fig. 3). Taken together, LRF/ZBTB7A is induced during the early phases of human and murine preadipocyte differentiation where it contributes to adipogenesis through influencing the switch from cellular proliferation to terminal differentiation by exhibiting thus a dual function of promoting both growth arrest and terminal adipocyte differentiation.

LRF/ZBTB7A switches 3T3-L1 preadipocytes’ fate from cellular proliferation to terminal differentiation. Human LRF/ZBTB7A expression in 3T3-L1 preadipocytes promotes cell growth arrest at the stage of mitotic clonal expansion via inhibition of Sp1-dependent activation of cyclin A, cyclin-dependent kinase 2, and p107, while indirectly (via E2F-4 inhibition] activates PPARγ and aP2 transcription factors, known to be involved in terminal differentiation of 3T3-L1 cells into adipocytes (in red color are cellular processes inhibited and in green color are cellular processes activated by LRF/ZBTB7A expression). Abbreviations: aP2, adipocyte fatty acid-binding protein; E2F-4, E2F transcription factor 4; PPARγ, peroxisome proliferator-activated receptor gamma; Sp1, specificity protein 1
ZBTB7A and insulin: a futile cycle
Type 1 diabetes (T1D) is a complex disease with a strong genetic background, and there are more than 40 loci identified to correlate with increased T1D susceptibility [120, 121]. Interestingly, insulin self-tolerance is, to a large extent, assured by the expression of small quantities of insulin by medullary thymic epithelial cells (mTECs) [122, 123]. The transcriptional regulation of thymic insulin gene expression was poorly understood until recently, where it has been confirmed the binding of specific TFs, namely LRF/ZBTB7A, AP-1 (JUN), and EWS RNA binding protein 1 (EWSR1), to the human insulin gene promoter alone or joined to a class I or class III VNTR allele, located 596 bp upstream of the insulin gene [124,125,126]. The shortest (class I) VNTR allele, consisting of 26 to 63 repeats, predisposes to T1D, while the long (class III) VNTR allele, consisting of 140 to 210 repeats, reduces 2- to 4-fold the risk for T1D compared to class I/I homozygotes [127, 128]. The selection for self-tolerant T cells takes place in the thymus and requires the AIRE, the autoimmune regulator, responsible for the expression of thousands of tissue-restricted antigens (TRAs) in a specialized subset of medullary thymic epithelial cells (mTECs) [129]. The AIRE-dependence of thymic insulin expression has been directly demonstrated in the mouse [130, 131] and indirectly in the human [132].
All three proteins (LRF/ZBTB7A, AP-1, and EWSR1) could induce insulin expression in transfected HEK-293 cells, but LRF/ZBTB7A provided the most robust results especially in the presence of the AIRE, with an additional 11-fold increase of the insulin mRNA levels from a co-transfected reporter driven by the class III VNTR allele. Consequently, LRF/ZBTB7A was identified as a strong candidate for the regulation of thymic insulin expression [133]. On the other hand, it has been shown that insulin stimulates the murine Zbtb7A promoter activity and enhances endogenous Zbtb7A mRNA and protein levels in a dose- and time-dependent manner. Furthermore, the PI3K/AKT cascade and transcription factor Sp1, known to recognize and bind methylated regions in genes’ promoter(s) [134], have been implicated to the observed insulin-induced Lrf/Zbtb7a expression in HepG2 cells [135].
Conclusions
LRF/ZBTB7A presents pleiotropic actions and is involved in the regulation of many fundamental physiological cell processes as follows: the terminal erythrocyte and adipocyte differentiation, the lineage cell fate decisions of pre-mature and mature B and T cells, the insulin self-tolerance, and also, pathophysiological conditions such as numerous cancer types.
Collectively, LRF/ZBTB7A is rising to a central player controlling proper cell proliferation and differentiation by participating in several molecular mechanisms and interacting with an impressive variety of binding partners in a tissue- and context-dependent manner to exert either oncogenic or oncosuppressive functions. Altered expression profile of LRF/ZBTB7A has been detected in several types of cancer, supporting the idea that it potentially bridges discrete downstream cell signaling pathways as well as intrinsic/extrinsic apoptotic pathways via regulation of mutual participants. To this end, LRF/ZBTB7A functions mainly as a co-operator with other TFs rather than a master or “pioneering” TF and does not directly activate or repress gene promoters as has been established for NF-κB or the Hypoxia Inducible Factor (HIF), but alternatively influences the accessibility of other TFs and DNA-binding proteins.
A critical LRF/ZBTB7A feature is its penchant for binding CG reach DNA sequences, located within regulatory gene regions and its evident ability to from homo- or hetero-dimmers capable to recruit chromatin remodeling complexes such as HDACs and NuRD. It remains to be clarified whether LRF/ZBTB7A belongs to the recently introduced generation of TFs, with the capability to sense epigenetic events and orchestrate reciprocal action as a response, which further transduces chromatin context and triggers changes in gene expression.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed.
Abbreviations
- AKT:
-
Akt serine/threonine kinase
- AR:
-
Androgen receptor
- AXL:
-
AXL receptor tyrosine kinase
- BCL2:
-
B-cell lymphoma 2
- BCL6:
-
B-cell lymphoma 6
- C/EBPα:
-
CCAAT/enhancer-binding protein alpha
- CDKs:
-
Cyclin-dependent kinases
- DAP5:
-
Death-associated protein 5
- DLK1:
-
Delta like non-canonical Notch ligand 1
- EGF:
-
Epidermal growth factor
- ELK1:
-
ETS Like-1 protein
- ERK:
-
Extracellular signal-regulated kinase
- ETS-1:
-
Protein C-ets-1
- FADD:
-
Fas-associated via death domain
- FAS receptor:
-
Fas cell surface death receptor
- HP1γ:
-
Heterochromatin protein 1γ
- ILF2/3:
-
Interleukin enhancer binding factors 2/3
- LncRNA GAS5:
-
Long non-coding RNA Growth arrest-specific transcript 5
- MCAM:
-
Melanoma cell adhesion molecule
- MEF2D:
-
Myocyte enhancer factor 2D
- MEK:
-
Mitogen-activated protein kinases
- miR-100:
-
microRNA-100
- miR-106b:
-
microRNA-106b
- miR-125:
-
microRNA-125
- miR-125a:
-
microRNA-125a
- miR-137:
-
microRNA-137
- miR-137:
-
microRNA-137
- miR-21:
-
microRNA-21
- miR-520e:
-
microRNA-520e
- miR-663a:
-
microRNA-663a
- MT1-MMP:
-
Membrane type 1-matrix metalloproteinase
- NF-κB:
-
Nuclear factor-κB
- p19Arf :
-
Cyclin-dependent kinase inhibitor 2A
- p21:
-
Cyclin-dependent kinase inhibitor 1A
- p27:
-
Cyclin-dependent kinase inhibitor 1B
- p53:
-
Tumor protein p53
- PI3K:
-
Phosphoinositide-3-kinase-protein kinase
- PVT1:
-
Plasmacytoma variant translocation 1
- RAF:
-
Raf-1 proto-oncogene, serine/threonine kinase
- RAS:
-
RAS oncogene family
- RUNX1/RUNX1T1:
-
RUNX family transcription factor 1/RUNX1 partner transcriptional co-repressor 1
- Smad4:
-
SMAD family member 4
- Sox9:
-
Sry-related HMG box 9
- SP1:
-
Specificity protein 1
- STRN4:
-
Striatin-4
- TGF-β:
-
Transforming growth factor beta
References
Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–812.
Chondrou V, Stavrou EF, Markopoulos G, Kouraklis-Symeonidis A, Fotopoulos V, Symeonidis A, et al. Impact of ZBTB7A hypomethylation and expression patterns on treatment response to hydroxyurea. Human genomics. 2018;12(1):45.
Ramos Pittol JM, Oruba A, Mittler G, Saccani S, van Essen D. Zbtb7a is a transducer for the control of promoter accessibility by NF-kappa B and multiple other transcription factors. PLoS Biology. 2018;16(5):e2004526.
Testa U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia. 2004;18(7):1176–99.
Maeda T, Ito K, Merghoub T, Poliseno L, Hobbs RM, Wang G, et al. LRF is an essential downstream target of GATA1 in erythroid development and regulates BIM-dependent apoptosis. Dev Cell. 2009;17(4):527–40.
Norton LJ, Funnell APW, Burdach J, Wienert B, Kurita R, Nakamura Y, et al. KLF1 directly activates expression of the novel fetal globin repressor ZBTB7A/LRF in erythroid cells. Blood Adv. 2017;1(11):685–92.
Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, et al. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Molecular Cell. 2009;36(4):682–95.
Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351(6270):285–9.
Rigano P, Pecoraro A, Calzolari R, Troia A, Acuto S, Renda D, et al. Desensitization to hydroxycarbamide following long-term treatment of thalassaemia intermedia as observed in vivo and in primary erythroid cultures from treated patients. British journal of haematology. 2010;151(5):509–15.
Martyn GE, Wienert B, Yang L, Shah M, Norton LJ, Burdach J, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. 2018;50(4):498–503.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33.
Mak TW, Saunders ME, Jett BD. B cell development, activation and effector functions. In: Mak TW, Saunders ME, Jett BD, editors. Primer to the Immune Response. 2nd ed. Boston: Acadenic Cell; 2014. p. 111–42.
Maeda T, Merghoub T, Hobbs RM, Dong L, Maeda M, Zakrzewski J, et al. Regulation of B versus T lymphoid lineage fate decision by the proto-oncogene LRF. Science. 2007;316(5826):860–6.
Lee SU, Maeda M, Ishikawa Y, Li SM, Wilson A, Jubb AM, et al. LRF-mediated Dll4 repression in erythroblasts is necessary for hematopoietic stem cell maintenance. Blood. 2013;121(6):918–29.
He X, He X, Dave VP, Zhang Y, Hua X, Nicolas E, et al. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 2005;433(7028):826–33.
Vacchio MS, Wang L, Bouladoux N, Carpenter AC, Xiong Y, Williams LC, et al. A ThPOK-LRF transcriptional node maintains the integrity and effector potential of post-thymic CD4+ T cells. Nat Immunol. 2014;15(10):947–56.
Carpenter AC, Grainger JR, Xiong Y, Kanno Y, Chu HH, Wang L, et al. The transcription factors Thpok and LRF are necessary and partly redundant for T helper cell differentiation. Immunity. 2012;37(4):622–33.
Carpenter AC, Wohlfert E, Chopp LB, Vacchio MS, Nie J, Zhao Y, et al. Control of regulatory T cell differentiation by the transcription factors Thpok and LRF. J Immunol. 2017;199(5):1716–28.
Sakurai N, Maeda M, Lee SU, Ishikawa Y, Li M, Williams JC, et al. The LRF transcription factor regulates mature B cell development and the germinal center response in mice. J Clin Invest. 2011;121(7):2583–98.
Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. 2012;247(1):172–83.
Suan D, Sundling C, Brink R. Plasma cell and memory B cell differentiation from the germinal center. Curr Opin Immunol. 2017;45:97–102.
Lunardi A, Guarnerio J, Wang G, Maeda T, Pandolfi PP. Role of LRF/Pokemon in lineage fate decisions. Blood. 2013;121(15):2845–53.
Maeda T. Regulation of hematopoietic development by ZBTB transcription factors. Int J Hematol. 2016;104(3):310–23.
DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metabol. 2016;23(1):27–47.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Tumoren. Biochemische Zeitschrift. 1924;152:319–44.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330(6009):1340–4.
Ma Y, Yu C, Mohamed EM, Shao H, Wang L, Sundaresan G, et al. A causal link from ALK to hexokinase II overexpression and hyperactive glycolysis in EML4-ALK-positive lung cancer. Oncogene. 2016;35(47):6132–42.
Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18(20):5546–53.
Lim SO, Li CW, Xia W, Lee HH, Chang SS, Shen J, et al. EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Research. 2016;76(5):1284–96.
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325(5947):1555–9.
Liu XS, Haines JE, Mehanna EK, Genet MD, Ben-Sahra I, Asara JM, et al. ZBTB7A acts as a tumor suppressor through the transcriptional repression of glycolysis. Genes Dev. 2014;28(17):1917–28.
Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009;9(9):1084–101.
Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metabol. 2013;18(5):617–33.
Zhang YQ, Xiao CX, Lin BY, Shi Y, Liu YP, Liu JJ, et al. Silencing of Pokemon enhances caspase-dependent apoptosis via fas- and mitochondria-mediated pathways in hepatocellular carcinoma cells. PloS One. 2013;8(7):e68981.
Maeda T, Hobbs RM, Merghoub T, Guernah I, Zelent A, Cordon-Cardo C, et al. Role of the proto-oncogene Pokemon in cellular transformation and ARF repression. Nature. 2005;433(7023):278–85.
Hartmann L, Dutta S, Opatz S, Vosberg S, Reiter K, Leubolt G, et al. ZBTB7A mutations in acute myeloid leukaemia with t(8;21) translocation. Nat Commun. 2016;7:11733.
Zhao Z, Wang J, Wang S, Chang H, Zhang T, Qu J. LncRNA CCAT2 promotes tumorigenesis by over-expressed Pokemon in non-small cell lung cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;87:692-7.
Zhang QL, Xing XZ, Li FY, Xing YJ, Li J. Pretreatment Pokemon level as a predictor of response to cisplatin and paclitaxel in patients with unresectable non-small cell lung cancer. Oncol Res Treat. 2015;38(10):496–502.
Zhao ZH, Wang SF, Yu L, Wang J, Chang H, Yan WL, et al. Overexpression of Pokemon in non-small cell lung cancer and foreshowing tumor biological behavior as well as clinical results. Lung Cancer. 2008;62(1):113–9.
Apostolopoulou K, Pateras IS, Evangelou K, Tsantoulis PK, Liontos M, Kittas C, et al. Gene amplification is a relatively frequent event leading to ZBTB7A (Pokemon) overexpression in non-small cell lung cancer. J Pathol. 2007;213(3):294–302.
Zhu M, Li M, Wang T, Linghu E, Wu B. MicroRNA-137 represses FBI-1 to inhibit proliferation and in vitro invasion and migration of hepatocellular carcinoma cells. Tumour Biol. 2016;37(10):13995–4008.
Kong J, Liu X, Li X, Wu J, Wu N, Chen J, et al. miR-125/Pokemon auto-circuit contributes to the progression of hepatocellular carcinoma. Tumour Biol. 2016;37(1):511–9.
Kong J, Liu X, Li X, Wu J, Wu N, Chen J, et al. Pokemon promotes the invasiveness of hepatocellular carcinoma by enhancing MEF2D transcription. Hepatol Int. 2016;10(3):493–500.
Hong X, Hong XY, Li T, He CY. Pokemon and MEF2D co-operationally promote invasion of hepatocellular carcinoma. Tumour Biol. 2015;36(12):9885–93.
Fang F, Yang L, Tao Y, Qin W. FBI-1 promotes cell proliferation and enhances resistance to chemotherapy of hepatocellular carcinoma in vitro and in vivo. Cancer. 2012;118(1):134–46.
Aggarwal H, Aggarwal A, Hunter WJ 3rd, Yohannes P, Khan AU, Agrawal DK. Expression of leukemia/lymphoma related factor (LRF/Pokemon) in human benign prostate hyperplasia and prostate cancer. Exp Mol Pathol. 2011;90(2):226–30.
Jiang F, Zheng Q, Chang L, Li X, Wang X, Gu X. Pro-oncogene Pokemon promotes prostate cancer progression by inducing STRN4 expression. J Cancer. 2019;10(8):1833–45.
Jiang L, Siu MK, Wong OG, Tam KF, Lam EW, Ngan HY, et al. Overexpression of proto-oncogene FBI-1 activates membrane type 1-matrix metalloproteinase in association with adverse outcome in ovarian cancers. Mol Cancer. 2010;9:318.
Zu X, Ma J, Liu H, Liu F, Tan C, Yu L, et al. Pro-oncogene Pokemon promotes breast cancer progression by upregulating survivin expression. Breast Cancer Res. 2011;13(2):R26.
Aggarwal A, Hunter WJ 3rd, Aggarwal H, Silva ED, Davey MS, Murphy RF, et al. Expression of leukemia/lymphoma-related factor (LRF/POKEMON) in human breast carcinoma and other cancers. Exp Mol Pathol. 2010;89(2):140–8.
Qu H, Qu D, Chen F, Zhang Z, Liu B, Liu H. ZBTB7 overexpression contributes to malignancy in breast cancer. Cancer Invest. 2010;28(6):672–8.
Shi DB, Wang YW, Xing AY, Gao JW, Zhang H, Guo XY, et al. C/EBPalpha-induced miR-100 expression suppresses tumor metastasis and growth by targeting ZBTB7A in gastric cancer. Cancer Lett. 2015;369(2):376–85.
Huang R, Xie T, Zhao Y, Yao CS. Attenuation of leukemia/lymphoma-related factor protein expression inhibits glioma cell proliferation and invasion. J Environ Pathol Toxicol Oncol. 2015;34(2):125–31.
Kumari R, Li H, Haudenschild DR, Fierro F, Carlson CS, Overn P, et al. The oncogene LRF is a survival factor in chondrosarcoma and contributes to tumor malignancy and drug resistance. Carcinogenesis. 2012;33(11):2076–83.
Zhang L, Wang Y, Zhang L, Xia X, Chao Y, He R, et al. ZBTB7A, a miR-663a target gene, protects osteosarcoma from endoplasmic reticulum stress-induced apoptosis by suppressing LncRNA GAS5 expression. Cancer Lett. 2019;448:105–16.
Zhao Y, Yao YH, Li L, An WF, Chen HZ, Sun LP, et al. Pokemon enhances proliferation, cell cycle progression and anti-apoptosis activity of colorectal cancer independently of p14ARF-MDM2-p53 pathway. Med Oncol. 2014;31(12):288.
Joo JW, Kim HS, Do SI, Sung JY. Expression of zinc finger and BTB domain-containing 7A in colorectal carcinoma. Anticancer Res. 2018;38(5):2787–92.
Zhu M, Li M, Zhang F, Feng F, Chen W, Yang Y, et al. FBI-1 enhances ETS-1 signaling activity and promotes proliferation of human colorectal carcinoma cells. PloS One. 2014;9(5):e98041.
Zhu M, Wang P, Feng F, Li MY. LRF inhibits p53 expression in colon cancer cells via modulating DAP5 activity. Cell Biochem Funct. 2017;35(7):401–6.
Wang L, Li Q, Ye Z, Qiao B. Pokemon/miR-137 auto-regulatory circuit promotes the progression of renal carcinoma. Oncol Res. 2019;27(9):1007–14.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Zhu X, Dai Y, Chen Z, Xie J, Zeng W, Lin Y. Knockdown of Pokemon protein expression inhibits hepatocellular carcinoma cell proliferation by suppression of AKT activity. Oncol Res. 2013;20(8):377–81.
Lin CC, Zhou JP, Liu YP, Liu JJ, Yang XN, Jazag A, et al. The silencing of Pokemon attenuates the proliferation of hepatocellular carcinoma cells in vitro and in vivo by inhibiting the PI3K/Akt pathway. PloS One. 2012;7(12):e51916.
Choi WI, Jeon BN, Yoon JH, Koh DI, Kim MH, Yu MY, et al. The proto-oncoprotein FBI-1 interacts with MBD3 to recruit the Mi-2/NuRD-HDAC complex and BCoR and to silence p21WAF/CDKN1A by DNA methylation. Nucleic Acids Res. 2013;41(13):6403–20.
Choi WI, Jeon BN, Yun CO, Kim PH, Kim SE, Choi KY, et al. Proto-oncogene FBI-1 represses transcription of p21CIP1 by inhibition of transcription activation by p53 and Sp1. J Biol Chem. 2009;284(19):12633–44.
Hojo N, Tatsumi N, Moriguchi N, Matsumura A, Morimoto S, Nakata J, et al. A Zbtb7a proto-oncogene as a novel target for miR-125a. Mol Carcinog. 2016;55(12):2001–9.
Jeon BN, Yoo JY, Choi WI, Lee CE, Yoon HG, Hur MW. Proto-oncogene FBI-1 (Pokemon/ZBTB7A) represses transcription of the tumor suppressor Rb gene via binding competition with Sp1 and recruitment of co-repressors. J Biol Chem. 2008;283(48):33199–210.
De Luca A, Maiello MR, D'Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancerpathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl2):S17–27.
Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189.
Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Lett. 2016;12(5):3045–50.
Vredeveld LC, Rowland BD, Douma S, Bernards R, Peeper DS. Functional identification of LRF as an oncogene that bypasses RASV12-induced senescence via upregulation of CYCLIN E. Carcinogenesis. 2010;31(2):201–7.
Wong M, Hyodo T, Asano E, Funasaka K, Miyahara R, Hirooka Y, et al. Silencing of STRN4 suppresses the malignant characteristics of cancer cells. Cancer Sci. 2014;105(12):1526–32.
Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415–24.
Chen L, Zhong J, Liu JH, Liao DF, Shen YY, Zhong XL, et al. Pokemon inhibits transforming growth factor beta-Smad4-related cell proliferation arrest in breast cancer through specificity protein 1. J Breast Cancer. 2019;22(1):15–28.
Yang Y, Cui J, Xue F, Zhang C, Mei Z, Wang Y, et al. Pokemon (FBI-1) interacts with Smad4 to repress TGF-beta-induced transcriptional responses. Biochimica et biophysica acta. 2015;1849(3):270–81.
Aggarwal H, Aggarwal A, Agrawal DK. Epidermal growth factor increases LRF/Pokemon expression in human prostate cancer cells. Exp Mol Pathol. 2011;91(2):496–501.
Zu X, Yu L, Sun Q, Liu F, Wang J, Xie Z, et al. SP1 enhances Zbtb7A gene expression via direct binding to GC box in HePG2 cells. BMC Res Notes. 2009;2:175.
Li W, Kidiyoor A, Hu Y, Guo C, Liu M, Yao X, et al. Evaluation of transforming growth factor-beta1 suppress Pokemon/epithelial-mesenchymal transition expression in human bladder cancer cells. Tumour Biol. 2015;36(2):1155–62.
Wang X, Cao P, Li Z, Wu D, Wang X, Liang G. EPAS-1 mediates SP-1-dependent FBI-1 expression and regulates tumor cell survival and proliferation. Int J Mol Sci. 2014;15(9):15689–99.
Liang X, Zhao Q, Geng T, Luo S, He Q. MiR-106b regulates the apoptosis and tumorigenesis of hepatocellular carcinoma via targeting zinc finger and BTB domain-containing protein 7A (Zbtb7a). J Biochem Mol Toxicol. 2018;32(8):e22169.
Jin XL, Sun QS, Liu F, Yang HW, Liu M, Liu HX, et al. microRNA 21-mediated suppression of Sprouty1 by Pokemon affects liver cancer cell growth and proliferation. J Cell Biochem. 2013;114(7):1625–33.
Zhijun Z, Jingkang H. MicroRNA-520e suppresses non-small-cell lung cancer cell growth by targeting Zbtb7a-mediated Wnt signaling pathway. Biochem Biophys Res Commun. 2017;486(1):49–56.
Canzio D, Larson A, Narlikar GJ. Mechanisms of functional promiscuity by HP1 proteins. Trends Cell Biol. 2014;24(6):377–86.
Alam H, Li N, Dhar SS, Wu SJ, Lv J, Chen K, et al. HP1gamma promotes lung adenocarcinoma by downregulating the transcription-repressive regulators NCOR2 and ZBTB7A. Cancer Res. 2018;78(14):3834–48.
Wang G, Lunardi A, Zhang J, Chen Z, Ala U, Webster KA, et al. Zbtb7a suppresses prostate cancer through repression of a Sox9-dependent pathway for cellular senescence bypass and tumor invasion. Nat Genet. 2013;45(7):739–46.
Thomsen MK, Ambroisine L, Wynn S, Cheah KS, Foster CS, Fisher G, et al. SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res. 2010;70(3):979–87.
Razzak M. Genetics: ZBTB7A suppresses castration-resistant prostate cancer. Nat Rev Clin Oncol. 2013;10(8):427.
Cui J, Yang Y, Zhang C, Hu P, Kan W, Bai X, et al. FBI-1 functions as a novel AR co-repressor in prostate cancer cells. Cell Mol Life Sci. 2011;68(6):1091–103.
Guarnerio J, Riccardi L, Taulli R, Maeda T, Wang G, Hobbs RM, et al. A genetic platform to model sarcomagenesis from primary adult mesenchymal stem cells. Cancer Discov. 2015;5(4):396–409.
Molloy ME, Lewinska M, Williamson AK, Nguyen TT, Kuser-Abali G, Gong L, et al. ZBTB7A governs estrogen receptor alpha expression in breast cancer. J Mol Cell Biol. 2018;10(4):273–84.
Sun G, Peng B, Xie Q, Ruan J, Liang X. Upregulation of ZBTB7A exhibits a tumor suppressive role in gastric cancer cells. Mol Med Rep. 2018;17(2):2635–41.
Liu XS, Genet MD, Haines JE, Mehanna EK, Wu S, Chen HI, et al. ZBTB7A Suppresses melanoma metastasis by transcriptionally repressing MCAM. Mol Cancer Res. 2015;13(8):1206–17.
Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18(5):309–24.
Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6910–24.
Zhang NN, Sun QS, Chen Z, Liu F, Jiang YY. Homeostatic regulatory role of Pokemon in NF-kappaB signaling: stimulating both p65 and IkappaBalpha expression in human hepatocellular carcinoma cells. Mol Cell Biochem. 2013;372(1-2):57–64.
Lee DK, Kang JE, Park HJ, Kim MH, Yim TH, Kim JM, et al. FBI-1 enhances transcription of the nuclear factor-kappaB (NF-kappaB)-responsive E-selectin gene by nuclear localization of the p65 subunit of NF-kappaB. J Biol Chem. 2005;280(30):27783–91.
Liu XS, Chandramouly G, Rass E, Guan Y, Wang G, Hobbs RM, et al. LRF maintains genome integrity by regulating the non-homologous end joining pathway of DNA repair. Nat Commun. 2015;6:8325.
Bielli P, Busa R, Di Stasi SM, Munoz MJ, Botti F, Kornblihtt AR, et al. The transcription factor FBI-1 inhibits SAM68-mediated BCL-X alternative splicing and apoptosis. EMBO Reports. 2014;15(4):419–27.
Guarnerio J, Zhang Y, Cheloni G, Panella R, Mae Katon J, Simpson M, et al. Intragenic antagonistic roles of protein and circRNA in tumorigenesis. Cell Res. 2019;29(8):628–40.
Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med. 2018;24(2):165–75.
Liu XS, Liu Z, Gerarduzzi C, Choi DE, Ganapathy S, Pandolfi PP, et al. Somatic human ZBTB7A zinc finger mutations promote cancer progression. Oncogene. 2016;35(23):3071–8.
Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-ordeath switch. Nat Rev Cancer. 2002;2:647–56.
Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121(5):671–4.
Scovassi AI, Poirier GG. Poly(ADP-ribosylation) and apoptosis. Mol Cell Biochem. 1999;199(1-2):125–37.
Lee SH, Shin MS, Lee HS, Bae JH, Lee HK, Kim HS, et al. Expression of Fas and Fas-related molecules in human hepatocellular carcinoma. Human Pathol. 2001;32(3):250–6.
Abd El-Ghany RM, Sharaf NM, Kassem LA, Mahran LG, Heikal OA. Thymoquinone triggers anti-apoptotic signaling targeting death ligand and apoptotic regulators in a model of hepatic ischemia reperfusion injury. Drug Discov Ther. 2009;3(6):296–306.
Okamoto K, Kobayashi T, Kobata T, Hasunuma T, Kato T, Sumida T, et al. Fas-associated death domain protein is a Fas-mediated apoptosis modulator in synoviocytes. Rheumatology. 2000;39(5):471–80.
Lin S, Liu X, Yin R, Kong D, Qu Y, Zhang Y. Inhibitory effects of short hairpin RNA against caspase-8 on apoptosis of murine hepatoma Hepa1-6 cells. Biosci Trends. 2009;3(2):53–7.
Fulda S. Caspase-8 in cancer biology and therapy. Cancer Lett. 2009;281(2):128–33.
Qi F, Li A, Inagaki Y, Xu H, Wang D, Cui X, et al. Induction of apoptosis by cinobufacini preparation through mitochondria- and Fas-mediated caspase-dependent pathways in human hepatocellular carcinoma cells. Food Chem Toxicol. 2012;50(2):295–302.
Nakae J, Kitamura T, Kitamura Y, Biggs WH 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell. 2003;4(1):119–29.
Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 1995;270(22):12953–6.
Digby JE, Montague CT, Sewter CP, Sanders L, Wilkison WO, O'Rahilly S, et al. Thiazolidinedione exposure increases the expression of uncoupling protein 1 in cultured human preadipocytes. Diabetes. 1998;47(1):138–41.
Laudes M, Christodoulides C, Sewter C, Rochford JJ, Considine RV, Sethi JK, et al. Role of the POZ zinc finger transcription factor FBI-1 in human and murine adipogenesis. J Biol Chem. 2004;279(12):11711–8.
Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell. 2002;3(1):39–49.
Laudes M, Bilkovski R, Oberhauser F, Droste A, Gomolka M, Leeser U, et al. Transcription factor FBI-1 acts as a dual regulator in adipogenesis by coordinated regulation of cyclin-A and E2F-4. J Mol Med. 2008;86(5):597–608.
Ounissi-Benkalha H, Polychronakos C. The molecular genetics of type 1 diabetes: new genes and emerging mechanisms. Trends Mol Med. 2008;14(6):268–75.
Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet. 2011;12(11):781–92.
Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet. 1997;15(3):289–92.
Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes. 2002;51(5):1383–90.
Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet. 1995;9(3):284–92.
Bell GI, Selby MJ, Rutter WJ. The highly polymorphic region near the human insulin gene is composed of simple tandemly repeating sequences. Nature. 1982;295(5844):31–5.
Vafiadis P, Ounissi-Benkalha H, Palumbo M, Grabs R, Rousseau M, Goodyer CG, et al. Class III alleles of the variable number of tandem repeat insulin polymorphism associated with silencing of thymic insulin predispose to type 1 diabetes. J Clin Endocrinol Metabol. 2001;86(8):3705–10.
Anjos S, Polychronakos C. Mechanisms of genetic susceptibility to type I diabetes: beyond HLA. Mol Genet Metabol. 2004;81(3):187–95.
Durinovic-Belló I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT. Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun. 2010;11(2):188–93.
Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immun. 2001;2(11):1032–9.
Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–401.
Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23(2):227–39.
Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322(26):1829–36.
St-Jean JR, Ounissi-Benkalha H, Polychronakos C. Yeast one-hybrid screen of a thymus epithelial library identifies ZBTB7A as a regulator of thymic insulin expression. Mol Immunol. 2013;56(4):637–42.
Cejas RB, Ferguson DC, Quinones-Lombrana A, Bard JE, Blanco JG. Contribution of DNA methylation to the expression of FCGRT in human liver and myocardium. Sci Reports. 2019;9(1):8674.
Tian J, Jiang Y. Insulin upregulates the expression of zinc finger and BTB domain-containing 7A in HepG2 cells. Mol Med Reports. 2012;6(6):1379–84.
Acknowledgements
We wish to thank Dr V. Fotopoulos for his contribution and technical assistance with the figures of the manuscript.
Funding
This work was supported by the PENER-EAP grant from the Hellenic Open University.
Author information
Authors and Affiliations
Contributions
CC and MS collected literature and drafted the manuscript, VC collected and discussed literature, GPP and AP edited the manuscript, and AS conceptualised the idea and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Constantinou, C., Spella, M., Chondrou, V. et al. The multi-faceted functioning portrait of LRF/ZBTB7A. Hum Genomics 13, 66 (2019). https://doi.org/10.1186/s40246-019-0252-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-019-0252-0