
Abstract
Acute kidney injury (AKI) is an important clinical issue that is associated with significant morbidity and mortality. Despite research advances over the past decades, the complex pathophysiology of AKI is not fully understood. The regulatory mechanisms underlying post-AKI repair and fibrosis have not been clarified either. Furthermore, there is no definitively effective treatment for AKI. MicroRNAs (miRNAs) are endogenous single-stranded noncoding RNAs of 19~23 nucleotides that have been shown to be crucial to the post-transcriptional regulation of various cellular biological functions, including proliferation, differentiation, metabolism, and apoptosis. In addition to being fundamental to normal development and physiology, miRNAs also play important roles in various human diseases. In AKI, some miRNAs appear to act pathogenically by promoting inflammation, apoptosis, and fibrosis, while others may act protectively by exerting anti-inflammatory, anti-apoptotic, anti-fibrotic, and pro-angiogenic effects. Thus, miRNAs have not only emerged as novel biomarkers for AKI; they also hold promise to be potential therapeutic targets.
Similar content being viewed by others

Background
Acute kidney injury
Acute kidney injury (AKI) is a complex syndrome that occurs in a variety of settings with clinical manifestations ranging from a minimal elevation in serum creatinine to anuric renal failure. AKI conveys significant morbidity and mortality, is a major risk factor of chronic kidney disease, and is thus associated with huge health and socioeconomic burdens [1, 2]. Despite research advances in the past decades, however, the complex pathophysiology of AKI is not fully understood. The regulatory mechanisms underlying post-AKI repair and fibrosis remain to be clarified. Furthermore, there is no definitively effective treatment for AKI.
MicroRNA biogenesis and function
MicroRNAs (miRNAs) are endogenous single-stranded noncoding mRNAs of 19~23 nucleotides. They were first discovered in Caenorhabditis elegans by Ambros’s group in 1993 [3] and show surprisingly high conservation across species. The evidence accumulated over the past two decades shows that miRNAs play a critical role in the post-transcriptional regulation of almost all biological cell functions, including proliferation, differentiation, metabolism, and apoptosis [4]. miRNAs, which are expressed in a tissue-specific manner, are fundamental to normal development and physiology [4] and are involved in the pathologic pathways of many disease models.
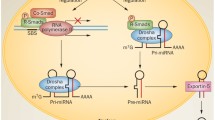
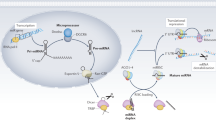
To date, more than 2000 miRNAs have been discovered in the human genome. The miRNA-encoded genes are found as either independent genes having their own promoters, or as sequences in the introns of protein-coding genes [5]. RNA polymerase II transcribes an miRNA gene into a primary transcript (called a pri-miRNA) of several kilobases that can encode either an individual miRNA or a polycistronic cluster of two or more miRNAs. The RNase III enzyme, DROSHA, and its cofactor DGCR8 (Di-George syndrome critical region gene 8 or Pasha), cleave a pri-miRNA at its stem-loop structure, generating an approximately 70-nucleotide intermediate called the pre-miRNA. Exportin-5 exports the pre-miRNA from the nucleus to the cytoplasm, and the RNase III enzyme, DICER, further cleaves it to yield a single-stranded mature miRNA. To perform its function, an miRNA is incorporated along with the argonaute (AGO) protein to form an effector complex called the RNA-induced silencing complex (RISC). RISC binds to the 3′-untranslated region (UTR) of a target messenger RNA (mRNA), leading to the repression of either protein translation or mRNA degradation. Unlike small interfering RNAs in plants, miRNAs do not require complete complementarity to bind their targets. Instead, the evidence suggests that the “seed sequence” (nucleotides 2 through 8 of the miRNA) is the most important region for the ability of an miRNA to bind and regulate its target gene(s). Once bound, miRNAs induce repression by blocking the initiation or elongation of translation or de-adenylating the mRNA transcripts. Because miRNAs do not require complete complementarity to repress gene expression, a given miRNA can regulate multiple mRNA transcripts and a given mRNA transcript can be repressed by multiple miRNAs. It is estimated that miRNAs regulate more than half of the protein-coding genes in human [6]. Moreover, miRNAs have been implicated in various human diseases [7, 8], including kidney diseases, such as polycystic kidney disease [9], renal cell carcinoma [10], diabetic nephropathy [11], lupus nephritis, [12] and renal allograft rejection [13]. In the past few years, researchers have begun to address the relevance of miRNAs to AKI.
miRNAs in acute kidney injury
The miRNAs that have been implicated in AKI are summarized in Tables 1 and 2, and those with potential pathological or protective roles are summarized in Table 3. The first evidence of miRNAs having pathological roles in AKI was reported by Wei et al. who developed a Dicer-knockout mouse model, in which Dicer was specifically deleted from proximal tubular cells. These mice exhibit a global down-regulation of microRNAs in the renal cortex. They have normal renal function and histology under control conditions but show resistance to the AKI that follows bilateral renal ischemia-reperfusion (IRI). Under the latter conditions, Dicer-null mice show significantly better renal function, less tissue damage, less tubular apoptosis, and better survival than their wild-type counterparts [14].
miR-10a is renal tubule-specific miRNA that is released from kidney tissues upon injury. In rodent models of renal IRI and streptozocin (STZ)-induced diabetic nephropathy, the levels of miR-10a are increased decreased in urine and kidney tissue, respectively [15, 16]. miR-10a is thought to exert protective actions during injury by targeting IL-12/IL-23p40 and the pro-apoptotic protein BIM [17]. In humans, decreased plasma levels of miR-10a have been shown to predict AKI in critical patients of intensive care units (ICUs) [18].
The members of the miR-17 family have been shown to be induced by pro-inflammatory cytokines, and their tissue expressions are increased in rodent models of renal IRI [19, 20].
miR-21 appears to play a dual role; on the one hand, it protects against injury by inhibiting apoptosis and inflammation; on the other hand, it may amplify the injury response and promote fibrosis. Studies have shown that miR-21 inhibits apoptosis by down-regulating programmed cell death protein 4 (PDCD4), down-regulating phosphatase and tensin homolog (PTEN), activating the AKT pathway, up-regulating B cell lymphoma 2 (BCL-2), and decreasing the levels of active caspase-3 and caspase-8 proteins [21, 22]. Up-regulation of miR-21 also inhibits inflammation by decreasing nuclear factor-kappaB (NF-kB), tumor necrosis factor (TNF), interleukin 6 (IL-6), and IL-18, and by increasing IL-10 [21]. Experimental up-regulation of miR-21 provides morphologic and functional renoprotection in animal models of AKI [21–23]. miR-21 is induced by transforming growth factor beta (TGF-β)/Smad, hypoxia inducible factor 1 alpha (HIF-1α), TNF, and fibroblast growth factor 2 (FGF-2) [24, 25], and this miRNA promotes fibrosis by targeting peroxisome proliferator-activated receptor alpha (Pparα) and altering lipid metabolism [26]. miR-21 also targets Mpv17l, a mitochondria inhibitor of reactive oxygen species (ROS) [26]. miR-21 inhibits autophagy by targeting Ras-related proteins in brain 11 a (Rab-11a), decreasing light chain 3-II (LC3-II), decreasing beclin-1, and increasing p62 [27]. In vivo blockade of miR-21 reduces renal fibrosis and macrophage infiltration in animal models. Moreover, increased urinary and plasma levels of miR-21 have been observed in various clinical AKI settings [26, 28, 29]. For example, urine and plasma miR-21 levels were shown to correlate with AKI severity and hospital mortality and to predict the need for postoperative renal replacement therapy [28]. Interestingly, one study found decreased, but not increased, expression of miR-21 in AKI patients. Lower baseline plasma levels of miR-21 have been demonstrated to predict cardiac surgery-associated AKI [30].
miR-24 is up-regulated in mouse kidney after IRI and in patients after kidney transplantation. This miRNA enhances apoptosis by down-regulating sphingosine-1-phosphate receptor 1 (S1PR1), H2A histone family member X (H2A.X), and heme oxygenase-1 (HO-1). Inhibition of miR-24 was shown to prevent renal injury in animal models [31].
miR-26a represses IL-6 expression to promote the expansion of regulator T cells (Tregs). The tissue levels of miR-26a is down-regulated in animal models of AKI, and experimental overexpression attenuates renal IRI and improves renal recovery [32]. miR-26b is down-regulated in the tissue and blood, yet up-regulated in the urine [18, 33]. Decreased blood levels of miR-26a and miR-27a predict AKI in the ICU. Decreased blood levels of miR-26a and miR-27a prior to cardiac surgery also predict AKI later on [18].
miR-29a is highly expressed in the kidney, where it acts against fibrosis by suppressing collagen expression in tubular cells. Decreased serum levels of miR-29a have been shown to predict AKI in ICU patients, and correlate with AKI severity [18].
miR-30c, which is essential for normal kidney homoeostasis, targets several genes important for kidney structure and function. miR-30c is up-regulated in the tissue, blood, and urine obtained from animal models of contrast nephropathy and IRI [34].
miR-30d, which is released to the urine from kidney tissues following injury, down-regulates the apoptotic proteins, caspase 3 and p53, and may provide protective effects during IRI [16].
miR-101-3p is highly expressed in the kidney, and decreased serum levels of this miRNA have been shown to predict AKI in the ICU [18].
miR-122 is down-regulated in the mice kidneys of mice subjected to cisplatin-induced AKI [35]. It exerts anti-apoptotic effects by down-regulating forkhead box O3 (Foxo3).
miR-127a, which is induced by HIF-1α, participates in protecting the cytoskeleton protection (by preventing actin depolmerization), maintaining cell-matrix and cell-cell adhesion maintenance (by preventing focal adhesion complexes disassembly and tight junctions disorganization), and promoting intracellular trafficking (by targeting kinesin family member 3B) [36]. Decreased blood levels of miR-127a were shown to predict AKI in the ICU. Decreased blood levels of miR-127a prior to cardiac surgery were found be predict AKI later on [18].
miR-146a is down- and up-regulated in the blood and kidney, respectively, during AKI. Decreased blood levels have been shown to predict AKI in the ICU and correlate with the severity of AKI [18]. It is induced by NF-kB and exerts anti-inflammatory effect by down-regulating TNF receptor-associated factor 6 (TRAF-6) and interleukin-1 receptor-associated kinase 1 (IRAK-1) [37].
miR-192 is enriched in kidneys and the small intestine. It is induced by TGF-β during the stress response. It promotes fibrosis by down-regulating SIP1. It also down-regulates E3 ubiquitin ligase and murine double-minute 2 (MDM2) and results in de-repression of p53 and G2/M arrest [38]. miR-194 is also enriched in kidneys and small intestine. It is induced during the stress response, and its levels in tissue, blood, and urine levels are increased during AKI [15, 38, 39].
miR-199a exerts anti-inflammatory effect by down-regulating inhibitor of NF-kB kinases b (IKKb) [40], exhibits anti-proliferatory effect by down-regulating the proto-oncogene MET [41], and confers anti-apoptosis effect by down-regulating extracellular signal–regulated kinase 2 (ERK-2) and HIF-1α [41, 42]. Therefore, it may help limit kidney injury.
miR-126 and miR-296 have been identified in microvesicles from endothelial progenitor cells and are thought to exert renoprotective effects via their abilities to decrease apoptosis and leukocyte infiltration, while promotes angiogenesis and tubular cell proliferation [43]. Hematopoietic overexpression of miR-126 enhances stromal cell-derived factor 1/chemokine receptor type 4 (CXCR4) -dependent vasculogenic progenitor cell mobilization and promotes vascular integrity and supports renal recovery after IRI [44]. Decreased serum levels of miR-126 have been shown to predict AKI in ICU patients, and correlate with the severity of AKI [18].
Members of the miR-200 family are highly expressed in tubular structures such as renal tubules, lungs, the small intestine, and various exocrine glands. miR-200b and miR-200c have been proposed to be anti-fibrotic. They down-regulate TGFβR1 and zinc finger E-box-binding homeobox (ZEB1/ZEB2), which are transcriptional repressors of E-cadherin, and thereby prevent the epithelial-to-mesenchymal transition (EMT) induced by TGF-β [45].
miR-210 is induced by HIF1-α and released by renal endothelial cell. It regulates angiogenesis by down-regulating ephrin-A3 and up-regulating vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor 2 (VEGFR2). It also regulates mitochondria ROS production. Increased blood levels of miR-210 was shown to predict post-AKI mortality in critically ill patients [46]. In another study, decreased blood levels of miR-210 were shown to predict AKI in the ICU and correlate with the severity of AKI [18].
miR-214 is induced by TGF-β and promotes fibrosis; it has been shown to down-regulate PTEN, up-regulate the AKT pathway and inhibit apoptosis of monocytes and macrophages. miR-214 is up-regulated in various models of AKI and renal fibrosis [24, 45, 47] as well as in monocytes of animal with chronic kidney disease. Experimental antagonism of miR-214 has been shown to ameliorate renal fibrosis [24].
miR-494 is up-regulated early in AKI, with increased urine levels detected in rodent models of renal IRI and patients with AKI. It has been reported to promote apoptosis and inflammation by down-regulating activating transcription factor 3 (ATF3) and increasing IL-6, monocyte chemoattractant protein-1 (MCP-1), p-selectin [48]. Pathway analysis has suggested that it also targets adiponectin receptor 2 (ADIPOR2), BCL-2 facilitator, and insulin-like growth factor 1 receptor (IGF1R), which would increase inflammation and lead to more damage. However, miR-494 also targets pro-apoptotic proteins in the AKT pathway, and to exert protective effects. The mechanism responsible for regulating the balance between these anti- and pro- apoptotic effects requires further study.
Finally, miR-687 is induced by HIF-1, and enhances apoptosis by down-regulating PTEN. Animal studies have shown that miR-687 blockade preserves PTEN expression and attenuates cell cycle activation and decreases apoptosis, resulting in protection against kidney injury [49].
Conclusions
Many miRNAs have been implicated in the AKI. Some of them contribute to the pathogenesis by regulating apoptosis and inflammation, to amplifying or reduce acute injury responses, while others regulate fibrosis and angiogenesis, to participate in renal recovery or the progression to fibrosis. The biological and pathological functions of many miRNAs in AKI are still not fully understood in AKI. Some studies have yielded inconsistent data regarding the expression pattern of miRNAs across different samples, species, disease models, and time points. These discrepancies warrant investigations.
In addition to their tissue expressions, miRNAs may be detected in various extracellular human body fluids, such as serum, urine, saliva, and cerebral spinal fluid. miRNAs are contained in exosomes and may remained stable over prolonged periods. They may be specifically up-regulated or down-regulated in response to injury signals and/or released into body fluids from resident tissues. Certain miRNAs have been investigated for their potential to serve as novel biomarkers for the early detection or prognostication of AKI. Given the complex pathophysiology and the dynamic nature of AKI, an miRNA panel may be more feasible rather than a single miRNA. Further validation studies are needed to evaluate the clinical utility of such a panel.
Some miRNAs may be potential therapeutic targets for AKI. Recently, an miRNA inhibitor has been proven to successfully suppress the replication of hepatitis C virus in a clinical trial [50]. Systemic or local administration of specific miRNAs mimics or antagonists in vivo could offer a strategy for preventing or ameliorating AKI or barring its progression to chronic kidney disease.
In the post-genome era, miRNAs are promising rising stars in translational medicine as they offer the potential to guide the individualized diagnosis and treatment of human diseases including AKI.
Abbreviations
- AA:
-
aristolochic acid
- ADIPOR2:
-
adiponectin receptor 2
- AGO:
-
argonaute
- AKI:
-
acute kidney injury
- ATF3:
-
activating transcription factor 3
- B:
-
blood
- BCL-2:
-
B cell lymphoma 2
- BUMPT-306 cell:
-
Boston University mouse proximal tubule cell clone 306
- CdCl2 :
-
cadmium chloride
- CRL-2753 cell:
-
rat mesangial cell line
- CKD:
-
chronic kidney disease
- CXCR4:
-
chemokine receptor type 4
- DGCR8:
-
Di-George syndrome critical region gene 8 or Pasha
- DM:
-
diabetes mellitus
- EMT:
-
epithelial-to-mesenchymal transition
- ER:
-
endoplasmic reticulum
- ERK-2:
-
extracellular signal-regulated kinase 2
- FGF-2:
-
fibroblast growth factor 2
- Foxo3:
-
forkhead box O3
- FSGS:
-
focal segmental glomerulosclerosis
- H2A.X:
-
H2A histone family member X
- HEK cell:
-
human embryonic kidney cell
- HepG2 cell:
-
human hepatocellular liver carcinoma cell line
- HIF-1α:
-
hypoxia-inducible factor 1 alpha, HK-2 cell, human kidney 2 cell
- HO-1:
-
heme oxygenase-1
- HPTEC:
-
human proximal tubular epithelial cell
- HUVEC:
-
human umbilical vein endothelial cell
- ICU:
-
intensive care units
- IGF1R:
-
insulin-like growth factor 1 receptor
- IL:
-
interleukin
- IKKb:
-
inhibitor of NF-kB kinases b
- IRAK-1:
-
interleukin-1 receptor-associated kinase 1
- IRI:
-
ischemia-reperfusion injury
- K2Cr2O7 :
-
potassium dichromate
- LC3-II:
-
light chain 3-II
- MCP-1:
-
monocyte chemoattractant protein-1
- MDM2:
-
murine double-minute 2
- miRNA:
-
microRNA
- mRNA:
-
messenger RNA
- NF-kB:
-
nuclear factor-kappaB
- NRK-52E cell:
-
rat renal proximal tubular cell line
- PDCD4:
-
programmed cell death protein 4
- Pparα:
-
peroxisome proliferator activated receptor alpha
- PTC:
-
proximal tubular cell
- PTEN:
-
phosphatase and tensin homolog
- Rab-11a:
-
Ras-related proteins in brain 11 a
- RISC:
-
RNA-induced silencing complex
- ROS:
-
reactive oxygen species
- S1PR1:
-
sphingosine-1-phosphate receptor 1
- SHRSP:
-
stroke-prone spontaneously hypertensive rat
- STZ:
-
streptozocin
- T:
-
tissue
- TEC:
-
tubular epithelial cell
- TEnC:
-
tubular endothelial cell
- TEpC:
-
tubular epithelial cell
- TNF:
-
tumor necrosis factor
- TGF-β:
-
transforming growth factor beta
- TRAF-6:
-
TNF receptor-associated factor 6
- Treg:
-
regulator T cell
- U:
-
urine
- UTR:
-
untranslated region
- UUO:
-
unilateral ureteral obstruction
- VEGF:
-
vascular endothelial growth factor
- VEGFR2:
-
vascular endothelial growth factor receptor 2
- ZEB1/ZEB2:
-
zinc finger E-box-binding homeobox
References
Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114(1):5–14. doi:10.1172/jci22353.
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care (London, England). 2004;8(4):R204–12. doi:10.1186/cc2872.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi:10.1038/nrg2843.
Bhatt K, Mi QS, Dong Z. microRNAs in kidneys: biogenesis, regulation, and pathophysiological roles. Am J Physiol Renal Physiol. 2011;300(3):F602–10. doi:10.1152/ajprenal.00727.2010.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33. doi:10.1016/j.cell.2009.01.002.
Huang JB, et al. MiR-196a2 rs11614913 T > C polymorphism is associated with an increased risk of Tetralogy of Fallot in a Chinese population. Acta Cardiol Sin. 2015;31(1):18–23.
Hsu A, Chen SJ, Chang YS, Chen HC, Chu PH. Systemic approach to identify serum microRNAs as potential biomarkers for acute myocardial infarction. Biomed Res Int. 2014;2014:418628. doi:10.1155/2014/418628.
Pandey P, et al. Microarray-based approach identifies microRNAs and their target functional patterns in polycystic kidney disease. BMC Genomics. 2008;9:624. doi:10.1186/1471-2164-9-624.
Nakada C, et al. Genome-wide microRNA expression profiling in renal cell carcinoma: significant down-regulation of miR-141 and miR-200c. J Pathol. 2008;216(4):418–27. doi:10.1002/path.2437.
Kato M, Natarajan R. MicroRNA circuits in transforming growth factor-beta actions and diabetic nephropathy. Semin Nephrol. 2012;32(3):253–60. doi:10.1016/j.semnephrol.2012.04.004.
Dai Y, et al. Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int. 2009;29(7):749–54. doi:10.1007/s00296-008-0758-6.
Sui W, et al. Microarray analysis of MicroRNA expression in acute rejection after renal transplantation. Transpl Immunol. 2008;19(1):81–5. doi:10.1016/j.trim.2008.01.007.
Wei Q, et al. Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2010;21(5):756–61. doi:10.1681/asn.2009070718.
Wang JF, et al. Screening plasma miRNAs as biomarkers for renal ischemia-reperfusion injury in rats. Med Sci Monit. 2014;20:283–9. doi:10.12659/msm.889937.
Wang N, et al. Urinary microRNA-10a and microRNA-30d serve as novel, sensitive and specific biomarkers for kidney injury. PLoS One. 2012;7(12):e51140. doi:10.1371/journal.pone.0051140.
Ho J, et al. The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J Am Soc Nephrol. 2011;22(6):1053–63. doi:10.1681/asn.2010080841.
Aguado-Fraile E, et al. A pilot study identifying a set of microRNAs as precise diagnostic biomarkers of acute kidney injury. PLoS One. 2015;10(6):e0127175. doi:10.1371/journal.pone.0127175.
Kaucsar T, et al. Activation of the miR-17 family and miR-21 during murine kidney ischemia-reperfusion injury. Nucleic Acid Ther. 2013;23(5):344–54. doi:10.1089/nat.2013.0438.
Ma L, et al. Changes of miRNA-17-5p, miRNA-21 and miRNA-106a level during rat kidney ischemia-reperfusion injury. Zhonghua yi xue za zhi. 2015;95(19):1488–92.
Hu H, Jiang W, Xi X, Zou C, Ye Z. MicroRNA-21 attenuates renal ischemia reperfusion injury via targeting caspase signaling in mice. Am J Nephrol. 2014;40(3):215–23. doi:10.1159/000368202.
Jia P, et al. Xenon protects against septic acute kidney injury via miR-21 target signaling pathway. Crit Care Med. 2015;43(7):e250–9. doi:10.1097/ccm.0000000000001001.
Xu X, et al. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 2012;82(11):1167–75. doi:10.1038/ki.2012.241.
Denby L, et al. MicroRNA-214 antagonism protects against renal fibrosis. J Am Soc Nephrol. 2014;25(1):65–80. doi:10.1681/asn.2013010072.
Zarjou A, Yang S, Abraham E, Agarwal A, Liu G. Identification of a microRNA signature in renal fibrosis: role of miR-21. Am J Physiol Renal Physiol. 2011;301(4):F793–801. doi:10.1152/ajprenal.00273.2011.
Chau BN, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4(121):121ra18. doi:10.1126/scitranslmed.3003205.
Liu X, et al. MiR-21 inhibits autophagy by targeting Rab11a in renal ischemia/reperfusion. Exp Cell Res. 2015;338(1):64–9. doi:10.1016/j.yexcr.2015.08.010.
Du J, et al. MicroRNA-21 and risk of severe acute kidney injury and poor outcomes after adult cardiac surgery. PLoS One. 2013;8(5):e63390. doi:10.1371/journal.pone.0063390.
Ramachandran K, et al. Human miRNome profiling identifies microRNAs differentially present in the urine after kidney injury. Clin Chem. 2013;59(12):1742–52. doi:10.1373/clinchem.2013.210245.
Gaede L, et al. Plasma microRNA-21 for the early prediction of acute kidney injury in patients undergoing major cardiac surgery. Nephrol Dial Transplant. 2016. doi:10.1093/ndt/gfw007
Lorenzen JM, et al. MicroRNA-24 antagonism prevents renal ischemia reperfusion injury. J Am Soc Nephrol. 2014;25(12):2717–29. doi:10.1681/asn.2013121329.
Liang S, Wang W, Gou X. MicroRNA 26a modulates regulatory T cells expansion and attenuates renal ischemia-reperfusion injury. Mol Immunol. 2015;65(2):321–7. doi:10.1016/j.molimm.2015.02.003.
Kanki M, et al. Identification of urinary miRNA biomarkers for detecting cisplatin-induced proximal tubular injury in rats. Toxicology. 2014;324:158–68. doi:10.1016/j.tox.2014.05.004.
Liu F, et al. Upregulation of microRNA-210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro. Kidney Blood Press Res. 2012;35(3):182–91. doi:10.1159/000331054.
Lee CG, et al. Discovery of an integrative network of microRNAs and transcriptomics changes for acute kidney injury. Kidney Int. 2014;86(5):943–53. doi:10.1038/ki.2014.117.
Aguado-Fraile E, et al. miR-127 protects proximal tubule cells against ischemia/reperfusion: identification of kinesin family member 3B as miR-127 target. PLoS One. 2012;7(9):e44305. doi:10.1371/journal.pone.0044305.
Godwin JG, et al. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci U S A. 2010;107(32):14339–44. doi:10.1073/pnas.0912701107.
Jenkins RH, et al. miR-192 induces G2/M growth arrest in aristolochic acid nephropathy. Am J Pathol. 2014;184(4):996–1009. doi:10.1016/j.ajpath.2013.12.028.
Kito N, Endo K, Ikesue M, Weng H, Iwai N. miRNA profiles of tubular cells: diagnosis of kidney injury. Biomed Res Int. 2015;2015:465479. doi:10.1155/2015/465479.
Chen R, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 2008;27(34):4712–23. doi:10.1038/onc.2008.112.
Kim S, et al. MicroRNA miR-199a* regulates the MET proto-oncogene and the downstream extracellular signal-regulated kinase 2 (ERK2). J Biol Chem. 2008;283(26):18158–66. doi:10.1074/jbc.M800186200.
Rane S, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1 alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104(7):879–86. doi:10.1161/circresaha.108.193102.
Cantaluppi V, et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012;82(4):412–27. doi:10.1038/ki.2012.105.
Bijkerk R, et al. Hematopoietic microRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J Am Soc Nephrol. 2014;25(8):1710–22. doi:10.1681/asn.2013060640.
Denby L, et al. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179(2):661–72. doi:10.1016/j.ajpath.2011.04.021.
Lorenzen JM, et al. Circulating miR-210 predicts survival in critically ill patients with acute kidney injury. Clin J Am Soc Nephrol. 2011;6(7):1540–6. doi:10.2215/cjn.00430111.
Cui R, Xu J, Chen X, Zhu W. Global miRNA expression is temporally correlated with acute kidney injury in mice. PeerJ. 2016;4:e1729. doi:10.7717/peerj.1729.
Lan YF, et al. MicroRNA-494 reduces ATF3 expression and promotes AKI. J Am Soc Nephrol. 2012;23(12):2012–23. doi:10.1681/asn.2012050438.
Bhatt K, et al. MicroRNA-687 induced by hypoxia-inducible factor-1 targets phosphatase and tensin homolog in renal ischemia-reperfusion injury. J Am Soc Nephrol. 2015;26(7):1588–96. doi:10.1681/asn.2014050463.
Janssen HL, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368(18):1685–94. doi:10.1056/NEJMoa1209026.
Muratsu-Ikeda S, et al. Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS One. 2012;7(7):e41462. doi:10.1371/journal.pone.0041462.
Gutierrez-Escolano A, Santacruz-Vazquez E, Gomez-Perez F. Dysregulated microRNAs involved in contrast-induced acute kidney injury in rat and human. Ren Fail. 2015;37(9):1498–506. doi:10.3109/0886022x.2015.1077322.
Nassirpour R, et al. Identification of tubular injury microRNA biomarkers in urine: comparison of next-generation sequencing and qPCR-based profiling platforms. BMC Genomics. 2014;15:485. doi:10.1186/1471-2164-15-485.
Wang IK, et al. MiR-20a-5p mediates hypoxia-induced autophagy by targeting ATG16L1 in ischemic kidney injury. Life Sci. 2015;136:133–41. doi:10.1016/j.lfs.2015.07.002.
Pavkovic M, Riefke B, Ellinger-Ziegelbauer H. Urinary microRNA profiling for identification of biomarkers after cisplatin-induced kidney injury. Toxicology. 2014;324:147–57. doi:10.1016/j.tox.2014.05.005.
Saikumar J, et al. Expression, circulation, and excretion profile of microRNA-21, -155, and -18a following acute kidney injury. Toxicol Sci. 2012;129(2):256–67. doi:10.1093/toxsci/kfs210.
Pellegrini KL, et al. Application of small RNA sequencing to identify microRNAs in acute kidney injury and fibrosis. Toxicol Appl Pharmacol. 2015. doi:10.1016/j.taap.2015.12.002.
Jia P, et al. miR-21 contributes to xenon-conferred amelioration of renal ischemia-reperfusion injury in mice. Anesthesiology. 2013;119(3):621–30. doi:10.1097/ALN.0b013e318298e5f1.
Shen B, et al. Revealing the underlying mechanism of ischemia reperfusion injury using bioinformatics approach. Kidney Blood Press Res. 2013;38(1):99–108. doi:10.1159/000355759.
Bhatt K, et al. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol Med (Cambridge, Mass). 2010;16(9-10):409–16. doi:10.2119/molmed.2010.00002.
Liu XJ, et al. MicroRNA-34a suppresses autophagy in tubular epithelial cells in acute kidney injury. Am J Nephrol. 2015;42(2):168–75. doi:10.1159/000439185.
Joo MS, Lee CG, Koo JH, Kim SG. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013;4:e899. doi:10.1038/cddis.2013.427.
Bijkerk R, et al. Silencing of miRNA-126 in kidney ischemia reperfusion is associated with elevated SDF-1 levels and mobilization of Sca-1+/Lin- progenitor cells. MicroRNA (Shariqah, United Arab Emirates). 2014;3(3):144–9.
Bussolati B, et al. Hypoxia modulates the undifferentiated phenotype of human renal inner medullary CD133+ progenitors through Oct4/miR-145 balance. Am J Physiol Renal Physiol. 2012;302(1):F116–28. doi:10.1152/ajprenal.00184.2011.
Ranganathan P, et al. MicroRNA-150 deletion in mice protects kidney from myocardial infarction-induced acute kidney injury. Am J Physiol Renal Physiol. 2015;309(6):F551–8. doi:10.1152/ajprenal.00076.2015.
Pellegrini KL, et al. MicroRNA-155 deficient mice experience heightened kidney toxicity when dosed with cisplatin. Toxicol Sci. 2014;141(2):484–92. doi:10.1093/toxsci/kfu143.
Qin W, Xie W, Yang X, Xia N, Yang K. Inhibiting microRNA-449 attenuates cisplatin-induced injury in NRK-52E cells possibly via regulating the SIRT1/P53/BAX pathway. Med Sci Monit. 2016;22:818–23.
Bellinger MA, et al. Concordant changes of plasma and kidney microRNA in the early stages of acute kidney injury: time course in a mouse model of bilateral renal ischemia-reperfusion. PLoS One. 2014;9(4):e93297. doi:10.1371/journal.pone.0093297.
Acknowledgements
Not applicable.
Funding
This work was supported by the Chang Gung Memorial Hospital Research Program grant CMRPG3D1452, CMRPG 3F0561, CMRPG1B0581 and CIRPG3B0042.; Ministry of Science and Technology 104-2314-B-182A-131 and 105-2314-B-182A-121.
Availability of data and materials
Not applicable.
Authors’ contributions
The manuscript was written by PCF. All authors critically revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fan, PC., Chen, CC., Chen, YC. et al. MicroRNAs in acute kidney injury. Hum Genomics 10, 29 (2016). https://doi.org/10.1186/s40246-016-0085-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-016-0085-z