
Abstract
Human dihydroorotate dehydrogenase (DHODH) is a flavin-dependent mitochondrial enzyme catalyzing the fourth step in the de novo pyrimidine synthesis pathway. It is originally a target for the treatment of the non-neoplastic diseases involving in rheumatoid arthritis and multiple sclerosis, and is re-emerging as a validated therapeutic target for cancer therapy. In this review, we mainly unravel the biological function of DHODH in tumor progression, including its crucial role in de novo pyrimidine synthesis and mitochondrial respiratory chain in cancer cells. Moreover, various DHODH inhibitors developing in the past decades are also been displayed, and the specific mechanism between DHODH and its additional effects are illustrated. Collectively, we detailly discuss the association between DHODH and tumors in recent years here, and believe it will provide significant evidences and potential strategies for utilizing DHODH as a potential target in preclinical and clinical cancer therapies.
Similar content being viewed by others


Introduction
Cellular metabolism is the basis of all biological activities. Metabolic dysregulation, an emerging hallmark of cancer, contributes to maintain cell proliferation, migration, and differentiation during tumorigenesis and progression [1]. The metabolic enzymes play a vital role in this process and become important targets for anti-cancer drug development [1].
Pyrimidine nucleotides play a significant role in tumor cell proliferation as precursors of RNA and DNA [2]. There are two ways for the synthesis of pyrimidine: the salvage synthesis pathway and the de novo synthesis pathway [2]. In resting or fully differentiated cells, pyrimidines are mainly provided by the former. While in highly proliferative cells like tumor cells, the latter is usually highly active to meet the increased demand for nucleic acid precursors and other cellular components [2]. Compared with normal proliferous cells, there is a significant imbalance of pyrimidine metabolism in cancer cells which is stringently linked with tumor transformation and progression [3].
Dihydroorotate dehydrogenase (DHODH), located in the inner membrane of mitochondria, is an iron-containing flavin-dependent enzyme and plays a crucial role in the de novo synthesis of pyrimidine [4]. DHODH catalyzes the conversion of dihydroorotate to orotate in a redox reaction, which is the fourth of six universally conserved enzymatic reactions in the pyrimidine de novo synthetic pathway [5]. The essential role of DHODH in pyrimidine synthesis and mitochondrial functions attracted much attention during past decades [6]. In 1959, its role in tumor progression was first reported, and increasing evidences indicated that DHODH expression and activity are directly related to cancer progression. Therefore, extensive efforts have been made to develop DHODH inhibitors for cancer treatment. The functional or biochemical role of DHODH in cancers or other diseases’ development as well as of their related inhibitors has been well reviewed in several papers [7,8,9]. However, it remains a hot topic and has been further investigated for the detailed mechanisms of this metabolic enzyme in multiple malignancies or other diseases. Therefore, due to the quick updates of some novel knowledge of this protein, especially in its inhibitor development, it is necessary to recap some new findings.
This review aims at providing an overview of recent findings in DHODH biology and inhibitor development. The relationship of DHODH with de novo pyrimidine metabolism and mitochondria system in various cancers is discussed. The broad-ranging progresses in recent years made in DHODH inhibitor development for cancer therapy are summarized.
Characteristic property of DHODH
DHODH gene, located in the open reading frame (ORF) of human chromosome 16q22 with full length of 1191 bp, encodes DHODH protein with 397 amino acid sequences [4]. The crystalline enzyme was deemed to contain flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and iron [10].
According to sequence similarity and subcellular location, DHODH is divided into Class 1 and Class 2 DHODHs. Soluble class 1 DHODHs are further classified into class 1A, class 1B, and class 1S, which are all located in cytoplasm. Class 1A are homodimeric proteins and found in Gram-positive bacteria. Class 1B DHODHs is a dimer of heterodimers and usually found in prevalent in Gram-positive bacteria, consisting of two distinct proteins. The S DHODH is a newly found type which is incapable of utilizing any of the natural electron acceptors. It uses serine as catalytic base, which is special for a cytosolic DHODH [5, 11]. Class 2 DHODHs are monomeric proteins that attach to the mitochondrial inner membrane in eukaryotes and some prokaryotes [11,12,13,14]. Class 1A and class 1B share approximately 30% sequence identity, whereas soluble class 1 and membrane-bound class 2 DHODHs share approximately 20% of sequence identity [11].
DHODH was first isolated in 1953 from extracts of Clostridium oroticum (CoDHODH) [11, 15]. The first crystal structure of flavin containing DHODH has been determined from Lactococcus lactis in 1997 (LlDHODH) [16], and the structure of human DHODH in complex with anti-proliferative agents was solved in 2000. Structural studies show that DHODH contains two domains, a large C-terminal domain and a smaller N-terminal domain, which are attached by an extended loop. The N-terminal extends approximately 40 residues folds into two α-helices (αA and αB) linked by a short loop and related to membrane association [11]. The cytosolic larger C-terminal contains the redox site, the N-terminal domain forms a tunnel inside the membrane that hides the FMN binding site [2, 9, 17]. Notably, the majority of the structural elements and residues relevant to both FMN and substrate binding are conserved in classe1 and 2 of DHODHs [11]. Ubiquinone, utilizing the tunnel to approach the FMN cofactor for participating in the redox reaction, can easily diffuse into the mitochondrial inner membrane [18, 19]. Ubiquinone Q10 binds the signal peptide located in the N-terminus of DHODH, which covers for mitochondrial import and is a transmembrane domain with a microdomain interacting with the mitochondrial inner membrane [20, 21].
DHODH regulates cancer progression
The de novo pyrimidine metabolism and DHODH
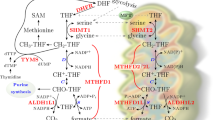
Pyrimidines are necessary for the biosynthesis of DNA, RNA, glycoproteins and phospholipids [9]. Pyrimidine nucleotides are synthesized through two pathways: the de novo synthesis pathway and the salvage pathway [22]. Pyrimidines are synthesized de novo from simple precursors with six steps (Fig. 1). The enzymes that catalyze uridine monophosphate (UMP) synthesis include carbamoylphosphate synthetase II (CPSII), aspartate transcarbamoylase (ATCase), dihydroorotase (DHOase), DHODH, and uridine monophosphate synthase (UMPS) [23]. Firstly, glutamine (Gln), ATP and HCO3− collectively form carbamoyl phosphate, which is catalyzed by CPSII, a vital enzyme located in the cytosol. ATCase contributes to the formation of the carbamoyl-aspartate following, the pyrimidine ring further is cyclized by DHOase to produce dihydroorotate. Secondly, the flavoenzyme DHODH converts dihydroorotate to orotate which takes place in mitochondria. Orotate further converts to UMP by the UMP synthase UMPS. UMP, a precursor of other pyrimidines and various biological processes, then converts to uridine triphosphate (UTP) by phosphorylation. UTP is subsequently converted to cytidine triphosphate (CTP) with the donor glutamine by CTP synthetase. Meanwhile, the uridine diphosphate (UDP) transforms to deoxyuridine diphosphate (dUDP) by ribonucleotide reductase, with its ribose moiety reducing to deoxyribose. Deoxyuridine monophosphate (dUMP) methylation generates deoxythymine nucleotides (dTMP or TMP) for DNA synthesis [2, 23,24,25]. In this process, DHODH catalyzes the fourth step in the de novo biosynthesis of pyrimidine by converting dihydroorotate into orotate in a redox reaction in the mitochondria with ubiquinone (CoQ) converting to ubiquinol (CoQH2), which is a substrate of respiratory complex III [2, 11, 26].

Pyrimidine de novo biosynthesis pathway
As DHODH plays a crucial part in pyrimidine synthesis, it exerts varying effects on different cell types and developmental stages owing to the relative contribution of de novo pyrimidine synthesis to maintaining proliferation. The de novo pyrimidine synthesis has gained a prominent position to satisfy increasing demand for nucleic acid precursors in rapidly proliferative cells such as activated T cells due to the rapidly increased demand for DNA replication and nucleic acid biosynthesis [9, 23, 27, 28]. While in resting or fully differentiated cells, they mainly obtain pyrimidines through the salvage pathway for proliferation [9, 28,29,30,31]. Thus, the effect of DHODH is more predominate in rapidly proliferating cells like cancer cells, which may be highly sensitive to inhibition of nucleotide synthesis [32,33,34,35].
Consistent with above observations, DHODH blockade by inhibitors or RNA interference exhibited anti-proliferation effect by pyrimidine depletion. Studies manifest that DHODH inhibitors have more potential to treat malignancies which are more dependent on de novo pyrimidine synthesis and have lower pyrimidine salvage activity. For instance, in PTEN-mutant cells which depend upon glutamine flux through the de novo pyrimidine synthesis pathway [29], inhibition of DHODH causes stalled forks due to inadequate nucleotide pools required to support replication. Sustained treatment with DHODH inhibitor further leads to Rad3-related kinase (ATR) activation, leading to a buildup of DNA damage and cell death [29]. Cancer cells are hypersensitive to DHODH inhibitors under the tumor hypoxia and nutrient-deprived microenvironment [27, 36]. Further, depletion of the pyrimidine nucleotide pool, especially UTP, resulting from DHODH inhibition, could impair biogenesis of ribosomes, which activates the tumor suppressor p53 pathway leading to cell cycle arrest [37, 38]. Moreover, DHODH is regulated by several critical transcription factors [24]. The predicted and known regulators of DHODH include E1A-binding protein p300, POU domain, class 3, transcription factor 2 (POU3F2), GATA-binding factor 2 (GATA-2), nuclear factor kappa-light-chain-enhancer of activated B cells 1 (NF-κB1), and proto-oncogene MYC [24]. It might be significant to inhibit DHODH through interfere with these regulatory factors, which can further contribute to pyrimidine depletion and induce death of tumor cells.
Apart from the direct blockade of pyrimidine synthesis, DHODH also exerts additional effects including O-linked N-acetylglucosaminylation (O-GlcNAc) [39], senescence [7], AND mRNA translation [40] indirectly by pyrimidine depletion. UMP, the downstream product of DHODH, is a precursor for components required for the assembly of various cellular macromolecules, including phospholipids, glycogen, hyaluronic acid, and proteoglycans, as well as for certain post-translational protein modifications [30, 41], which may account for the above extensive influences of DHODH. For example, O-GlcNAc, a post-translational modification of proteins, can add N-acetylglucosamine (GlcNAc) on UDP [42]. UMP reduction by DHODH suppression may result in integral decreases in protein N-acetyl glycosylation in acute myeloid leukemia (AML) [43], and reduction of UMP-GlcNAc promotes myeloid differentiation [43]. Given that O-GlcNAc plays a crucial role in myeloid differentiation [44,45,46], it may explain the mechanism that DHODH inhibition promotes the myeloid differentiation in AML to some extent. Collectively, DHODH inhibition decreases the de novo pyrimidine synthesis and affects many biological processes.
Mitochondria function and DHODH
Mitochondria plays important roles in energy metabolism in eukaryotic cells by produce ATP via electron transport chain enzyme complexes (I-IV) which are located in the mitochondrial inner membrane [27]. Complexes I and II transfer reducing equivalents from NADH and succinate to complex III via the ubiquinone pool, respectively, and complex III further transfers these equivalents to complex IV through cytochrome c [36]. Electrons are eventually transferred to dioxygen, subsequently producing water [36]. ATP synthase generates ATPs by oxidative phosphorylation utilizing the transmembrane electrochemical gradient maintained by proton pumping activities of complexes I, III, and IV [36] (Fig. 2). The human DHODH protein resembles mitochondrial-targeting pre-sequences in its N-terminal segment and contains a bipartite signal that governs import and correct insertion into the mitochondrial inner membrane [2, 20]. As being located in mitochondria inner membrane, DHODH has inextricable link with mitochondria function.

DHODH and mitochondrial respiratory chain. The mammalian mitochondrial electron transport chain comprises four enzyme complexes located in the mitochondrial inner membrane: complexes I, II, III, and IV. Complexes I and II transfer reducing equivalents from NADH and succinate to complex III via the ubiquinone pool, respectively, and complex III further transfers these equivalents to complex IV through cytochrome c. Electrons from complex IV are eventually transferred to dioxygen, subsequently producing water. ATP synthase generates ATPs by oxidative phosphorylation utilizing the transmembrane electrochemical gradient maintained by proton pumping activities of complexes I, III, and IV [36]. DHODH converts dihydroorotate to orotate, generating electrons that are transferred via redox-cycling of ubiquinone to complex III [2]. Thus, the de novo synthesis of pyrimidine nucleotides is coupled to the mitochondrial respiratory chain via DHODH [47]. Abbreviations: CI, complex I; CII, complex II; CIII, complex III; CIV, complex IV; CV, complex IV; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide. Q, coenzyme Q (CoQ), so known as ubiquinone; QH2, the hydroquinone (antioxidant) form of CoQ, also known as ubiquinol; UMP, uridine monophosphate
DHODH is located between the complex II and complex III [4] (Fig. 2) and involved in electros production when it converts dihydroorotate to orotate [2]. Further, DHODH physically interacts with complexes II and III though the specific mechanism of this interaction awaits to be elucidated [22, 27]. Thus, inhibition of DHODH results in impairment of complex II and III function under hypoxia and nutrient-deprived conditions [27, 35, 48,49,50], followed by dysfunction of oxidative phosphorylation (OXPHOS) and aerobic glycolysis in activated T cells [51]. Meanwhile, inhibition of mitochondrial complex III also can suppress the function of DHODH by reducing turnover of ubiquinone which is essential for DHODH activity, thus reducing the pyrimidine de novo biosynthesis [52].
Mitochondria produce cellular energy by OXPHOS [53]. Essential protein subunits of OXPHOS complexes are assembled from both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) genes [53]. The reduction of pyrimidine nucleotide synthesis by inhibition of DHODH results in shortage of nDNA and mtDNA for OXPHOS complexes. Depletion of mtDNA induces mitochondria devoid of a functional respiratory chain, further decreases ATP production and leads to cell proliferation inhibition [54, 55]. For example, DHODH inhibition affects ATP depletion in breast cancer cells [56]. Conversely, these studies also revealed that DHODH knockout cells feature normal levels of OXPHOS-derived ATP and bioenergetics [57]. It is found that DHODH only contributed to no more than 5–10% of conventional (ATP-coupled) oxygen consumption in OXPHOS in 4T1 and B16 cells, suggesting that the direct contribution of DHODH to ATP generation may be negligible at baseline [7]. While the mechanism why DHODH inhibition makes ATP levels unchanged remains to be studied.
ROS production and DHODH
Reactive oxygen species (ROS), which includes superoxide (O2∙), hydrogen peroxide (H2O2), and the hydroxyl radical (OH∙) are formed by the chemical reduction of O2 [58]. Mitochondria are a major source of ROS production within cells (mROS). There are at least 10 sites in the mitochondrial electron transport chain and matrix that are able to produce superoxide/H2O2 at measurable rates [48]. Cancer cells struggle to achieve a delicate redox balance to ensure survival [54]. Mounting evidence shows that mROS are critical for intracellular redox signaling by which they contribute to a plethora of cellular processes such as proliferation [56, 59]. ROS can induce DNA damage, activate oncogenes, block the function of tumor suppressors, and drive migratory signaling, which further results in tumorigenesis [56, 59]. On the other hand, ROS also can induce oxidation stress leading to cell apoptosis [54]. DHODH is a major contributor to mitochondrial oxygen consumption and ROS production in malignant tumor such as leukemia [47]. DHODH can generate superoxide and/or H2O2 directly at low rates and is capable of indirect production at higher rates from other sites through its ability to reduce the ubiquinone pool [14]. Conversely, recent studies prove that DHODH inhibition may also contribute to increased ROS. DHODH depletion partially inhibits the mitochondria complex III, decreases the mitochondrial membrane potential, and increases the generation of ROS [22]. Meanwhile, there is also a study that finds DHODH inhibitor-sensitive breast cancer cells reveal unchanged ROS production [56, 57]. Possible reasons are that DHODH induces ROS production which generally arises from other mitochondrial sites, and the function of these mitochondrial sites may affect the efficacy of ROS production under DHODH inhibition [14]. Moreover, the effect of DHODH in ROS level is relevant with some specific conditions [7]. For example, when cells are treated with anti-cancer agent fenretinide which can induce ROS production, DHODH suppression significantly reduces fenretinide-induced ROS generation [7, 55]. Taken together, the connection between DHODH and ROS in cancer is not fully understood and remains to be further verified.
The various malignances and DHODH
The overexpression of DHODH are observed in multiple malignance cells, including AML [60], skin cancer [61], colorectal cancer (CRC) [62], pancreatic cancer [63], breast cancer [56], lung cancer [64], multiple myeloma cells [65], neuroblastoma cells [48], renal cell carcinoma (RCC) [49], cervical cancer [50], and glioblastoma stem cells (GSCs) [25]. Especially, hepatocellular carcinoma exhibits the highest DHODH mRNA level among above malignance (Fig. 3). The overexpression of DHODH is associated with malignance growth and invasion.

DHODH mRNA level in different malignancy types. The chart shows DHODH mRNA level in different kinds of malignancies. A circle represents a sample. All these data are concluded in Cbioportal, from TCGA Pan-Cancer Atlas Studies which cover 10967 samples from 32 studies
AML
AML, belonging to leukemia which ranks fifteen of 36 cancers in 185 countries in 2020 [66], is a clinically devastating disease with a quite variable prognosis and a high mortality rate [67]. Its 5-year overall survival is lesser than 50%, and in elderly patients only 20% will survive 2 years after diagnosis [67]. AML cells are found to be particularly sensitive to DHDOH depletion [68]. When in condition of pyrimidine starvation by DHODH inhibition, the AML blasts revealed cell death and differentiation with changes of morphology, cell surface marker expression, and gene expression [68]. Moreover, it is unraveled that inhibition of DHODH enables myeloid differentiation in human and mouse AML models via downregulating MYC which is a key transcription factor correlated with tumor cell differentiation [68, 69]. Besides, the differentiation effects by DHODH inhibition may include suppression of nucleic acid synthesis, cell cycle arrest, and influences in post-translational glycosylation of crucial proteins [39]. The mechanism of DHODH inhibition and myeloid differentiation still remains to be further manifested.
So far, two newly patented human DHODH inhibitors are currently being investigated for AML treatment: ASLAN003, currently being evaluated in Phase II clinical trial (clinical trial identifier: NCT03451084); BAY2402234, a compound by Bayer entered Phase I clinical trials in January 2018 (clinical trial identifier: NCT03404726) [60]. Meanwhile, isobavachalcone, a novel DHODH inhibitor can directly inhibit human DHODH and induce apoptosis and differentiation of AML cells [70]. PTC299, another novel DHODH inhibitor, has broad and potent activity against hematologic cancer cells in preclinical models [40]. DHODH inhibition also has strong effects in mixed-lineage leukemia gene MLL-fused leukemia cell lines [71]. Taken together, DHODH inhibitors promote differentiation of leukemia cells, decrease levels of leukemia-initiating cells, and improve survival in vivo, which demonstrates DHODH inhibition as a strategy for AML treatment [39].
Breast cancer
Breast cancer, the fifth leading cause of cancer mortality worldwide, with 685,000 deaths [66], has now surpassed lung cancer as the leading cause of global cancer incidence in 2020, with an estimated 2.3 million new cases, representing 11.7% of all cancer cases [66]. DHODH is overexpressed in human breast cancer tissues (Fig. 3), and breast cancer cells expressing high DHODH show high sensitivity to DHODH inhibitors [56]. DHODH inhibitors lead to ATP depletion with maintained ROS level, S phase arrest and upregulate the expression of p53, p65, and STAT6 proteins in sensitive T-47D and MDA-MB-231 breast cancer cells, compared with non-sensitive MDA-MB-436 and W3.006 breast cancer cells [56]. PTEN-mutant human breast cancer cell lines display increased cell death overtime upon treatment with DHODH inhibitor leflunomide [29]. DHODH suppression induces sensitivity to TRAIL and replication stress in MCF-7 breast cancer cells [37, 72]. Furthermore, DHODH blockade in p53-mutant MDA-MB-231 and p53-deficient 4T1 breast cancer cells displays pyrimidine depletion on the cell cycle [37]. It reminds us of distinguishing the sensitivity of breast cancer cells to DHODH inhibitor by considering different molecular mechanism and DHODH inhibition as an alternative method for breast cancer treatment.
CRC
CRC ranks third in terms of incidence of 6%, but second in terms of mortality of 5.8% in global cancer statics of 2020 [66] with 5-year relative survival ranges from greater than 90% in patients with stage I disease to slightly greater than 10% in patients with stage IV disease [73, 74]. DHODH shows significantly higher expression in CRC tumor tissue compared with normal samples using oncomine dataset analysis, which is consistent with a study that cell lines derived from the small and large intestine malignancies are most sensitive to DHODH knockdown [8]. As DHODH overexpression is associated with proliferation of CRC cells, inhibition of DHODH exhibited anti-proliferative effect against CRC cells. Moreover, the sensitivity of CRC cells to DHODH inhibitors are associated with the gene expression of cells. KRAS mutation is among the most prominent molecular markers in metastatic CRC patients, and mutations of KRAS render cells responsive to anti-cancer therapy such as anti-EGFR antibody treatment [73]. However, KRAS mutant CRC cells exhibit proliferation suppression in response to DHODH inhibitor brequinar (BRQ) treatment, which includes an increase in the steady-state concentrations of fructose 1,6-bisphosphate and change of metabolite levels including decreased glutamine and glutamate [63]. DHODH blockade and impairment of de novo pyrimidine biosynthesis by leflunomide trigger apoptosis in human CRC cells expressing transcriptionally active p53, which is consequential to the inhibition of the electron transport chain complex III [75]. When HCT116 CRC cells were treated with leflunomide, cell cycle arrest as well as increased switch to senescence is observed [75]. In addition, leflunomide effectively decreases proliferation of CRC cells via induction of replication and ribosomal stress in a p53- and checkpoint kinase 1 (Chk1)-dependent manner [37]. DHODH inhibition by leflunomide also strongly impairs CRC liver metastatic colonization compared with primary tumor growth [76]. HT29 cell lines are shown to be sensitized to TRAIL by BRQ [72]. Moreover, BRQ potentiates 5-fluorouracil antitumor activity in a murine model colon cancer by tissue-specific modulation of uridine nucleotide pools [77]. Similar results are found in MC38 colon carcinoma cells [56], which further display combined effect of DHODH inhibitors and 5-fluorouracil. Moreover, DHODH expression, with a predictive miR-502 binding site at position 245 to 251 in 3′-UTR of its mRNA, is directly regulated by miR-502 in CRC [62]. Ectopic expression of miR-502 precursor in human CRC cells HCT116 leads to significant decrease in DHODH mRNA and protein levels compared with negative miRNA transfection [62]. All these evidences prove that DHODH blockade could be a potential target in CRC treatment, especially in KRAS mutant, p53-deficient, or other genetic context CRC cells.
Lung cancer
Lung cancer, the second most commonly diagnosed cancer and the leading cause of cancer death in 2020, approximately 11.4% in cancers diagnosed and 18.0% in deaths, is also the leading cause of cancer morbidity and mortality in men [66], whereas its incidence ranks third after breast cancer and CRC, and its mortality ranks second after breast cancer in women [66]. It mainly includes small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) in terms of biological characteristics [78,79,80]. It is manifested that lung cancer cell lines show higher DHODH expression than normal lung cells and DHODH knockout exerts significant effects on lung cancer cell proliferation inhibition, suggesting that lung cancer cell lines are sensitive to DHODH inhibitors [8]. A recent study demonstrates that the sensitivity of SCLC cells toward disruption of the pyrimidine biosynthesis pathway enhanced, and DHODH inhibition by BRQ suppresses SCLC tumor growth and extends mice survival in vivo [72]. Downregulation of DHODH sensitizes SCLC cell U1690 to TRAIL-mediated apoptosis, and BRQ possesses significantly lower toxicity toward U1690 cells, which would be optimal for a combinatorial therapy [41, 69]. Besides, selective sensitivity to blockade of the de novo purine synthesis pathway is revealed in the variant subtype of SCLC [81], though the mechanism on how the variant subtype of SCLC exhibits sensitivity toward DHODH inhibition remains to be further elucidated [41]. It might show distinctions of sensitivity to DHODH inhibition in different gene context and tumors, which is an interesting direction to be further explored. Accordingly, under different genetic conditions in vivo, the decisive limitation of pyrimidine biosynthesis in tumors remains to be determined. Though related researches are not numerous, it seems to be a meaningful start for the future application of DHODH inhibitor in lung cancer therapy.
Pancreatic cancer
Pancreatic cancer, with the occurrence of 2.6% and mortality of 4.7% [66], accounts for almost 466,000 deaths and 496,000 new cases because of its poor prognosis and is the seventh leading cause of cancer death in both sexes [66] with the overall average 5-year survival of 9% [74]. It is predicted that pancreatic cancer will surpass breast cancer as the third leading cause of cancer death by 2025 in 28 European countries [66]. DHODH reveals higher mRNA level in human pancreatic cancer tissues compared with normal pancreatic tissue [8]. A recent study reveals a greater increase in concentrations of newly synthesized UMP over time in human pancreatic ductal adenocarcinoma (PDAC) cell lines, suggesting that PDAC cells exhibit higher flux through the de novo pyrimidine synthesis pathway [41]. It manifests that DHODH may play a vital role in pancreatic cancer and its depletion might contribute to anti-cancer efficiency. Notably, DHODH inhibition induces cell cycle S phase arrest and mitochondrial depolarization in KP-4 human pancreatic cancer cells. In vivo preclinical studies demonstrate strong anti-cancer activity upon DHODH inhibition in a pancreatic tumor xenograft model [63]. Moreover, DHODH inhibitor teriflunomide not only shows in vitro anti-proliferative activity but also synergizes with the chemotherapeutic gemcitabine in pancreatic cancer cells, which is explained by interfering with PIM-3 kinase which is important in cell proliferation and protein synthesis of pancreatic cancer cells [82]. DHODH inhibition by Ter may exert its effect on PIM-3 and its downstream target leading to apoptosis [82]. Collectively, DHODH inhibitor may provide a novel way for pancreatic cancer treatment.
Skin cancer
Skin cancers are one of the most common malignancies, particularly in the white population. The three commonest skin cancer types are basal cell carcinomas (BCC), squamous cell carcinomas (SCC) (also referred as nonmelanocytic skin cancer, NMSC) and cutaneous malignant melanoma (CM) (also designed as malignant melanoma of the skin or melanoma) [83]. Nonmelanoma skin cancer is responsible for over one million new cases of 6.2% and 64,000 deaths of 0.6% in 2020. While melanoma accounts for only 2% of all skin cancers, it causes most skin cancer deaths [84]. It is demonstrated that DHODH is mainly expressed in the epidermal hyperplasia, actinic keratoses (AK, the promotion phase), and Bowen’s disease, and invasive SCC specimens also display diffuse distribution [85]. Of note, DHODH activity is roughly twice higher in skin cancers than normal skin [85]. Recently, a study shows that DHODH, playing a critical role in UVB-induced energy metabolism reprogramming, is upregulated mainly resulting from transcriptional activation by signal transducer and activator of transcription 3 (STAT3) in a multistage model of UVB radiation-induced skin cancer. Importantly, inactivated DHODH or impaired electron transport chain blocks neoplastic transformation of keratinocytes [86]. DHODH blockade could be significant in the prevention or therapy of nonmelanoma skin cancers and possibly other hyperproliferative keratinocytic diseases [85]. Chronic inhibition of DHODH by leflunomide blocks UVB-induced cutaneous SCC initiation [87]. Moreover, it is found that BRQ and doxorubicin synergistically inhibited melanoma xenografts [88], and this combination significantly downregulated cell cycle regulatory proteins, cyclin B1, and upregulated its binding partner pcdc-2 and p21 [88]. It is demonstrated that DHODH inhibition abrogates the transcriptional elongation of genes, including Myc target genes and mitfa [89]. These genes are required for melanoma and multipotent neural crest cells which is an important precursor involved in melanoma pathogenesis proliferation [89]. Further, a DHODH inhibitor (R)-HZ00, which induced the accumulation of cells in S phase, an increase in p53 synthesis by inhibiting p53 degradation, and enhancement of the antitumor effect in human melanoma cell line ARN8, is uncovered [61]. Leflunomide abrogates the effective transcription elongation of genes required for neural crest development and melanoma growth in vivo, and nucleotide depletion reduces the chromatin occupancy of the RNA helicase protein DDX21 in human A375 melanoma cells [90]. DHODH inhibitor HZ-05 can suppress ARN8 melanoma cell growth and viability and induce induction DNA damage through activating p53-dependent transcription factor activity [35]. Given these observations found, DHODH inhibitor might be a bright method in the targeted therapy of melanoma.
DHODH and other neoplastic diseases
DHODH inhibition can inhibit a variety of cancer cells, including multiple myeloma cells [65], neuroblastoma cells [48], RCC [49], cervical cancer [50], and GSCs [25].
Teriflunomide induces G1 cell cycle arrest via modulation of cyclin D2 and pRb expression and decreases phosphorylation of protein kinase B (PKB), p70S6K, and eukaryotic translation initiation factor 4E-binding protein-1 in multiple myeloma cells [65]. Leflunomide induces S phase cell cycle arrest and promotes cell apoptosis, further to suppress neuroblastoma cell proliferation in vitro and in vivo [48]. Leflunomide at high concentrations targets the canonical WNT/β-catenin signaling and induces the nucleocytoplasmic shuttling of β-catenin, subsequently promoting its proteasome-dependent proteolysis. Thus, DHODH inhibition can interrupt the canonical WNT/β-catenin signaling to induce RCC cell growth arrest and apoptosis [49]. Further, an fluorescence detection assay for measuring DHODH activity is developed in cultured HeLa cervical cancer cells, and in stage III stomach cancer and adjacent normal tissues from the same patient. Results indicate significantly DHODH shows higher activity in malignant tumor tissue than in adjacent normal tissue [50]. DHODH expression is also associated with increased grade and stage in glioma patients from the TCGA database [84]. Higher expression of pyrimidine synthesis genes including DHODH portends poor prognosis of patients with glioblastoma [25]. Targeting the pyrimidine synthetic rate-limiting step enzyme CAD or the critical downstream enzyme DHODH markedly inhibits GSC survival, self-renewal, and in vivo tumor initiation through the depletion of the pyrimidine nucleotide supply in rodent models [25].
Taken together, DHODH is releasing increasing potential abilities in various cancer therapies and it is worthy of expecting its more powerful utilization in other tumors.
Co-expression network of DHODH
Co-expressed genes with DHODH
Various genes are found to be associated with DHODH through Coexpedia, which is a database widely applied for gene co-expression network analysis. Genes co-expressed with DHODH which were reported before are listed (Fig. 4a). Relationship among all these genes and different kinds of cancers are displayed (Fig. 4b). Lymphoma holds an important portion in these malignancies, which is in accord with the inhibitory effect of DHODH in T lymphocyte. Moreover, the top 20 co-expressed genes with DHODH are ranked according to the Lake Louise Score (LLS) score (Fig. 4c), including acrosomal vesicle protein 1 (ACRV1), cytochrome P450 family 2 subfamily C member 9 (CYP2C9), pappalysin 2 (PAPPA2), and neurotrophic tyrosine kinase, receptor, type 3 (NTRK3). GO analysis reveals the positively correlated top 20 pathways to DHODH co-expression genes (Fig. 4d), including type I interferon signaling pathway, positive regulation of ERK1 and ERK2 cascade, inflammatory response, positive regulation of extrinsic apoptotic signaling pathway via death domain receptors, MAPK cascade, positive regulation of phosphatidylinositol 3-kinase signaling, activation of protein kinase B activity, positive regulation of ROS metabolic process, drug metabolic process, and positive regulation of adenylate cyclase activity involved in G-protein coupled receptor signaling pathway.

DHODH co-expressed genes. a Co-expression genes network with DHODH. b Relationship between DHODH co-expressed genes and different cancers. c The top 20 co-expressed genes with DHODH are ranked by sum of their LLS score. d Positively correlated top 20 pathways to DHODH co-expression genes by GeneSet analysis. Enriched terms by p value < 0.05. All these data are concluded from database Coexpedia. Abbreviations: ACRV1, acrosomal vesicle protein 1; CYP2C9, cytochrome P450 family 2 subfamily C member 9; PAPPA2, pappalysin 2; NTRK3, neurotrophic tyrosine kinase, receptor, type 3; DCT, dopachrome tautomerase; CA12, arbonic anhydrase XII; DRD2, dopamine receptor D2; WNT6, wingless-type MMTV integration site family member 6; PAX8, paired box 8; CYP2A6, cytochrome P450 family 2 subfamily A member 6; HFE, hemochromatosis; UBE2D4, ubiquitin conjugating enzyme E2D 4 (putative); EGFR, epidermal growth factor receptor; SIGLEC6, sialic acid binding Ig-like lectin 6; ETV1, ets variant 1; MID2, midline 2; EXPH5, exophilin 5; ATP10B, ATPase, class V, type 10B; SLC30A3, solute carrier family 30 (zinc transporter), member 3; ALDOB, aldolase, fructose-bisphosphate B
Co-expressed proteins with DHODH
A number of proteins which may be co-expressed with DHODH are found by STRING dataset. Here we mainly display the top 20 relative proteins with DHODH, as is depicted in Table 1 and Fig. 5.

DHODH co-expressed proteins. a Co-expressed proteins with DHODH. b GO analysis concludes the top 20 biological process of proteins/genes co-expressed with DHODH. c KEGG analysis reveals the related pathways of proteins/genes co-expressed with DHODH. All these data are from STRING, which is a website used for protein-protein interaction network and functional enrichment analysis
Mutation and alternation of DHODH
This section displays a series of charts that show the distribution of different types of mutations for DHODH.
Post-translational modification sites (PTMC) of DHODH protein are shown (Fig. 6b), which include phosphorylation at 63, 121, 146, 217, 356, and 359, acetylation at 99, and ubiquitination at 185, 306, and 344 [91,92,93,94]. In the total unique 217 samples, some verified DHDOH mutations are summarized, which includes nonsense substitution (0.92%), missense distribution (46.54%), synonymous substitution (12.90%), frameshift insertion (1.38%), frameshift deletion (0.92%), and other mutations (5.07%) (Fig. 6c).

Mutation and alternation of DHODH. a All these data are from Cbioportal. b DHODH post-translational modifications (PTMs) sites, which are available for the Ensembl transcript ENST00000219240. Mutation diagram circles are colored with respect to the corresponding mutation types. In case of different mutation types at a single position, color of the circle is determined with respect to the most frequent mutation type. Mutation types and corresponding color codes are as follows: green, missense mutations; black, truncating mutations (nonsense, nonstop, frameshift deletion, frameshift insertion, splice site); brown, inframe mutations (inframe deletion, inframe insertion). All these data are from Cbioportal. c An overview of the types of DHODH mutation distribution. All these data are from COSMIC
DHODH and non-neoplastic diseases
DHODH not only plays a vital part in malignancy therapy, but also in many other non-neoplastic diseases. DHODH inhibitors have been suggested for treating diseases such as adjuvant arthritis [95], lymphoproliferation and systemic lupus erythematosus [96], graft-versus-host disease [97], tubulointerstitial nephritis [98], proteoglycan-induced arthritis [99], glomerulonephritis [99], myasthenia gravis [100], diabetes [101], psoriasis, autoimmune diseases, plasmodium, bacterial infections [102], rheumatoid arthritis [103] and multiple sclerosis (MS) [51, 104, 105], Miller syndrome [106], edema [107], and hippocampal networks related epilepsy [108]. Moreover, DHODH inhibitors exhibited efficacy against virus, including respiratory syncytial virus [109], rotavirus [110], foot-and-mouth disease virus infection [111], flavivirus which contains dengue virus, Zika virus, and Japanese encephalitis virus [112], and notably, against RNA viruses including newly emerged coronavirus SARS-CoV-2 [113]. DHODH inhibitors are also found to be effective against oomycete phytophthora infestans [114], schistosomiasis [115, 116], invasive fungal infections [117], eumycetoma [118], and malaria [119,120,121,122].
In addition, DHODH, which mediates the de novo pyrimidine synthesis in actively proliferating T and B lymphocytes, also displays immunoregulation properties. Interference with immune cell proliferation represents a successful treatment strategy in T cell-mediated autoimmune diseases [51, 104]. DHODH inhibitor teriflunomide causes selective changes in T cell subset composition and T cell receptor repertoire diversity in patients with relapsing-remitting MS [51]. DHODH inhibition modulates T cell mitochondrial respiration with affinity-dependent effects in multiple sclerosis [104]. Further, human DHODH represents an important target for the treatment of hyperproliferative and inflammatory diseases, and its inhibition is beneficial to immunosuppressant and anti-proliferative effects in diseases such as RA. DHODH inhibitors BRQ and leflunomide show immunosuppressive and anti-proliferative activities, most pronounced in T cells [18]. Additionally, DHODH suppression might exert anti-inflammatory activities in autoimmune disease treatment by preventing the generation of proinflammatory Th1 effectors and promoting Th2 cell differentiation. The DHODH dysfunction directly inhibits NF-kB activity and disrupted cell migration, diminished cellular proliferation, and increased apoptosis, which contributes to Miller syndrome [22, 106].
A recent study displays that DHODH is a regulator of activity set points in hippocampal networks, and its inhibition can attenuate susceptibility to seizures. The DHODH inhibitor teriflunomide stably suppresses mean firing rates via synaptic and intrinsic excitability mechanisms by modulating mitochondrial Ca2+ buffering and spare respiratory capacity [108]. DHODH inhibition by teriflunomide is effective in suppressing FMDV infection [111]. Related DHODH inhibitors are currently in development now. DSM265, a triazolo-pyrimidine-based inhibitor of DHODH developed by Philips, is the first DHODH inhibitor able to reach clinical research for the treatment of malaria [123]. DHDOH point mutations, which are in the DSM265 binding site in Plasmodium falciparum, confer in vitro resistance to the clinical candidate DSM265 [71]. Significantly, DHODH blockade can be significantly realized in fighting against a broad spectrum of viruses especially in emerging serve coronavirus SARS-CoV-2, which may bring hope to the patients now suffering from severe COVID-19 and other infectious diseases caused by emerging and re-emerging viruses [113, 124]. It also widens the potential impact of DHODH inhibition on additional disease phenotypes. Diverse functions of DHODH uncover its value in multiple diseases not used exclusively for cancer.
DHODH inhibitors
As for the importance of DHODH in tumorigenesis and metastasis, DHODH becomes an attractive target for anti-malignance drug development. Until now, many DHODH inhibitors with different structures have been reported [7], such as canonical DHODH inhibitors BRQ [125], leflunomide [2], teriflunomide [126], ALASN003 [127], BAY2202234 [43], and other novel inhibitors which show DHODH inhibition effects including viral growth inhibitory factor [128], PARP inhibitor [129], myeloid differentiation inducing agent [60], p53 activating factor [61], a natural product isobavachalcone [70], or vascular endothelial growth factor A (VEGF-A) mRNA translation inhibitor [40]. Over 90 patent applications involving DHODH inhibitors have been filed in the last decade [107, 130]. Notably, there is only one product, ASLAN003, that has been awarded orphan drug designation from the Food and Drug Administration (FDA) up to now [131].
As more and more novel DHODH inhibitors are emerging, it is significant to explore the binding modes between DHODH and DHODH inhibitors. Studies reveal that most DHODH inhibitors bind to the CoQ binding channel and display favorable activities [9]. For instance, BRQ bind to the CoQ binding site, and its biphenyl fragment forms a strong hydrophobic interaction with Leu46, Met43, Leu42, Leu68, and Met111, and the biphenyl fragment interacts with Tyr38 and Phe62 of DHODH [9].
Besides the binding modes between DHODH and DHODH inhibitors, some other factors also affect the efficiency of DHODH inhibitors like membrane lipid interactions [40, 132] and hypoxia conditions [36]. It is revealed that distinctive membrane-associated characteristic and mitochondrial location of DHODH may be associated with efficiency of DHODH inhibitors [133]. DHODH binds charged phospholipids which stabilize the flexible substrate- and drug-binding site to adhere to the mitochondrial membrane [40, 132]. The respiratory chain-coupled DHODH and its interplay with the surrounding lipids of the inner mitochondrial membrane provide a crucial direction for the development of DHODH-targeted inhibitors [133]. For example, the reason that DHODH inhibitor PTC299 displays potent activity is properly owing to the lipid bilayer of mitochondrial membrane facilitate the lipophilic PTC299 interacting with the DHODH binding pocket [40]. Thus, considering the impact of membrane lipid interactions on the drug-binding site may be another effective strategy to the design and development of novel DHODH inhibitors [132]. Furthermore, studies suggest the favorable targeting to DHODH in cancer cells living under hypoxia; it is worthwhile to dig into whether DHODH inhibitors with distinct scaffold also exhibit hypoxia-selective anti-cancer activity [36]. Taken together, all of these provide feasible methods for development of DHODH inhibitors (Table 2).
All the information above originated from ClinicalTrials.gov (https://clinicaltrials.gov/). In note, this table only refers to currently ongoing clinical trials in which DHODH inhibitors are applied alone, but do not cover previous failed or completed clinical trials.
Conventional DHODH inhibitors
BRQ
BRQ is originally developed for the therapy of organ transplant rejection, and the preclinical and clinical development of BRQ as an anti-cancer agent in 1997 [9, 18, 134]. Earlier investigation has revealed that BRQ is a powerful inhibitor predominantly for the dehydrogenase and not for the oxidase activity [135], and then BRQ is discovered to be one of the strongest known inhibitors of human DHODH with an IC50 value of 1.8 nM [125, 136]. Meanwhile, it is shown that both BRQ and its analog are located in the DHODH proposed ubiquinone binding channel [18]. BRQ can compete with ubiquinone and its core part, 2-phenyl 5-quinoline carboxylic acid (PQC), which also has a competitive inhibitory effect on ubiquinone [126]. BRQ structural analog not only interacts with DHODH through a carboxylic acid group, but also binds extensively to serum albumin, which alters their free unbound plasma concentration [61]. The in vivo study shows BRQ activates a p53-dependent reporter in cells at low concentrations and inhibits tumor growth. BRQ exhibits strong in vivo antitumor activity through DHODH depletion in a KRAS mutant pancreatic xenograft model and induces a decrease in steady-state glutamate concentration [63]. It also significantly sensitizes U1690 cells to TRAIL-mediated apoptosis [72].
BRQ is evaluated in the Phase I and II trials of patients with advanced solid tumor malignancies [107], such as squamous cell carcinoma of the head and neck [137], lung cancer [64], gastrointestinal cancer [138], and breast cancer and melanoma [139, 140]. However, it does not get different Phase II clinical trials where it demonstrated only very modest anti-cancer effects on various solid tumors. Moreover, its narrow therapeutic window further hinders clinical application [141]. Increased toxicity of BRQ and its analogs is revealed when given in combination with cyclosporine. The side effects observed include leukocytopenia, thrombocytopenia, reduced body weight gain or body weight loss, thymic atrophy, cellular depletion of bone marrow and splenic white pulp, and villous atrophy in jejunum [134]. In 2018, BRQ has been reapplied in clinical trials of AML, which reveals that BRQ may have potential in malignancy therapy.
Leflunomide
Leflunomide (HR486) is an isoxazole derivative that originated from an anti-inflammatory drug development program at Hoechst during the 1980s in order to control autoimmune diseases [30, 35]. Leflunomide binds with DHODH within the tunnel which can directly lead to the bound FMN and provide access to ubiquinone so as to block ubiquinone to the active site [2]. It exhibits both anti-inflammatory and immunomodulatory properties with a different structure from other known immunomodulatory drugs [142]. The anti-inflammatory effects of leflunomide are associated with inhibited de novo synthesis of pyrimidine nucleotides (and consequently DNA and RNA), inhibited oxidative damage, and reduced proinflammatory cytokine expression in immune response cells [143, 144]. Leflunomide exerts effects on murine models of lymphoproliferation and systemic lupus erythematosus [96], graft-versus-host disease [97], proteoglycan-induced arthritis [99], and diabetes [101], and on rat models of adjuvant arthritis [95], tubulointerstitial nephritis [98], glomerulonephritis due to anti-basement membrane antibody [99], and the anti-acetylcholine receptor-induced model of myasthenia gravis [100]. In 1999, leflunomide was approved by the FDA and first used as an oral disease-modifying antirheumatic drug (DMARD) for the treatment of active RA refractive to methotrexate.
In addition to above efficiency, leflunomide shows antitumor potency against various malignancies. DHODH inhibition by leflunomide induces upregulation of p53 in Hela cells [52], inhibits proliferation of PTEN-mutant human breast cancer cell lines [29, 145], and contributes to RCC S phase arrest and autophagy [49]. Leflunomide induces S phase arrest and accumulation of cyclin E in breast cancer cells, which is further supported in DHODH-silenced cells [37]. DHODH suppression by leflunomide decreased cell motility in breast cancer cells, which could explain the anti-metastatic effect of leflunomide [37]. Leflunomide-treated mice show decreased DHODH activity and fail to develop pre-malignant and malignant lesions [86]. DHODH inhibition by leflunomide also leads to an almost complete abrogation of neural crest development in zebrafish and to a reduction in the self-renewal of mammalian neural crest stem cells [89]. However, severe side effects of leflunomide are observed including diarrhea, abnormal liver functions, hypertension, myelosuppression, nausea, and hair loss [107] during its administration. In 2010, the FDA gave leflunomide a black box warning due to its causing of acute liver failure in humans though the underlying mechanism of its toxicity remains unclear. The severe side effects hinder its further clinical use.
Teriflunomide
In 1979, teriflunomide (A77-1726, Ter), the active metabolite of leflunomide, was originally found as a drug for rheumatoid arthritis and multiple sclerosis treatment. Leflunomide is rapidly converted into its active metabolite teriflunomide via a non-enzymatic, base-caused isoxazole ring-opening reaction in the plasma and intestinal mucosa [146]. Teriflunomide is a non-competitive inhibitor with ubiquinone with an IC50 value of approximately 600 nM against DHODH activity [143]. Compared with leflunomide, teriflunomide is well tolerated, has a long half-life of more than 2 weeks, and can be delivered into the brain, which supports that teriflunomide is a particularly attractive choice of pyrimidine-targeting strategy [36, 38].
Teriflunomide exhibits antitumor effects by inhibiting DHODH activity in different malignances. It significantly reduces proliferation of primary microglia [147], GSCs [25], etc. Teriflunomide treatment of GSCs with either EGFR amplification or PTEN deletion inhibits pyrimidine biosynthesis and cells proliferation. Moreover, teriflunomide displays combinatorial function with either lapatinib or BKM120 in reduction of pyrimidine synthesis [25]. The in vivo mechanism studies have demonstrated that teriflunomide can inhibit adhesion molecules, cytokines, protein tyrosine kinases, nuclear factor-kB (NF-kB) activation, and cyclooxygenase 2 activity, suggesting that teriflunomide in addition to its anti-proliferative effects may also impact signal transduction, migration, and inflammatory processes [147,148,149]. Besides inhibiting malignancies, teriflunomide contributes to apoptosis in normal and transformed human keratinocytes [85], suggesting its potential toxicity against normal tissues. The common adverse events including headache, nausea, diarrhea, fatigue, elevated alanine aminotransferase levels, hair thinning or decreased hair density, and urinary tract infection are observed in its clinical use [107, 150, 151].
BAY2402234
BAY2402234 is a novel, potent, selective, and orally bioavailable DHODH inhibitor that shows monotherapy efficacy and differentiation induction across multiple AML subtypes [43]. It is developed by Bayer and has entered Phase I clinical trials in January 2018 (Clinical trial identifier: NCT03404726). In May 2018, the patent containing the BAY2402234 structure and a description of the activity profile has been published [60]. BAY2402234, binding the ubiquinone binding site of DHODH between the N-terminal helices, can concentration-dependently inhibit human full-length DHODH enzyme with an IC50 value of 1.2 nM [43]. Further, BAY 2402234 has moderate plasma protein binding, low blood clearance, and a high volume of distribution [43]. Additionally, BAY2402234 can downregulate key enzymes in UDP GlcNAc metabolism Glucosamine-Phosphate N-Acetyltransferase 1 and UDP-N-Acetylglucosamine Pyrophosphorylase 1, which may account for previous observation of global decreases in protein N-acetyl glycosylation after DHODH inhibition [39, 43]. As so far, the researches related to BAY2402234 are not abundant for its new discovery in 2018. It is shown that BAY2402234 induces differentiation and inhibits proliferation in AML cell lines across multiple AM subtypes by inhibiting DHODH, including ten leukemia cell lines representing diverse AML subtypes, with a low range of IC50 [43]. Significantly, BAY2402234 induces profound transcriptional changes, prolongs survival and induces differentiation in AML PDX models [43]. However, the side effects and anti-proliferation effects in other tumors remain to be explored.
ASLAN003
ASLAN003 (LAS186323), a bioavailable and novel DHODH inhibitor, was first discovered by Almirall, S.A., and global rights to the compound have been granted to ASLAN Pharmaceuticals Singapore in 2012. It is currently being evaluated in Phase II clinical trial (clinical trial identifier: NCT03451084) in AML patients [60, 127]. ASLAN003 is an effective inhibitor of human DHODH with an IC50 value of 35 nM. It can induce differentiation of AML cells in vitro and in vivo and trigger apoptosis, inhibit protein synthesis, and activate AP-1 transcription. Meanwhile, gene expression of four members of eukaryotic translation initiation factor (eIF) family, EEF1B2, EIF4B, EIF3L, EEF1B2P3, were significantly decreased in ASLAN003-treated cells. It has been proven tolerable in the first phase of single and multiple escalating dose clinical trials in healthy volunteers [127], while its side effects and antitumor functions in other cancers need to be further manifested.
Other novel DHODH inhibitors
In recent years, novel DHODH inhibitors are massively emerging, which include analogs of BRQ [60, 102], IMU-838, and its active metabolite vidofludimus [152,153,154], PTC299 [40, 155], DD264 [128], plant extract Celastrol [156], protein kinase inhibitors OSU-03012 and TAK-632 [112, 130, 157], isobavachalcone [70], tetrahydroindazole (HZ) analogs [35, 61], alkaloid cerpegin-related P1788 [158], compound 11 [79], etc. (Table 3).
Benzimidazole derivatives have shown a wide variety of pharmacological activities including dual PARP and DHODH inhibition. It is considerable to further study the binding mechanisms and enhance their potential as inhibitors of PARP-1 and DHODH [129]. IMU-838, a novel DHODH inhibitor, acts on activated T and B cells without affecting other immune cells, which allows the immune system to keep functioning [160]. Vidofludimus, the active moiety and free acid form of IMU-838, is a potent and selective inhibitor DHODH [152,153,154]. It can inhibit the intracellular metabolism of activated immune cells by blocking DHODH and inhibit colonic interleukin-17 and improve colitis in rats [154]. Lucas-Hourani et al. find a novel DHODH inhibitor DD264 with dual effects of immune-stimulation and antiviral through screening 41353 compounds. However, the antiviral property of pyrimidine biosynthesis inhibitors is not a direct consequence of pyrimidine deprivation on the virus machinery, but rather involves the induction of cellular immune response [128]. Antiviral activity of DD264 is found strictly dependent on cellular gene transcription and nuclear export machinery and required interferon regulatory transcription factor 1 (IRF1) transcription factor. Celastrol, extracted from “Thunder of God Vine,” is a DHODH inhibitor and promising anti-cancer natural product. Celastrol could induce apoptosis of acute promyelocytic leukemia (APL) cells via p53-activated mitochondrial pathway [156]. Recently, Abt et al. substantiate that two protein kinase inhibitors OSU-03012 and TAK-632 found by metabolic modifier screen, competing with ubiquinone, engage in DHODH through crystallography studies. Notably, OSU-03012 and TAK-632 bind in the same hydrophobic tunnel of DHODH as known inhibitors BRQ and teriflunomide. By competitively inhibiting the binding of ubiquinone, these compounds prevent DHODH from completing its redox cycle and effectively abrogate its activity [130]. Meanwhile, OSU-03012 and analogs can inhibit pyrimidine biosynthesis in host cells to inhibit viral replication, specifically illustrating regulation of DHODH activity [112, 130]. Isobavachalcone is a novel DHODH inhibitor which derived from traditional Chinese medicine psoralea corylifolia. It directly inhibits human DHODH and triggers apoptosis and differentiation of AML cells. Oral administration of isobavachalcone suppresses subcutaneous HL60 xenograft tumor growth without obvious toxicity. Importantly, combining isobavachalcone with adriamycin prolong mice survival in an intravenous HL60 leukemia model [70]. Ladds et al. identified over 100 small molecules activating p53 in cells and found active enantiomer (R)-HZ00 can inhibit DHODH through target deconvolution. Twelve other DHODH inhibitor chemotypes are detailed among the p53 activators, which identifies DHODH as a frequent target for structurally diverse compounds [61]. Recently, several HZ analogs are further to be optimized and display anti-proliferation effect on ARN8 melanoma cells [35]. PTC299 is a novel, orally bioavailable small molecule that selectively inhibits VEGF receptor protein synthesis at the post-transcriptional level. Recent study demonstrated that PTC299 is a more potent inhibitor of DHODH in isolated mitochondria. PTC299 has advantages over previously reported DHODH inhibitors, including greater potency, good oral bioavailability, and lack of off-target kinase inhibition and myelosuppression. PTC299 is currently in Phase I clinical trial against AML patients (clinical trial identifier: NCT03761069) and thus may be useful for the targeted treatment of hematologic malignancies [40]. 10580, a novel DHODH inhibitor, found through high-throughput drug screening, decreases pyrimidine nucleotide levels and enhances sex-determining region Y–box 2 nuclear export by antagonizing the enzyme activity of DHODH [165]. P1788, a compound chemically related to the alkaloid cerpegin, is a new class of DHDOH inhibitors. Pyrimidine depletion by P1788 amplifies cellular response to both type I and type II interferons and induces DNA damage [158]. Compound 11l and 11k, newly found in 2020, can significantly induce ROS production and mitochondrial dysfunction and inhibit leukemia cells and solid tumor cell proliferation [164].
Prospects of DHODH inhibitors in cancer therapy
DHODH is a promising target in multiple malignancy therapies. When applying in specific genetic context, DHODH inhibitors might exert more targeted and meaningful effects on malignancy therapy. Recent studies have illustrated that DHODH inhibitors are more effective against malignancies with specific genetic context, such as melanomas carrying BRAF (V600E) mutation [89] and glioblastoma stem cells [25] and breast cancer with PTEN deletion [25, 29] and pancreatic cancer with KRAS mutation [63]. Therefore, the specific genetic context for DHODH inhibitor treatment need to be further explored.
Meanwhile, combination of DHODH inhibitors with other anti-cancer drugs is a promising approach to achieving clinical benefits in various tumor therapies as DHODH inhibitors alone have limited therapeutic windows and may not achieve desired outcomes. Studies verify that combination of DHODH inhibitors with traditional cell toxicity anti-cancer drugs synergistically inhibits tumor proliferation. Gemcitabine, a first-line choice for pancreatic cancer, is an ideal combination partner for DHODH inhibitors [63]. As gemcitabine treatment leads to increased de novo pyrimidine synthesis in pancreatic cancer cells, combination of DHODH inhibitor Ter with gemcitabine reversed gemcitabine-induced nucleoside synthesis and thus exhibits synergistic effect against pancreatic cancer proliferation [82]. Furthermore, teriflunomide combined with the genotoxic agents such as 5-fluorouracil (5-FU) [87], melphalan, treosulfan, doxorubicin, dexamethasone, and bortezomib reveal the synergistic and additive activity in multiple myeloma [65] and UVB-induced cSCC [87] proliferation suppression. Combination treatment of BRQ and doxorubicin results in significant anti-proliferation effects on melanoma cells both in vitro and in vivo [88].
Besides traditional cell toxicity of anti-cancer drugs, DHODH inhibitors in combination with novel targeted anti-cancer drugs attracted much attention. Combination of DHODH inhibitors with mitochondria complex I inhibitors can interfere two crucial steps of pyrimidine biosynthesis, which may provide a broad-spectrum and effective antitumor strategy [57]. Combination of leflunomide and the Chk1 inhibitor reduces proliferation and induces massive cell apoptosis of p53-deficient breast tumors compared with leflunomide treatment alone [37]. Given that p53 is generally mutated in more than 50% of tumors, the simultaneous inactivation of DHODH and Chk1 kinase is a potential method for the treatment of refractory p53 mutant tumors [37]. As salvage and de novo pyrimidine pathways are suppressed by ATR blockade [157], combination of ATR inhibitor and OSU-03012, a DHODH inhibitor, reveals synergy against pancreatic ductal adenocarcinoma [130]. These observations suggested that DHODH inhibitors may have utility in conjunction with DNA damage response pathway inhibitors [130, 157]. Meanwhile, in case of DHODH inhibitors ineffective due to efficient uridine salvage synthesis in certain cells, combination DHODH inhibitors with non-competitive pyrimidine salvage enzyme uridine-cytidine kinase 2 (UCK2) inhibitors was able to suppress nucleoside synthesis and inhibit cell proliferation [166]. Deoxycytidylate deaminase, converting deoxycytidine monophosphate (dCMP) to dUMP, is an enzyme that supplies the vast majority of dUMP for cancer cells [41, 167], and cells with high expression of DCTD are better able to maintain dUMP pools when the de novo pyrimidine synthesis pathway is inhibited, suggesting that combination of DHODH and DCTD inhibitors maybe a significant antitumor strategy, though the utility requires further investigation [41, 168, 169].
Although combined targeting strategies offer powerful methods for cancer therapy, the limitations are still existent, including alternate concentrations of inhibitors which are required to achieve therapeutic doses in order to satisfy distribution in different organs in the clinical trial. Combination BQR with other immuno-suppression inhibitors shows altered pharmacokinetics, including increased drug toxicity and decreased drug efficacious doses, suggesting pharmacokinetics and the therapeutic window in combination treatment may be a significant focus [134]. Taken together, DHODH inhibitors and anti-cancer drug combination may be a promising method of cancer therapy.
Conclusions
DHODH, a key enzyme in the de novo pyrimidine synthesis pathway, plays a crucial role in tumorigenesis via influencing pyrimidine metabolism and mitochondria function. Inhibiting DHODH has gradually become a hit spot in malignancy therapy. Novel DHODH inhibitors are springing up at an accelerated pace in recent decades. Significantly, combined target therapy seems to be a particularly promising strategy, especially under specific genetic contexts, due to the re-sensitive of tumors to traditional chemotherapy. Meanwhile, the highly specific DHODH inhibitors and more appropriate administration dosage may improve efficacy and adverse side effects associated with traditional DHODH treatment, which is required to further investigate for the development of DHODH-targeted inhibitors in the near future. Collectively, DHODH, revealing multiple biological functions in neoplastic and non-neoplastic diseases, has broad-spectrum prospective remained to be explored.
Availability of data and materials
Not applicable.
Abbreviations
- 5-FU:
-
5-Fluorouracil
- AK:
-
Actinic keratoses
- ALDOB:
-
Aldolase, fructose-bisphosphate B
- AML:
-
Acute myeloid leukemia
- ACRV1:
-
Acrosomal vesicle protein 1
- ATR:
-
Ataxia telangiectasia and Rad3-related kinase
- ATP10B:
-
ATPase, class V, type 10B
- APPA2:
-
Pappalysin 2
- BCC:
-
Basal cell carcinomas
- CA12:
-
Arbonic anhydrase XII
- CAD:
-
Carbamoylphosphate synthetase II (CPSII), aspartate transcarbamoylase (ATCase), dihydroorotase (DHOase)
- CHK1:
-
Checkpoint kinase 1
- CM:
-
Cutaneous malignant melanoma
- CoQ:
-
Coenzyme Q
- COVID-19:
-
Coronavirus disease 2019
- cSCCs:
-
Cutaneous squamous cell carcinomas
- CTP:
-
Cytidine triphosphate
- CYP2A6:
-
Cytochrome P450 family 2 subfamily A member 6
- CYP2C9:
-
Cytochrome P450 family 2 subfamily C member 9
- dCMP:
-
Deoxycytidine monophosphate
- DCTD:
-
Deoxycytidylate deaminase
- DCT:
-
Dopachrome tautomerase
- dCTP:
-
Deoxycytidine triphosphate
- DHODH:
-
Dihyrdroorotate dehydrogenase
- DHO:
-
Dihydroorotate
- dUDP:
-
Deoxyuridine diphosphate
- DRD2:
-
Dopamine receptor D2
- dUMP:
-
Deoxyuridine monophosphate
- EGFR:
-
Epidermal growth factor receptor
- eIF:
-
Eukaryotic translation initiation factor
- ER:
-
Endoplasmic reticulum
- ETC:
-
Electron transport chain
- ETV1:
-
ets variant 1
- EXPH5:
-
Exophilin 5
- FMN:
-
Flavin mononucleotide
- GATA-2:
-
GATA-binding factor 2
- GlcNAc:
-
N-acetylglucosaminylation
- Gln:
-
Glutamine
- GSCs:
-
Glioblastoma stem cells
- HFE:
-
Hemochromatosis
- HIF1:
-
Hypoxia-inducible factor 1
- HZ:
-
Tetrahydroindazoles
- IBC:
-
Isobavachalcone
- IRF:
-
Interferon regulatory transcription factor 1
- KRAS:
-
Kirsten rat sarcoma viral oncogene
- LLS:
-
Lake Louise Score
- MLL:
-
Mixed-lineage leukemia gene
- MID2:
-
Midline 2
- mROS:
-
Mitochondria reactive oxygen species
- mtDNA:
-
Mitochondrial DNA
- NAD+ :
-
Oxidized nicotinamide adenine dinucleotide
- NADH:
-
Reduced nicotinamide adenine dinucleotide
- nDNA:
-
Nuclear DNA
- NMSC:
-
Nonmelanocytic skin cancer
- NSCLC:
-
Non-small cell lung cancer
- NTRK3:
-
Neurotrophic tyrosine kinase, receptor, type 3
- ODC:
-
Orotidine-5'-decarboxylase
- O-GlcNAc:
-
O-linked N-acetylglucosaminylation
- OMP:
-
Orotidine 5'-monophosphate
- OPRT:
-
Orotate phosphoribosyltransferase
- ORF:
-
Open reading frame
- OXPHOS:
-
Oxidative phosphorylation
- PAX8:
-
Paired box 8
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PIMs:
-
The PIM protein serine-threonine kinase family
- PKB:
-
Phosphorylation of protein kinase B
- PLK:
-
Polo-like kinase
- PTEN:
-
Phosphatase and tensinhomolog
- POU3F2:
-
POU domain, class 3, transcription factor 2
- RB:
-
Retinoblastoma protein
- RCC:
-
Renal cell carcinoma
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SCCs:
-
Squamous cell carcinomas
- SCLC:
-
Small cell lung cancer
- SIGLEC6:
-
Sialic acid binding Ig-like lectin 6
- SLC30A3:
-
Solute carrier family 30 (zinc transporter), member 3
- STAT3:
-
Signal transducer and activator of transcription 3
- TFMA:
-
Mitochondrial transcription factor A
- TM:
-
Transmembrane domain
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- UBE2D4:
-
Ubiquitin conjugating enzyme E2D 4 (putative)
- UCK2:
-
Uridine-cytidine kinase 2
- UDP:
-
Uridine diphosphate
- UMP:
-
Uridine monophosphate
- UMPS:
-
Uridine monophosphate synthase
- UTP:
-
Uridine triphosphate
- WNT6:
-
Wingless-type MMTV integration site family member 6
References
Counihan JL, Grossman EA, Nomura DK. Cancer metabolism: current understanding and therapies. Chem Rev. 2018;118:6893–923 American Chemical Society.
Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–8.
Weber G. Reciprocal regulation: recognition of pattern of gene expression in cancer cells. Adv Enzym Regul. 2002;42:83–100.
Barnes T, Parry P, Hart I, Jones C, Minet M, Patterson D. Regional mapping of the gene encoding dihydroorotate dehydrogenase, an enzyme involved in UMP synthesis, electron transport, and superoxide generation, to human chromosome region 16q22. Somat Cell Mol Genet. 1993;19:405–11.
Sørensen PG, Dandanell G. A new type of dihydroorotate dehydrogenase, type 1S, from the thermoacidophilic archaeon Sulfolobus solfataricus. Extremophiles. 2002;6(3):245–51.
Smith LH, Baker FA. Pyrimidine metabolism in man. I. The biosynthesis of orotic acid. J Clin Investig. 1959;38(5):798.
Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, Rohlena J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim Biophys Acta Mol basis Dis. 2020:165759.
Madak JT, Bankhead A, Cuthbertson CR, Showalter HD, Neamati N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol Ther. 2019;195:111–31.
Vyas V, Ghate M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) Inhibitors. Mini-Rev Med Chem. 2011;11:1039–55.
General I. Dihydroorotate Dehydrogenase. J Biol Chem. 1967;18:4087–96.
Reis RAG, Calil FA, Feliciano PR, Pinheiro MP, Nonato MC. The dihydroorotate dehydrogenases: past and present. Arch Biochem Biophys. 2017;632:175–91.
Björnberg O, Grüner AC, Roepstorff P, Jensen KF. The activity of Escherichia coli dihydroorotate dehydrogenase is dependent on a conserved loop identified by sequence homology, mutagenesis, and limited proteolysis. Biochemistry. 1999;38:2899–908.
Palfey BA, Björnberg O, Jensen KF. Insight into the chemistry of flavin reduction and oxidation in Escherichia coli dihydroorotate dehydrogenase obtained by rapid reaction studies. Biochemistry. 2001;40:4381–90.
Hey-Mogensen M, Goncalves RLS, Orr AL, Brand MD. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic Biol Med. 2014;72:149–55.
Nielsen FS, Andersen PS, Jensen KF. The B form of dihydroorotate dehydrogenase from Lactococcus lactis consists of two different subunits, encoded by the pyrDb and pyrK genes, and contains FMN, FAD, and [FeS] redox centers. J Biol Chem. 1996;271:29359–65.
Rowland P, Nielsen FS, Jensen KF, Larsen S. The crystal structure of the flavin containing enzyme dihydroorotate dehydrogenase A from Lactococcus lactis. Structure. 1997;5:239–52.
Leban J, Kralik M, Mies J, Gassen M, Tentschert K, Baumgartner R. SAR, species specificity, and cellular activity of cyclopentene dicarboxylic acid amides as DHODH inhibitors. Bioorg Med Chem Lett. 2005;15:4854–7.
Baumgartner R, Walloschek M, Kralik M, Gotschlich A, Tasler S, Mies J, et al. Dual binding mode of a novel series of DHODH inhibitors. J Med Chem. 2006;49(4):1239–47.
Rich PR, Maréchal A. The mitochondrial respiratory chain. Essays Biochem. 2010;47:1–23.
Rawls J, Knecht W, Diekert K, Lill R, Löffler M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur J Biochem. 2000;267:2079–87.
Rodriguez JMO, Krupinska E, Wacklin-Knecht H, Knecht W. Preparation of human dihydroorotate dehydrogenase for interaction studies with lipid bilayers. Nucleosides Nucleotides Nucleic Acids. 2020;0:1–14.
Fang J, Uchiumi T, Yagi M, Matsumoto S, Amamoto R, Takazaki S, et al. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci Rep. 2013;33:217–27.
Löffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA. Pyrimidine pathways in health and disease. Trends Mol Med. 2005;11:430–7.
Lane AN, Fan TWM. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–85.
Wang X, Yang K, Wu Q, Kim LJY, Morton AR, Gimple RC, et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci Transl Med. 2019;11:1–15.
Löffler M, Jöckel J, Schuster G, Becker C. Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol Cell Biochem. 1997;174:125–9.
Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–16.
Bofill M. Importance of Ribonucleotide Availability to Proliferating Tlymphocytes from Healthy Humans. J Biol Chem. 1995;270:29682–9.
Mathur D, Stratikopoulos E, Ozturk S, Steinbach N, Pegno S, Schoenfeld S, et al. PTEN regulates glutamine flux to pyrimidine synthesis and sensitivity to dihydroorotate dehydrogenase inhibition. Cancer Discov. 2017;7:380–90.
Lewis TA, Sykes DB, Law JM, Muñoz B, Rustiguel JK, Nonato MC, et al. Development of ML390: a human DHODH inhibitor that induces differentiation in acute myeloid leukemia. ACS Med Chem Lett. 2016;7(12):1112–7.
Mascia L, Turchi G, Bemi V, Ipata PL. Uracil salvage pathway in PC12 cells. Biochim Biophys Acta, Gen Subj. 2000;1524:45–50.
Huisman WH, Raivio KO, Becker MA. Simultaneous determination of rates of purine and pyrimidine synthesis in cultured human lymphoblasts and fibroblasts. Adv Exp Med Biol. 1979;122(B):223–9.
Jackson RC, Lui MS, Boritzki TJ, Weber G, Morris HP. Purine and pyrimidine nucleotide patterns of normal, differentiating, and regenerating liver and of hepatomas in rats. Cancer Res. 1980;40:1286–91.
Sigoillot FD, Berkowski JA, Sigoillot SM, Kotsis DH, Guy HI. Cell cycle-dependent regulation of pyrimidine biosynthesis. J Biol Chem. 2003;278:3403–9.
Popova G, Ladds MJGW, Johansson L, Saleh A, Larsson J, Sandberg L, et al. Optimization of tetrahydroindazoles as inhibitors of human dihydroorotate dehydrogenase and evaluation of their activity and in vitro metabolic stability. J Med Chem. 2020;63:3915–34.
Miyazaki Y, Inaoka DK, Shiba T, Saimoto H, Sakura T, Amalia E, et al. Selective cytotoxicity of dihydroorotate dehydrogenase inhibitors to human cancer cells under hypoxia and nutrient-deprived conditions. Front Pharmacol. 2018;9:1–13.
Hubackova S, Davidova E, Boukalova S, Kovarova J, Bajzikova M, Coelho A, et al. Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors. Cell Death Dis. 2020;11.
Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–77.
Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–86.e15.
Cao L, Weetall M, Trotta C, Cintron K, Ma J, Kim MJ, et al. Targeting of hematologic malignancies with PTC299, a novel potent inhibitor of dihydroorotate dehydrogenase with favorable pharmaceutical properties. Mol Cancer Ther. 2019;18:3–16.
Li L, Ng SR, Colón CI, Drapkin BJ, Hsu PP, Li Z, et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci Transl Med. 2019;11:517.
Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: a new cancer hallmark? Front Endocrinol. 2013;4:1–15.
Christian S, Merz C, Evans L, Gradl S, Seidel H, Friberg A, et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia. 2019;33:2403–15.
Ishihara K, Takahashi I, Tsuchiya Y, Hasegawa M, Kamemura K. Characteristic increase in nucleocytoplasmic protein glycosylation by O-GlcNAc in 3 T3–L1 adipocyte differentiation. Biochem Biophys Res Commun. 2010;398:489–94.
Andrés-Bergós J, Tardio L, Larranaga-Vera A, Gómez R, Herrero-Beaumont G, Largo R. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J Biol Chem. 2012;287:33615–28.
Sun C, Shang J, Yao Y, Yin X, Liu M, Liu H, et al. O-GlcNAcylation: a bridge between glucose and cell differentiation. J Cell Mol Med. 2016;20(5):769–81.
Vélez J, Hail N, Konopleva M, Zeng Z, Kojima K, Samudio I, et al. Mitochondrial uncoupling and the reprograming of intermediary metabolism in leukemia cells. Front Oncol. 2013;3:1–8.
Zhu S, Yan X, Xiang Z, Ding HF, Cui H. Leflunomide reduces proliferation and induces apoptosis in neuroblastoma cells in vitro and in vivo. PLoS One. 2013;8:1–7.
Chen Y, Huang Q, Zhou H, Wang Y, Hu X, Li T. Inhibition of canonical WNT/β-catenin signaling is involved in leflunomide (LEF)-mediated cytotoxic effects on renal carcinoma cells. Oncotarget. 2016;7:50401–16.
Yin S, Kabashima T, Zhu Q, Shibata T, Kai M. Fluorescence assay of dihydroorotate dehydrogenase that may become a cancer biomarker. Sci Rep. 2017;7:1–7.
O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–303.
Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci U S A. 2010;107:12828–33.
Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–62.
Idelchik M, Begley U, Begley TJ, Melendez JA. Mitochondrial ROS control of cancer. Semin Cancer Biol. 2017;47:57–66.
Hail N, Chen P, Kepa JJ, Bushman LR, Shearn C. Dihydroorotate dehydrogenase is required for N-(4-hydroxyphenyl)retinamide-induced reactive oxygen species production and apoptosis. Free Radic Biol Med. 2010;49:109–16.
Mohamad Fairus AK, Choudhary B, Hosahalli S, Kavitha N, Shatrah O. Dihydroorotate dehydrogenase (DHODH) inhibitors affect ATP depletion, endogenous ROS and mediate S-phase arrest in breast cancer cells. Biochimie. 2017;135:154–63.
Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab. 2019;29:399–416.e10.
Glasauer A, Chandel NS. ROS. Curbio. 2013;23:R100–2 Available from: https://ac.els-cdn.com/S0960982212014510/1-s2.0-S0960982212014510-main.pdf?_tid=9a1a3c38-1eba-4239-b4cf-f525551ae16e&acdnat=1534850536_409ccb71c248b51c378946d838f88eb7.
Diebold L, Chandel NS. Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med. 2016;100:86–93.
Sainas S, Pippione AC, Lupino E, Giorgis M, Circosta P, Gaidano V, et al. Targeting myeloid differentiation using potent 2-hydroxypyrazolo[1,5- a]pyridine scaffold-based human dihydroorotate dehydrogenase inhibitors. J Med Chem. 2018;61(14):6034–55.
Ladds MJGW, Van Leeuwen IMM, Drummond CJ, Chu S, Healy AR, Popova G, et al. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat Commun. 2018;9:1–4.
Zhai H, Song B, Xu X, Zhu W, Ju J. Inhibition of autophagy and tumor growth in colon cancer by miR-502. Oncogene. 2013;32:1570–9.
Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, et al. Dependence on the pyrimidine biosynthetic enzyme DHODH is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem Biol. 2018;25:705–17.e11.
Maroun J, Ruckdeschel J, Natale R, Morgan R, Dallaire B, Sisk R, et al. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Cancer Chemother Pharmacol. 1993;32:64–6.
Baumann P, Mandl-Weber S, Völkl A, Adam C, Bumeder I, Oduncu F, et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009;8:366–75.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;68:caac.21660.
Prada-Arismendy J, Arroyave JC, Röthlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2017;31:63–76.
Sykes DB. The emergence of dihydroorotate dehydrogenase (DHODH) as a therapeutic target in acute myeloid leukemia. Expert Opin Ther Targets. 2018;22:893–8.
Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–46.
Wu D, Wang W, Chen W, Lian F, Lang L, Huang Y, et al. Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica. 2018;103:1472–83.
White J, Dhingra SK, Deng X, El Mazouni F, Lee MCS, Afanador GA, et al. Identification and mechanistic understanding of dihydroorotate dehydrogenase point mutations in Plasmodium falciparum that confer in vitro resistance to the clinical candidate DSM265. ACS Infect Dis. 2019;5:90–101.
He T, Haapa-Paananen S, Kaminskyy VO, Kohonen P, Fey V, Zhivotovsky B, et al. Inhibition of the mitochondrial pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase by doxorubicin and brequinar sensitizes cancer cells to TRAIL-induced apoptosis. Oncogene. 2014;33:3538–49.
Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–502.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Khutornenko AA, Dalina AA, Chernyak BV, Chumakov PM, Evstafieva AG. The role of dihydroorotate dehydrogenase in apoptosis induction in response to inhibition of the mitochondrial respiratory Chain complex III. Acta Nat. 2014;6:69–75.
Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, Janjigian YY, et al. PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife. 2019;8:189–92.
Pizzorno G, Wiegand RA, Lentz SK, Handschumacher RE. Brequinar potentiates 5-fluorouracil antitumor activity in a murine model colon 38 tumor by tissue-specific modulation of uridine nucleotide pools. Cancer Res. 1992;52:1660–5.
Demedts IK, Vermaelen KY, Van Meerbeeck JP. Treatment of extensive-stage small cell lung carcinoma: Current status and future prospects. Eur Respir J. 2010;35:202–15.
Califano R, Abidin AZ, Peck R, Faivre-Finn C, Lorigan P. Management of small cell lung cancer: Recent developments for optimal care. Drugs. 2012;72:471–90.
Byers LA, Rudin CM. Small cell lung cancer: where do we go from here? Cancer. 2015;121:664–72.
Huang F, Ni M, Chalishazar MD, Huffman KE, Kim J, Cai L, et al. Inosine monophosphate dehydrogenase dependence in a subset of small cell lung cancers. Cell Metab. 2018;28:369–82.e5.
Buettner R, Morales C, Wu X, Sanchez JF, Li H, Melstrom LG, et al. Leflunomide synergizes with gemcitabine in growth inhibition of PC cells and impairs c-Myc signaling through PIM kinase targeting. Mol Ther Oncolytics. 2019;14:149–58.
Simões MCF, Sousa JJS, Pais AACC. Skin cancer and new treatment perspectives: a review. Cancer Lett. 2015;357:8–42.
Linares MA, Zakaria A, Nizran P. Skin Cancer. Prim Care. 2015;42(4):645–59.
Hail N, Chen P, Kepa JJ, Bushman LR. Evidence supporting a role for dihydroorotate dehydrogenase, bioenergetics, and p53 in selective teriflunomide-induced apoptosis in transformed versus normal human keratinocytes. Apoptosis. 2012;17(3):258–68.
Hosseini M, Dousset L, Mahfouf W, Serrano-Sanchez M, Redonnet-Vernhet I, Mesli S, et al. Energy metabolism rewiring precedes UVB-induced primary skin tumor formation. Cell Rep. 2018;23:3621–34.
Hosseini M, Dousset L, Michon P, Mahfouf W, Muzotte E, Bergeron V, et al. UVB-induced DHODH upregulation, which is driven by STAT3, is a promising target for chemoprevention and combination therapy of photocarcinogenesis. Oncogenesis. 2019;8(10):52.
Dorasamy MS, Aravind B, Nellore K, Wong PF. Synergistic inhibition of melanoma xenografts by Brequinar sodium and Doxorubicin. Biomed Pharmacother. 2019;110:29–36.
White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–22.
Santoriello C, Sporrij A, Yang S, Flynn RA, Henriques T, Dorjsuren B, et al. RNA helicase DDX21 mediates nucleotide stress responses in neural crest and melanoma cells. Nat Cell Biol. 2020;22:372–9.
Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40(D1):D261–70.
Ginter E. New findings on essential amino acids. Cesk Fysiol. 1990;39:13–25 Czech Republic.
Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–40.
Raijmakers R, Kraiczek K, de Jong AP, Mohammed S, Heck AJR. Exploring the human leukocyte phosphoproteome using a microfluidic reversed-phase-TiO2-reversed-phase high-performance liquid chromatography phosphochip coupled to a quadrupole time-of-flight mass spectrometer. Anal Chem. 2010;82:824–32.
Bartlett R, Schleyerbach R. Immunopharmacological profile of a novel isoxazol derivative, HWA 486, with potential antirheumatic activity — I. Disease modifying action on adjuvant arthritis of the rat. Int J Immunopharmacol. 1985;7(1):7–18.
Popovic S, Bartlett R, Popovic S, Bartlett RR. Disease modifying activity of HWA 486 on the development of SLE in MRL/l-mice. Agents Actions. 1987;19:313–4.
Popovic S, Bartlett R. The use of the murine chronic graft Vs host (CGVH) disease, a model for systemic lupus erythematosus (SLE), for drug discovery. Agents Actions. 1987;21:284–6.
Thoenes GH, Sitter T, Langer KH, Bartlett RR, Schleyerbach R. Leflunomide (HWA 486) inhibits experimental autoimmune tubulointerstitial nephritis in rats. Int J Immunopharmacol. 1989;11:921–9.
Ogawa T, Inazu M, Gotoh K, Inoue T, Hayashi S. Therapeutic effects of leflunomide, a new antirheumatic drug, on glomerulonephritis induced by the antibasement membrane antibody in rats. Clin Immunol Immunopathol. 1991;61:103–18.
Vidic-Dankovic B, Kosec D, Damjanovic M, Apostolski S, Isakovic K, Bartlett R. Leflunomide prevents the development of experimentally induced myasthenia gravis. Int J Immunopharmacol. 1995;17:273–81.
Stosić-Grujicić S, Dimitrijević M, Bartlett RR. A novel immunomodulating agent--leflunomide inhibits experimental autoimmune diabetes in mice. Transplant Proc. 1996;28:3072–3.
Walse B, Dufe VT, Svensson B, Fritzson I, Dahlberg L, Khairoullina A, et al. The structures of human dihydroorotate dehydrogenase with and without inhibitor reveal conformational flexibility in the inhibitor and substrate binding sites. Biochemistry. 2008;47:8929–36.
Wiese MD, Hopkins AM, King C, Wechalekar MD, Lee A, Spargo L, et al. Precision Medicine with leflunomide: consideration of DHODH haplotype and plasma teriflunomide concentration can substantially modify outcomes in patients with rheumatoid arthritis. Arthritis Care Res. 2020:acr.24236.
Klotz L, Eschborn M, Lindner M, Liebmann M, Herold M, Janoschka C, et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci Transl Med. 2019;11:490.
Liu Z, Hu Q, Wang W, Lu S, Wu D, Ze S, et al. Natural product piperine alleviates experimental allergic encephalomyelitis in mice by targeting dihydroorotate dehydrogenase. Biochem Pharmacol. 2020;177:114000.
Kinoshita F, Kondoh T, Komori K, Matsui T, Harada N, Yanai A, et al. Miller syndrome with novel dihydroorotate dehydrogenase gene mutations. Pediatr Int. 2011;53:587–91.
Lolli ML, Sainas S, Pippione AC, Giorgis M, Boschi D, Dosio F. Use of human dihydroorotate dehydrogenase (hDHODH) inhibitors in autoimmune diseases and new perspectives in cancer therapy. Recent Pat Anticancer Drug Discov. 2018;13:86–105.
Styr B, Gonen N, Zarhin D, Ruggiero A, Atsmon R, Gazit N, et al. Mitochondrial regulation of the hippocampal firing rate set point and seizure susceptibility. Neuron. 2019;102(1009–1024):e8.
Bonavia A, Franti M, Keaney EP, Kuhen K, Seepersaud M, Radetich B, et al. Identification of broad-spectrum antiviral compounds and assessment of the druggability of their target for efficacy against respiratory syncytial virus (RSV). Proc Natl Acad Sci U S A. 2011;108:6739–44.
Chen S, Ding S, Yin Y, Xu L, Li P, Peppelenbosch MP, et al. Suppression of pyrimidine biosynthesis by targeting DHODH enzyme robustly inhibits rotavirus replication. Antivir Res. 2019;167:35–44.
Mei-jiao G, Shi-fang L, Yan-yan C, Jun-jun S, Yue-feng S, Ting-ting R, et al. Antiviral effects of selected IMPDH and DHODH inhibitors against foot and mouth disease virus. Biomed Pharmacother. 2019;118:1–7.
Yang CF, Gopula B, Liang JJ, Li JK, Chen SY, Lee YL, et al. Novel AR-12 derivatives, P12-23 and P12-34, inhibit flavivirus replication by blocking host de novo pyrimidine biosynthesis. Emerg Microbes Infect. 2018;7:1–11.
Xiong R, Zhang L, Li S, Sun Y, Ding M, Wang Y, et al. Novel and potent inhibitors targeting DHODH, a rate-limiting enzyme in de novo pyrimidine biosynthesis, are broad-spectrum antiviral against RNA viruses including newly emerged coronavirus SARS-CoV-2. Protein Cell. 2020;11(10):723–39.
Garavito MF, Narvaez-Ortiz HY, Pulido DC, Löffler M, Judelson HS, Restrepo S, et al. Phytophthora infestans dihydroorotate dehydrogenase is a potential target for chemical control - a comparison with the enzyme from solanum tuberosum. Front Microbiol. 2019;10:1–14.
Nonato MC, de Pádua RAP, David JS, Reis RAG, Tomaleri GP, D’Muniz Pereira H, et al. Structural basis for the design of selective inhibitors for Schistosoma mansoni dihydroorotate dehydrogenase. Biochimie. 2019;158:180–90.
Calil FA, David JS, Chiappetta ERC, Fumagalli F, Mello RB, Leite FHA, et al. Ligand-based design, synthesis and biochemical evaluation of potent and selective inhibitors of Schistosoma mansoni dihydroorotate dehydrogenase. Eur J Med Chem. 2019;167:357–66.
du Pré S, Birch M, Law D, Beckmann N, Sibley GEM, Bromley MJ, et al. The dynamic influence of olorofim (F901318) on the cell morphology and organization of living cells of aspergillus fumigatus. J Fungi. 2020;6:1–14.
Lim W, Eadie K, Konings M, Rijnders B, Fahal AH, Oliver JD, et al. Madurella mycetomatis, the main causative agent of eumycetoma, is highly susceptible to olorofim. J Antimicrob Chemother. 2020;75(4):936–41.
Kokkonda S, Deng X, White KL, Coteron JM, Marco M, De Las HL, et al. Tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidines targeting Plasmodium falciparum dihydroorotate dehydrogenase display potent and selective antimalarial activity. J Med Chem. 2016;59:5416–31.
Mandt REK, Lafuente-Monasterio MJ, Sakata-Kato T, Luth MR, Segura D, Pablos-Tanarro A, et al. In vitro selection predicts malaria parasite resistance to dihydroorotate dehydrogenase inhibitors in a mouse infection model. Sci Transl Med. 2019;11:1–14.
Kokkonda S, Deng X, White KL, El Mazouni F, White J, Shackleford DM, et al. Lead optimization of a pyrrole-based dihydroorotate dehydrogenase inhibitor series for the treatment of malaria. J Med Chem. 2020;63:4929–56.
Ling L, Mulaka M, Munro J, Dass S, Mather MW, Riscoe MK, et al. Genetic ablation of the mitoribosome in the malaria parasite Plasmodium falciparum sensitizes it to antimalarials that target mitochondrial functions. J Biol Chem. 2020;295:7235–48 jbc.RA120.012646.
Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. 2015;7(296):296ra111.
Xiong R, Zhang L, Li S, Sun Y, Ding M, Wang Y, et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell. 2020;11:723–39.
Peters G, Sharma SL, Laurensse E, Pinedo H. Inhibition of pyrimidine de novo synthesis by DUP-785 (NSC 368390). Investig New Drugs. 1987;5:235–44.
McLean JE, Neidhardt EA, Grossman TH, Hedstrom L. Multiple inhibitor analysis of the brequinar and leflunomide binding sites on human dihydroorotate dehydrogenase. Biochemistry. 2001;40:2194–200.
Zhou J, Quah JY, Ng Y, Chooi J-Y, Toh SH-M, Lin B, et al. ASLAN003, a potent dihydroorotate dehydrogenase inhibitor for differentiation of acute myeloid leukemia. Haematologica. 2018;132:4047.
Lucas-Hourani M, Dauzonne D, Jorda P, Cousin G, Lupan A, Helynck O, et al. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog. 2013;9:e1003678.
Abdullah I, Chee CF, Lee YK, Thunuguntla SSR, Satish Reddy K, Nellore K, et al. Benzimidazole derivatives as potential dual inhibitors for PARP-1 and DHODH. Bioorg Med Chem. 2015;23:4669–80.
Abt ER, Rosser EW, Durst MA, Lok V, Poddar S, Le TM, et al. Metabolic modifier screen reveals secondary targets of protein kinase inhibitors within nucleotide metabolism. Cell Chem Biol. 2020;27:197–205.e6.
Thompson E, Uhl R. ASLAN Pharmaceuticals announces publication of preclinical data on ASLAN003 in AML in Haematologica related articles; 2021. p. 7–9.
Costeira-Paulo J, Gault J, Popova G, Ladds MJ, van Leeuwen IM, Sarr M, et al. Lipids shape the electron acceptor-binding site of the peripheral membrane protein dihydroorotate dehydrogenase. Cell Chem Biol. 2018;25:309–317.e4.
Löffler M, Carrey EA, Knecht W. The pathway to pyrimidines: the essential focus on dihydroorotate dehydrogenase, the mitochondrial enzyme coupled to the respiratory chain. Nucleosides Nucleotides Nucleic Acids. 2020;0:1–25.
Pally C, Smith D, Jaffee B, Magolda R, Zehender H, Dorobek B, et al. Side effects of brequinar and brequinar analogues, in combination with cyclosporine, in the rat. Toxicology. 1998;127:207–22.
Löffler M, Becker C, Wegerle E, Schuster G. Catalytic enzyme histochemistry and biochemical analysis of dihydroorotate dehydrogenase/oxidase and succinate dehydrogenase in mammalian tissues, cells and mitochondria. Histochem Cell Biol. 1996;105:119–28.
Cappelli A, Gl G, Gallelli A, Rizzo M, Anzini M, Vomero S, et al. Design, synthesis, structural studies, biological evaluation, and computational simulations of novel potent AT1 angiotensin II receptor antagonists based on the 4-phenylquinoline structure. J Med Chem. 2004;47:2574–86.
Urba S, Doroshow J, Cripps C, Robert F, Velez-Garcia E, Dallaire B, et al. Multicenter phase II trial of brequinar sodium in patients with advanced squamous-cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 1992;31:167–9.
Moore M, Maroun J, Robert F, Natale R, Neidhart J, Dallaire B, et al. Multicenter phase II study of brequinar sodium in patients with advanced gastrointestinal cancer. Investig New Drugs. 1993;11:61–5.
Schwartsmann G, Dodion P, Vermorken JB, ten Bokkel Huinink WW, Joggi J, Winograd B, et al. Phase I study of Brequinar sodium (NSC 368390) in patients with solid malignancies. Cancer Chemother Pharmacol. 1990;25:345–51.
Natale R, Wheeler R, Moore M, Dallaire B, Lynch W, Carlson R, et al. Short report: multicenter phase II trial of brequinar sodium in patients with advanced melanoma. Ann Oncol. 1992;3(8):659–60.
Munier-Lehmann H, Vidalain PO, Tangy F, Janin YL. On dihydroorotate dehydrogenases and their inhibitors and uses. J Med Chem. 2013;56:3148–67.
Brazelton TR, Morris RE. Mecanismos De Acción Inmunosupresores. Curr Opin Immunol. 1996;24:710–20.
Fox RI. Mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol. 1999;93:198–208.
Alldred A, Emery P. Leflunomide: a novel DMARD for the treatment of rheumatoid arthritis. Expert Opin Pharmacother. 2001;2:125–37.
Siemasko K, Chong A, Williams J, Bremer E, Finnegan A. Regulation of B cell function by the immunosuppressive agent leflunomide. Transplantation. 1996;61:635–42.
Gutteridge WE, Dave D, Richards WH. Conversion of dihydroorotate to orotate in parasitic protozoa. Biochim Biophys Acta. 1979;582:390–401.
Wostradowski T, Prajeeth CK, Gudi V, Kronenberg J, Witte S, Brieskorn M, et al. In vitro evaluation of physiologically relevant concentrations of teriflunomide on activation and proliferation of primary rodent microglia. J Neuroinflammation. 2016;13:1–12.
Hamilton LC, Vojnovic I, Warner TD. A771726, the active metabolite of leflunomide, directly inhibits the activity of cyclo-oxygenase-2 in vitro and in vivo in a substrate-sensitive manner. Br J Pharmacol. 1999;127:1589–96.
Papadopoulou A, Kappos L, Sprenger T. Teriflunomide for oral therapy in multiple sclerosis. Expert Rev Clin Pharmacol. 2012;5:617–28.
He D, Xu Z, Dong S, Zhang H, Zhou H, Wang L, et al. Teriflunomide for multiple sclerosis. Cochrane Database Syst Rev. 2016;12(12):CD009882.
Khachanova NV, Gorokhova TV. Extending the potential of the treatment of multiple sclerosis with a new agent for oral use – teriflunomide (Aubagio). Neurosci Behav Physiol. 2017;47:112–6.
Kulkarni OP, Sayyed SG, Kantner C, Ryu M, Schnurr M, Sárdy M, et al. 4SC-101, a novel small molecule dihydroorotate dehydrogenase inhibitor, suppresses systemic lupus erythematosus in MRL-(Fas)lpr mice. Am J Pathol. 2010;176:2840–7.
Fitzpatrick LR, Deml L, Hofmann C, Small JS, Groeppel M, Hamm S, et al. 4SC-101, a novel immunosuppressive drug, inhibits IL-17 and attenuates colitis in two murine models of inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1763–77.
Fitzpatrick LR, Small JS, Doblhofer R, Ammendola A. Vidofludimus inhibits colonic interleukin-17 and improves hapten-induced colitis in rats by a unique dual mode of action. J Pharmacol Exp Ther. 2012;342:850–60.
Packer RJ, Rood BR, Turner DC, Stewart CF, Fisher M, Smith C, et al. Phase I and pharmacokinetic trial of PTC299 in pediatric patients with refractory or recurrent central nervous system tumors: a PBTC study. J Neuro-Oncol. 2015;121:217–24.
Zhang X, Yang J, Chen M, Li L, Huan F, Li A, et al. Metabolomics profiles delineate uridine deficiency contributes to mitochondria-mediated apoptosis induced by celastrol in human acute promyelocytic leukemia cells. Oncotarget. 2016;7:46557–72.
Le TM, Poddar S, Capri JR, Abt ER, Kim W, Wei L, et al. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat Commun. 2017;8(1):241.
Hayek S, Pietrancosta N, Hovhannisyan AA. Alves de Sousa R, Bekaddour N, Ermellino L, et al. Cerpegin-derived furo[3,4-c]pyridine-3,4(1H,5H)-diones enhance cellular response to interferons by de novo pyrimidine biosynthesis inhibition. Eur J Med Chem. 2020;186:1–13.
Deans RM, Morgens DW, Ökesli A, Pillay S, Horlbeck MA, Kampmann M, et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol. 2016;12:361–6.
Muehler A, Kohlhof H, Groeppel M, Vitt D. Safety, tolerability and pharmacokinetics of vidofludimus calcium (IMU - 838) after single and multiple ascending oral doses in healthy male subjects. Eur J Drug Metab Pharmacokinet. 2020.
Diedrichs-Möhring M, Leban J, Strob S, Obermayr F, Wildner G. A new small molecule for treating inflammation and chorioretinal neovascularization in relapsing-remitting and chronic experimental autoimmune uveitis. Investig Ophthalmol Vis Sci. 2015;56:1147–57.
Ullrich A, Knecht W, Fries M, Löffler M. Recombinant expression of n-terminal truncated mutants of the membrane bound mouse, rat and human flavoenzyme dihydroorotate dehydrogenase: A versatile tool to rate inhibitor effects? Eur J Biochem. 2001;268:1861–8.
Hurt DE, Sutton AE, Clardy J. Brequinar derivatives and species-specific drug design for dihydroorotate dehydrogenase. Bioorg Med Chem Lett. 2006;16:1610–5.
Zuo Z, Liu X, Qian X, Zeng T, Sang N, Liu H, et al. Bifunctional Naphtho[2,3- d][1,2,3]triazole-4,9-dione compounds exhibit antitumor effects in vitro and in vivo by inhibiting dihydroorotate dehydrogenase and inducing reactive oxygen species production. J Med Chem. 2020;63:7633–52.
Echizenya S, Ishii Y, Kitazawa S, Tanaka T, Matsuda S, Watanabe E, et al. Discovery of a new pyrimidine synthesis inhibitor eradicating glioblastoma-initiating cells. Neuro-Oncology. 2020;22:229–39.
Okesli-Armlovich A, Gupta A, Jimenez M, Auld D, Liu Q, Bassik MC, et al. Discovery of small molecule inhibitors of human uridine-cytidine kinase 2 by high-throughput screening. Bioorg Med Chem Lett. 2019;29:2559–64.
Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–74.
Bianchi V, Pontis E, Reichard P. Regulation of pyrimidine deoxyribonucleotide metabolism by substrate cycles in dCMP deaminase-deficient V79 hamster cells. Mol Cell Biol. 1987;7:4218–24.
Maley F, Maley GF. Nucleotide interconversions. II. Elevation of deoxycytidylate deaminase and thymidylate synthetase in regenerating rat liver. J Biol Chem. 1960;235:2968–70.
Acknowledgements
Not applicable.
Funding
This work was supported by Project of the National S&T Major project (2018ZX09201018), National Natural Science Foundation of China (81773198).
Author information
Authors and Affiliations
Contributions
YZ wrote the manuscript and prepared the figures. YLZ supervised the design of the review and revised the manuscript. XZ, ZZZ, JG, XCL, YZ, YZ, CQL, NS, HL, JZ, KG, and XWY edited the part of their own research area. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agree.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, Y., Tao, L., Zhou, X. et al. DHODH and cancer: promising prospects to be explored. Cancer Metab 9, 22 (2021). https://doi.org/10.1186/s40170-021-00250-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40170-021-00250-z