
Abstract
In the past decade, the traditional view of cancers as a homogeneous collection of malignant cells is being replaced by a model of ever increasing complexity suggesting that cancers are complex tissues composed of multiple cell types. This complex model of tumorigenesis has been well supported by a growing body of evidence indicating that most cancers including those derived from blood and solid tissues display a hierarchical organization of tumor cells with phenotypic and functional heterogeneity and at the apex of this hierarchy are cells capable of self-renewal. These “tumor imitating cells” or “cancer stem cells” drive tumorigenesis and contribute to metastasis, treatment resistance and tumor relapse. Although tumor stem cells themselves may display both genetic and phenotypic heterogeneity, recent studies have demonstrated that cancer stem cells maintain plasticity to transition between mesenchymal-like (EMT) and epithelial-like (MET) states, which may be regulated by the tumor microenvironment. These stem cell state transitions may play a fundamental role in tumor progression and treatment resistance. In this review, we discuss the emerging knowledge regarding the plasticity of cancer stem cells with an emphasis on the signaling pathways and noncoding RNAs including microRNAs (miRNA) and long non-coding RNAs (lncRNAs) in regulation of this plasticity during tumor growth and metastasis. Lastly, we point out the importance of targeting both the EMT and MET states of CSCs in order to eliminate these lethal seeds of cancers.
Similar content being viewed by others


Introduction
Despite significant advances over the last decade in the diagnosis and treatment of cancer, the fact remains that, once metastatic, the disease remains almost universally incurable. There is now substantial evidence that many cancers, including breast cancer, are hierarchically organized and driven by a population of cells that display stem cell properties [1]-[3]. These cells have been referred to as tumor-initiating cells (TICs) or cancer stem cells (CSCs). Subsequent studies have provided further evidence that CSCs mediate tumor metastasis and are associated with therapeutic resistance [4]-[7]. On this basis, targeting CSCs holds great potential for limiting tumor growth and metastasis as well as preventing therapeutic resistance and cancer relapse.
The high degree of phenotypic and functional plasticity has been increasingly recognized as one of the significant properties of CSCs driving therapeutic resistance. We recently reported that breast CSCs display two reversible phenotypic and functional states: an EMT, epithelial-to-mesenchymal transition state and an MET, mesenchymal-to-epithelial transition state [8]. The transition between these two states in breast CSCs may facilitate breast cancer growth, dissemination and formation of distal metastasis. In this review, we will discuss emerging aspects regarding the plasticity of CSCs, the signaling pathways that regulate this plasticity during tumor growth and the role of CSC plasticity in tumor progression and metastasis. We will also discuss the clinical significance of targeting CSC plasticity.
Review
Cancer stem cells and plasticity
The cancer stem hypothesis
The cancer stem hypothesis posits that the majority of human cancers are initiated and maintained by a small population of cells that display stem cell properties including unlimited self-renewal as well as the ability to recapitulate the cellular distribution of the original tumor [9],[10]. These cells were first described in human acute myeloid leukemia [11]. The first evidence for CSCs in solid tumors was presented in 2003 by Al-Hajj et al. who prospectively enriched a population of breast cancer cells with high tumorigenic properties [12]. These breast CSCs were characterized by virtue of their expression profile of specific cell surface markers, including EpCAM+, CD24- and CD44+. As few as 100 cells bearing this phenotype were capable of forming tumors in immune deficient NOD/SCID mice, whereas 10,000 cells without this cell surface phenotype were non-tumorigenic. An in vitro cell culture assay under non-adherent conditions for quantitating the stem/progenitor cell proportion in human mammary epithelial cells has also been described [13]. In this assay, only the cells with stemness are able to proliferate and generate mammosphere structures. More recently, it has been demonstrated that cells high in aldehyde dehydrogenase (ALDH) activity are enriched in breast CSCs, as determined by using the Aldefluor assay(Stem Cell Technologies) [14]. The cancer stem hypothesis and the prospective isolation and characterization of cancer stem-like populations from leukemia, breast cancer and a wide variety of other solid malignancies including that of the brain [15], prostate [16],[17], colon [18],[19], pancreas [20], liver [21],[22], lung [23], and head and neck [24] in the past decade has been one of the major advances in current cancer research. Increasing studies have shown that CSCs display treatment resistance to chemotherapy and radiation therapy [4]-[6],[25], while clinical neoadjuvant chemotherapy expanded the proportion of CSCs [20],[26].
Epithelial-mesenchymal plasticity of CSCs
In the case of breast cancer, CSC populations identified by the markers CD24-CD44+ or ALDH+ were characterized as minimally overlapping, largely separate cell populations, each capable of initiating tumors in immune deficient mice [14]. However, whether these different phenotypic populations identify distinct or independent CSCs in the tumor remained to be resolved. To further characterize these distinct breast CSC populations, we prospectively isolated these distinct subsets of breast cancer cells from a total of 30 human breast cancer samples. These tumor samples were digested in collagenase to obtain single tumor cells. Following tumor cell disassociation, tumor cell samples were incubated with anti-CD44, anti-CD24, and anti-lineage mixture antibodies (PE-conjugated anti-CD2, CD3, CD10, CD16, CD18, CD31, and CD 140b), and then labeled by Aldefluor assay, and analyzed using MoFlo Astrios flow cytometry. Side and forward scatter were used to eliminate debris and cell doublets, and the Lin- tumor cells were further analyzed and sorted for ALDH+, ALDH-CD24-CD44+ and bulk (non-ALDH+CD24-CD44+) tumor cell populations. Using gene expression profiling of ALDH+ and CD24-CD44+ BCSCs (comparing to bulk tumor cells) isolated across different subtypes of human breast cancer tissues together with multi-marker immunofluorescence including CD24, CD44 and ALDH1, we have recently shown that the CD24-CD44+ and ALDH+ cell populations identify anatomically distinct breast CSCs with distinct EMT (epithelial-to-mesenchymal transition) and MET (mesenchymal-to-epithelial transition) gene-expression profiles respectively [8]. The EMT-like CD24-CD44+ breast CSCs are primarily quiescent and localized at the tumor invasive front, while the MET-like ALDH+ breast CSCs are proliferative cells located mainly in the central part of tumors. Importantly, the epithelial and mesenchymal states of breast CSCs are not static; instead they display a cellular plasticity allowing them to transit between EMT and MET states [8]. This reversible, metastable epithelial-mesenchymal plasticity of breast CSCs builds upon the current model of cancer metastasis postulating that EMT drives tumor cell dissemination while subsequent MET drives metastatic colonization [27].
Targeting two states of cancer stem cells
Cancer stem cell and drug resistance
A body of literature has shown the EMT type of CSC plays a critical role in drug resistance and cancer metastasis, which could partially explain why it has traditionally been difficult to cure cancer using cytotoxic chemotherapy. For instance, recent studies have shown that EMT breast CSCs were particularly resistant to treatments of either chemotherapy or radiation therapy in cell lines and patient-derived mouse xenografts [6],[25],[28],[29]. The molecular mechanisms by which the CSCs possess increased drug resistance are still not completely clear. The proteins of the ABC transporter family are believed to function as drug efflux pumps and increased levels of these proteins in CSCs could therefore be contributing to the protection of CSCs from cytotoxic chemotherapy [30]-[33]. For example, Osteosarcoma CSCs were enriched for ABCG2, a member of ABC transporter, and showed increased drug resistance and metastasis [34],[35]. Hu et al confirmed this finding by showing that high expression levels of ABCG2 in ovarian CSCs contributed significantly to Cisplatin resistance [36]. Another member of ABC transporters, MDR1/ABCCB1, has been reported to be associated with a drug-resistant profile and high clonogenic activity of the stem cell population in small-cell lung carcinoma [37]. A recent study showed that transcription factor Oct1 directly regulates ABC transporter genes Abcg2, Abcb1 and Abcb4 in CSCs [38]. In this study, the authors found an elevated protein level of Oct1, but not mRNA, highly correlated with the frequency of CD24-CD44+ EMT breast CSCs or ALDH+ MET breast CSCs.
There is mounting evidence suggesting that CSCs employ alternative DNA repair pathways to maintain genomic stability and prevent damage from radiotherapy. A previous study reported CD24-CD44+ EMT breast CSCs are a relatively radioresistant subpopulation and increase in numbers after short courses of fractionated irradiation [5]. Another group reported that CD24-CD44+ EMT breast CSCs isolated from either MCF7 or MDA-MB231 breast cancer cell lines, or primary culture of patient tumors, displayed increased capacity for sphere formation and resistant to radiation compared to the non- CD24-CD44+ subpopulation through the enhanced activation of DNA damage response [39]. CD133+ brain tumor stem cells in glioblastoma have reduced sensitivity to radiation-induced apoptosis [15],[40] and similar findings were made by Bao et al that CD133+ tumor stem cells isolated from both human glioma xenografts and primary patient glioblastoma specimens preferentially activate the DNA damage response after radiation treatment and repair radiation-induced DNA damage more effectively than CD133- bulk tumor cells [41]. These results also suggest that the DNA repair machinery may be an effective target to eliminate CSCs, leading to increased patient survival.
Aldehyde dehydrogenase1(ALDH1) is a detoxification enzyme involved in the oxidization of intracellular aldehydes to carboxylic acids [42]. Elevated expression level of ALDH1 in MET type of breast CSCs is thought to be responsible for drug resistance to cytotoxic agents, such as cyclophosphamide [43]. Studies have shown that ALDH+ MET breast CSCs exhibit similarly enhanced metastatic and drug resistant features with EMT type of breast CSCs [14]. In this study, the ALDH+ CSC population was found to be a prognostic indicator for high tumor grade, negative ER/PR status, ERBB2 overexpression, and expression of basal-like cytokeratins. Beside those findings, ALDH1 activity in breast CSCs was considered as a predictor of poor clinical outcome [14]. Consistent with those observations, a clinical study examining ALDH1 expression level in primary breast tissue treated with Paclitaxel and Epirubicin revealed that ALDH1 activity was strongly associated with a low pathologically complete response and high resistance to chemotherapy [26]. Most recently, it has been shown that ALDH enzymes play a directed role in chemotherapy resistance [44]. Therefore, ALDH1 not only serves as a functional marker of CSCs, but also plays important role in resistance to chemotherapy.
Targeting signaling pathways of cancer stem cells
The Wnt pathway
The Wnt signaling pathway is a key developmental pathway involved in a variety of biological processes including cell proliferation, survival and differentiation [45]. The well characterized canonical Wnt/β-catenin signaling pathway is initiated with ligand-receptor binding resulting in transcriptional activation of a subset of response genes, which have been suggested to play a critical role in tumor initiation in many tissues. The Wnt/β-catenin signaling pathway is often aberrantly activated in CSCs, which are responsible for generation of metastasis and decreased survival of patients [46]. Therefore, targeting the Wnt/β-catenin signaling pathway may potentially reduce the number of, or even eradicate, CSCs. To this end, a number of small-molecule inhibitors of Wnt signaling are being studied including existing drugs such as nonsteroidal anti-inflammatory drugs (NSAID), new molecular-targeted agents, including many that are currently in the discovery, preclinical, or clinical testing stages [47]. Among them, CBP/β-catenin antagonist ICG-001ICG-001, was reported to be capable of eliminating imatinib-resistant leukemic stem cells both in vitro and mouse xenografts. In Wilm’s tumor, the stem cell properties of sphere formation and clonogenicity can be largely abrogated after the application of anti-FZD7, an antibody for Wnt receptor Frizzled 7 [48]. Moreover, application of Dkk1 has been shown to decrease mammosphere formation in primary breast cancer cells and MCF-7 cells and drive CD24-CD44+ EMT breast CSCs into differentiation at a high concentration by preventing the formation of FZD-Wnt-LRP complex [49]. Recently, it has been demonstrated that salinomycin is able to dramatically reduce the number of CSCs in triple negative breast cancer [50] by promoting the degradation of Wnt co-receptor LRP6 [51]. Thus, targeting the components in Wnt signaling pathway is a promising approach to eliminate CSCs, especially the CSCs with EMT characteristics.
The Hedgehog pathway
Recent studies have suggested that the Hedgehog (Hh) pathway is involved in the maintenance of CSCs in a number of tumors, including pancreatic cancer [52], gastric cancer [53], colorectal cancer [54], and glioma [55]. An Hh pathway inhibitor GDC-0449(Vismodegib) has been developed to inhibit the signaling component Smoothened (SMO) and has shown promise in clinical trials of advanced basal cell carcinoma and advanced metastatic medulloblastoma [56],[57]. However, mutations in SMO in patients conferred a drug resistance to this inhibitor [58]. An active compound in green tea, (-)-epigallocatechin-3-gallate (EGCG), has been proposed to inhibit self-renewal capacity of pancreatic CSCs through blocking Hh pathway [52]. In this study, the expression levels of sonic Hh signaling components Patched (Ptch), SMO, Gli1 and Gli2 were downregulated, and the transcriptional activities of Gli1 and Gli2 were also inhibited after application of EGCG. However, the direct target(s) of EGCG in CSC inhibition are still unclear. A small-molecule Hh pathway inhibitor, IPI-269609, has been tested both in vitro and in vivo model systems of pancreatic cancer and the results showed that this SMO inhibitor is effective in reducing ALDH+ MET CSCs and preventing metastasis [59]. Moreover, treatment with the Hh pathway inhibitor cyclopamine combined with gemcitabine and rapamycin showed significant inhibitory effects on CD133+ CSCs of human pancreatic cancer [60], suggesting that combined targeted treatment may have therapeutic efficacy. Bmi-1 is an oncogene and may cooperate with Twist1 to regulate EMT state of CSCs and metastasis. Bmi-1 has been shown to play an important role in the regulation of stem cell self-renewal in breast cancer, where the activity of Bmi-1 is increased by Hh pathway activation [61]. A recent study demonstrated that a small molecular inhibitor, PTC-209, is capable of inhibiting Bmi-1, further blocking self-renewal of colon cancer initiating cells (CICs) [62],[63], which provides a therapeutic rationale for exploring the efficacy of PTC-209 in targeting CSCs.
The Notch pathway
The role for Notch signaling in cancer was demonstrated by the identification of activating mutations as well as amplification of Notch pathway components in a number of tumors [64],[65]. Among them, activating mutations of Notch-1 have been identified in more than 50% of human T-cell acute lymphoblastic leukemia patients (T-ALL) [66]. Overexpression of receptor Notch-1, or its ligand Jagged-1, has been shown to predict poor survival of breast cancer patients [67],[68], suggesting a significant role of the Notch pathway in this malignant disease. Moreover, Notch activated HER2 overexpression is essential for self-renewal of breast CSCs in patients and is associated with poor prognosis [69]. Blocking the activity of Notch intracellular domain (NICD) is currently one of the practical approaches to inhibiting Notch signaling to eliminate CSCs, and this therapeutic inhibition of Notch signaling can be achieved using γ-secretase inhibitors (GSIs) to block the release of NICD. GSIs were shown to reduce glioblastoma CSCs [70] and breast CSCs [71] through inhibition of Notch pathway. In another study, GSI MRK-003 has been shown to inhibit CSCs in a mouse model of breast cancer [72]. A blocking antibody or siRNA knockdown of Notch-4 has been shown to reduce the stem cell population, inhibiting tumor formation from EpCAM+CD24-CD44+ CSCs and that this effect was greater than Notch-1 inhibition [71],[73], this suggests that Notch4-targeted therapies may be more effective than Notch1-targeted therapies in targeting breast CSCs.
PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway has been shown to play an important role in the regulation of CSCs. PTEN acts as a negative regulator of PI3K/Akt/mTOR signaling through dephosphorylating PIP3, a product of PI3K, resulting in pathway inhibition [74]-[77]. Loss of PTEN activity leads to constitutive PI3K/Akt pathway activation, which results in increased stem cells in breast cancer. Treatment with Akt or mTOR inhibitors has been shown to increase sensitivity of CSCs to irradiation and chemotherapy, respectively [78],[79]. Accordingly, inhibition of AKT with Perifosine reduced CSC activity as accessed by mammosphere formation. These results suggest the PTEN/PI3K/AKT/mTOR pathway could be an effectively therapeutic target in sensitizing and eliminating CSCs.
Targeting cytokines through STAT3 and NF-κB pathways
The STAT3 and NF-κB pathways play a pivotal role in the induction and maintenance of inflammatory microenvironment of malignant diseases and tumors [80]-[82], and these pathways are activated by numerous cytokines including IL-6 and IL-8 secreted by a variety of inflammatory cells [83],[84]. In cancer patients, high levels of IL-6 and IL-8 are associated with poor outcome. IL-6 activated JAK2/STAT3 pathway was proposed to support the maintenance of CD24-CD44+ breast CSCs [85]. STAT3 and AKT activation causes the activation of NF-κB pathways in inflammatory cells leading to increased levels of Lin28 and decreased levels of miRNA let7. This in turn leads to increased IL-6 and IL-8 production in the tumor microenvironment [86],[87]. A recent study revealed that IL-6 activated Jagged1/Notch1 signaling contributes to bone metastasis of breast cancer [88], indicating that multiple pathways may be involved in the IL-6-mediated CSC regulation. Moreover, we previously reported that the IL-8 receptor CXCR1 is selectively expressed in breast CSCs, and the addition of IL-8 promotes proliferation and self-renewal of breast CSCs [89]. These results suggest blocking IL-6, IL-8 and their receptors IL-6R and CXCR1 may be an attractive therapeutic strategy.
Approximately 20-25% of human breast cancers are HER2-positive and almost 50% of patients with HER2-amplified cancer will develop trastuzumab resistance after 1-2 years of treatment [90]. Notably, PTEN deletion was identified in over 40% of those HER2-positive trastuzumab resistant breast cancer patients [91]. In a recent study, we found that PTEN inactivation in HER2-positive breast cancer cells activates an IL-6, STAT3, AKT and NF-κB involved inflammatory feedback loop, which expands EMT type of breast CSCs [87]. Moreover, it has been shown that activation of the IL6 inflammatory loop in p53−PTEN− cancer cells is associated with reduced SOCS3 expression which serves as a negative regulator of IL6-mediated signaling, and forced expression of SOCS3 or using IL6-R blockade inhibited tumor proliferation and metastasis in mouse xenografts [92]. Through the IL6 feedback loop, trastuzumab resistance is acquired and EMT-CSCs are significantly enriched. Based on this observation, we treated HER2-positive, PTEN deleted, tumor xenografts with a combined treatment of trastuzumab and tocilizumab, a monoclonal antibody directed against IL-6R. This treatment completely overcame de novo and acquired trastuzumab resistance of HER2-positive PTEN-deleted breast cancer. Furthermore, reparaxin, an inhibitor of the IL-8 receptor CXCR1, is currently in a phase I clinical trial, to evaluate the strategy of adding repertaxin to paclitaxel to block the chemotherapy-induced increase in CSCs.
Non-coding RNAs in CSC plasticity
Non-coding RNAs (ncRNAs) are RNA molecules which are transcribed from the genome, but do not encode proteins. One class of ncRNAs are microRNAs which are highly-conserved small non-coding RNAs with a length of ~22 nucleotides that serve as regulatory inhibitors for protein expression [93]. They modulate gene function through their binding to complementary regions within the 3’-UTRs of target mRNAs, leading to mRNA degradation or repression of translation. MicroRNAs have been extensively studied and regulate expression of a variety of target genes involved in different developmental processes and diseases including cancer [94]. MicroRNAs can function as oncogenes [95],[96] or tumor suppressors [97],[98]. There is mounting evidence that microRNAs are potential targets for cancer diagnosis and therapy [99]-[102]. Long non-coding RNAs(lncRNAs) are a newly identified class of non-coding RNAs longer than 200 nuceotides [93]. Although relatively few lncRNAs are well characterized compared to microRNAs, lncRNAs function in physiological and pathological processes through a variety of mechanisms such as interacting with microRNAs, mRNAs, proteins and genomic DNA [103]. LncRNAs have been implicated in many aspects of epigenetic, transcriptional and translational regulation [104]. Emerging evidence suggests that non-coding RNAs might serve as potential targets for anti-CSC therapies.
MicroRNA regulation of cancer stem cell plasticity
The most established regulatory network of microRNAs in CSCs is the miR-200 family, which includes miR-200a, b, c, miR-141 and miR-429. Two prominent targets of the miR-200 family are ZEB1 and ZEB2 which are involved in the regulation of epithelial/mesenchymal transitions. E-cadherin is one of the key epithelial genes, and downregulation of E-cadherin is generally associated with EMT. miR200-family members act to maintain E-cadherin expression through directly suppressing the negative regulators ZEB1 and ZEB2 in EMT CSCs. Forced expression of miR-200 family members was shown to prevent a TGF-β induced mesenchymal phenotype [105], and block tumorigenicity of CD24-CD44+ CSCs [106]. Consistent with this result, miR-200 family members were found to be strongly suppressed in CD24-CD44+ breast CSCs which is associated with an EMT state [106],[107]. In addition, inhibition of miR-200 family members was also shown to promote CSC formation and maintenance [108]. miR-200c inhibited the polycomb gene Bmi-1 in breast CSCs, leading to reduced self-renewal of CSCs [109]. In contrast, loss of miR-200b promotes the expression of the E-cadherin suppressor Suz12, which resulted in increased numbers of EMT CSCs [108]. In breast cancer patients, the expression of miR-200 family members inversely correlated with the proportion of CD24-CD44+ EMT breast CSCs. Furthermore, forced expression of miR-200 genes in those EMT CSCs resulted in a conversion of these CSCs to an epithelial MET state [110].
The microRNA let-7 family regulates stem cell self-renewal and differentiation and acts as a tumor suppressor through targeting oncogenic RAS and HMGA2, which are involved in the EMT state of CSCs. Overexpression of let-7 leads to suppression of mammosphere formation, proliferation, and reduced proportion of breast CSCs, suggesting let-7 inhibits cancer growth through regulation of the EMT state of CSCs [111]. Similar to the let-7 family, overexpression of miR-30 in breast CSCs resulted in reduced self-renewal and anoikis resistance and increased apoptosis via decreased Ubc9 levels and silencing of ITGB3. When both let-7 and miR-30 were expressed in breast cancer, a much more significant inhibition of self-renewal of breast CSCs was observed compared to expression of either miRNA alone [112], indicating multiple miRNAs could be used to eliminate CSCs in anti-cancer therapy. miR-34c has been identified as a tumor suppressor because it has inhibitory activity in regulating self-renewal and EMT of breast CSCs through targeting Notch4. The miR-181 family which interacts with the TGF-β pathway functions in the regulation of EMT breast CSCs [113]. Finally, miR-21 functions as an oncogenic miRNA and promotes the EMT-like breast CSCs through AKT/ERK1/2 inactivation by targeting PTEN [114].
Recently miR-93 [115], miR-100 [116], and miR-221 [114] were shown to be important regulators of the transition between EMT and MET CSC states. Low mir-93 expression is associated with increased tumor-initiating capacity, while overexpression diminishes the presence of CSCs. Forced expression of miR-93 in claudinlow SUM159 and basal HCC1954 cell lines, as well as NOD/SCID mouse xenografts, dramatically reduced the proportion of ALDH+ MET-like breast CSCs and overexpression of miR-100 in breast cancer cell lines and tumor xenografts also modulates the MET and EMT breast CSC states [115]-[116]. A number of targets of miR-93, including JAK1, SOX4, STAT3, AKT, EZH1, and HMGA2 are known regulators of stem cell-renewal. Expression of miR-93 was also found to suppress the TGF-β signaling pathway through targeting TGF-βR2 and SMAD5 to promote the conversion of EMT to MET, resulting in an increased proportion of ALDH+ MET breast CSCs. In contrast, forced expression of miR-100 and miR-221in MCF10A, a non-tumorigenic breast cell line, as well as a variety of tumorigenic breast cancer cell lines resulted in the induction of CD24-CD44+ EMT stem cells and decreased proportion of ALDH+ MET stem cells [115].
LncRNA regulation of cancer stem cell plasticity
Hotair (Hox transcript antisense intergenic RNA) is one of the first lncRNAs whose function has been elucidated. It plays a pivotal role in the polycomb repressive complex 2 (PRC2), which directs epigenetic regulation of target genes through histone H3 lysine 27 trimethylation (H3K27me3) [117],[118]. High expression level of Hotair contributes to metastasis and poor survival in breast cancer patients [117]. Hotair is also highly expressed in CD133+CD44+ colon cancer cells which have been characterized as EMT CSCs [120]. Overexpression of Hotair resulted in increased expression of EMT inducing genes such as ZEB1, Snai1, Twist, β-catenin, vimentin and fibronectin in breast CSCs [118],[120]. Those results suggest Hotair regulates EMT CSCs and promotes cancer metastasis through global reprogramming of chromatin states.
Xist, one of the lncRNAs involved in X chromosome inactivation(XCI), was demonstrated to be a tumor suppressor and a stem cell regulator [121]. Using mathematical modeling, SUZ12 and EZH2, regulators of CSCs, were predicted to show high binding affinity to Xist RNA [122], which is further evidence that Xist may function in regulation of CSCs [123]. Salvado et al. validated that low Xist expression in patient derived xenografts is associated with drug response and a significant decrease of the ALDH+ breast CSC population after treatment with a histone deacetylase inhibitor (HDACi) abexinostat. In contrast, high expression of Xist in patient derived xenografts increased the proportion of breast CSCs. This result indicates that Xist’s regulation of the proportion of CSCs may be dependent on CSC states. Our laboratory identified BRCA1 as a regulator of ALDH+ CSCs [124], while a previous study showed that Xist RNA concentration in XCI was increased by BRCA1 in breast cancer cells [125], suggesting Xist regulation of breast CSCs is in a BRCA1 dependent manner.
Translational regulatory RNA (treRNA) was found to function in the nucleus as a cis element to upregulate the expression of Snail while suppress cytoplasmic E-cadherin expression promoting EMT in cancer cells [126],[127]. Overexpression of lncRNA treRNA in MCF7 breast cancer cells suppressed E-cadherin and other epithelial proteins and increased the protein levels of mesenchymal proteins, resulting in increased cell migration and invasion [127]. These results suggest treRNA may play a role in the regulation of EMT CSCs and may thus be an appropriate therapeutic target. The metastasis–associated lung adenocarcinoma transcript 1(MALAT 1) was found to be associated with highly metastatic tumors and correlated with poor patient outcome [128]. In bladder cancer, downregulation of MALAT-1 led to the inhibition of the EMT associated genes ZEB1, ZEB2, and Slug, and the activation of E-cadherin. In addition, increased expression of MALAT-1 was also found to activate the Wnt pathway to promote EMT and human bladder cancer cell metastasis [129].
LncRNA H19, together with its partner miR-675, a miRNA embedded in its first exon, were proposed to regulate EMT through multiple signaling pathways, one of them being the PI3K–AKT pathway [130]. Over-expression of H19 in lung cancer lines abolished the expression of E-cadherin by activation of Slug in a miR-675 dependent manner, suggesting a biological function of H19 in the regulation of EMT. A recent study showed that H19 can serve as a molecular sponge for the let-7 microRNA family in muscle tissue, suggesting an alternative function of lncRNAs in the transition between EMT to MET of CSCs through blocking microRNAs [131]. Linc-ROR was first characterized in induced pluripotent stem cells (iPSCs) [132] and recently it was proposed to act as a sponge for mir-145 serving to de-repress a number of mir-145 targets, including OCT4, SOX2, and Nanog [133]. More recently, linc-ROR has been demonstrated to play an important role in the regulation of breast cancer metastasis and EMT CSCs. Overexpression of linc-ROR in mammary epithelial cells resulted in an increase in the CD24-CD44+ stem cell population with strong upregulation of the mesenchymal markers such as Vimentin, α-SMA, N-cadherin and Fibronetin, while the epithelial markers E-cadherin and Occludin were dramatically suppressed [134]. In this study, linc-ROR was suggested to have a role as a sponge for mir-205 thereby preventing the degradation of its targets such as ZEB1 and ZEB2 in breast cancer to regulate CD24-CD44+ EMT stem cells. Recently, lncRNA-activated by TGF-β (lncRNA-ATB) was demonstrated to competitively bind the miR-200 family and sequester them from their targets, ZEB1 and ZEB2, thereby inducing EMT and invasion in hepatocellular carcinoma. On the other hand, lncRNA-ATB also interacts with, and increases the stability of IL-11 mRNA, which results in the activation of the IL-11-STAT3 signaling pathway and enhanced colonization in hepatocellular carcinoma [135]. These findings suggest that lncRNA-ATB might act as a key regulator of TGF-β signaling by targeting both EMT and MET states of CSCs. Future studies will further elucidate the role of lncRNAs in CSC signaling and plasticity.
Conclusions
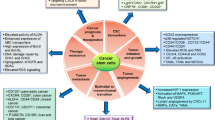
The current model of CSC plasticity between EMT and MET states suggests that CSCs may not constitute fixed populations but rather a dynamic, plastic and phenotypic state that can be acquired as a function of dynamic tumor microenvironment changes such as growth factor and inflammatory signaling, stromal cell interactions, and tumor hypoxia/metabolic reprograming. This epithelial-mesenchymal plasticity of CSCs adds another layer of complexity in terms of therapeutic strategies to target these lethal seeds of tumors. However, no matter how plastic and dynamic, CSCs are still regulated by and are depended on specific stem cell signaling pathways, and thus may be amenable to CSC specific therapies. The plasticity of CSCs is regulated by a plethora of factors, such as many signaling pathways, transcription factors, miRNAs and lncRNAs. Thus, targeting both EMT and MET states of CSCs may prove to be the most effective therapeutic strategy for anti-cancer treatment (Figure 1). Emerging evidence has shown that lncRNAs may play roles in the regulation of the EMT and MET states of CSCs. However, lncRNAs have been characterized to be involved in diverse physiological and pathological processes through multiple targets and distinct molecular mechanisms. Therefore, the therapeutic potential of lncRNAs in control of EMT and MET states of CSCs needs to be validated in future studies.

Potential therapeutic targets of CSC plasticity. MET and EMT CSCs can readily transition back and forth between the two cell states. Potential targets for eliminating these populations include targeting each specific population with particular drugs such as Trastuzumab(MET CSCs) or Tocilizumab (EMT CSCs) or developing new drugs to target the pathways and ncRNAs involved in the transition between the populations.
Author’s contributions
YZ and ML wrote the manuscript. MB created Figure 1. All the authors read, approved and edited the final manuscript.
Abbreviations
- TIC:
-
Tumor-initiating cells
- CSC:
-
Cancer stem cell
- EMT:
-
Epithelial-to-mesenchymal transition
- MET:
-
Mesenchymal-to-epithelial transition
- ALDH:
-
Aldehyde dehydrogenase
- ABC:
-
ATP-binding cassette
- MDR:
-
Multi-drug resistance
- ncRNA:
-
Non-coding RNA
- lncRNA:
-
Long non-coding RNA
References
Wicha MS, Liu S, Dontu G: Cancer stem cells: an old idea–a paradigm shift. Cancer Res 2006, 66(4):1883–1890. discussion 1895–1886 discussion 1895-1886
Liu S, Wicha MS: Targeting breast cancer stem cells. J Clin Oncol 2010, 28(25):4006–4012.
Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS: Cancer stem cells in breast: current opinion and future challenges. Pathobiology 2008, 75(2):75–84.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, et al.: Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Nat Cancer Inst 2008, 100(9):672–679.
Phillips TM, McBride WH, Pajonk F: The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Nat Cancer Inst 2006, 98(24):1777–1785.
Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY: Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res 2008, 68(9):3243–3250.
Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H: CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 2006, 8(5):R59.
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al.: Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep 2014, 2(1):78–91.
Beck B, Blanpain C: Unravelling cancer stem cell potential. Nat Rev Cancer 2013, 13(10):727–738.
Enderling H, Hlatky L, Hahnfeldt P: Cancer stem cells: a minor cancer subpopulation that redefines global cancer features. Front Oncol 2013, 3. Enderling H, Hlatky L, Hahnfeldt P: Cancer stem cells: a minor cancer subpopulation that redefines global cancer features.Front Oncol 2013, 3.
Bonnet D, Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997, 3(7):730–737.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proceed Nat Acad Sci 2003, 100(7):3983–3988.
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS: In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003, 17(10):1253–1270.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al.: ALDH1 Is a Marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1(5):555–567.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB: Identification of human brain tumour initiating cells. Nature 2004, 432(7015):396–401.
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ: Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005, 65(23):10946–10951.
Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, et al.: Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25(12):1696–1708.
O/'Brien CA, Pollett A, Gallinger S, Dick JE: A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445(7123):106–110.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R: Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445(7123):111–115.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM: Identification of pancreatic cancer stem cells. Cancer Res 2007, 67(3):1030–1037.
Ma S, Chan KW, Hu L, Lee TKW, Wo JYH, Ng IOL, Zheng BJ, Guan XY: Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2004, 132(7):2542–2556.
Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, Ng I, Man K, Wong N, To K-F, et al.: miR-130b Promotes CD133+ liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 2010, 7(6):694–707.
Kim CFB, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T: Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121(6):823–835.
Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE: Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proceed Nat Acad Sci 2007, 104(3):973–978.
Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM: WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A 2007, 104(2):618–623.
Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S: Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res 2009, 15(12):4234–4241.
Brabletz T: To differentiate or not — routes towards metastasis. Nat Rev Cancer 2012, 12(6):425–436.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al.: Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458(7239):780–783.
Zielske SP, Spalding AC, Wicha MS, Lawrence TS: Ablation of breast cancer stem cells with radiation. Translat Oncol 2011, 4(4):227–233.
Dean M, Fojo T, Bates S: Tumour stem cells and drug resistance. Nat Rev Cancer 2005, 5(4):275–284.
Styczynski J, Drewa T: Leukemic stem cells: from metabolic pathways and signaling to a new concept of drug resistance targeting. Acta Biochim Pol 2007, 54: 717–726.
Zhou S, Schuetz JD, Bunting KD, Colapietro A-M, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, et al.: The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 2001, 7(9):1028–1034.
Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG: Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res 2005, 65(14):6207–6219.
Adhikari AS, Agarwal N, Wood BM, Porretta C, Ruiz B, Pochampally RR, Iwakuma T: CD117 and Stro-1 Identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res 2010, 70(11):4602–4612.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C: Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1(3):313–323.
Hu L, McArthur C, Jaffe RB: Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br J Cancer 2010, 102(8):1276–1283.
Gutova M, Najbauer J, Gevorgyan A, Metz MZ, Weng Y, Shih C-C, Aboody KS: Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLoS One 2007, 2: e243.
Maddox J, Shakya A, South S, Shelton D, Andersen JN, Chidester S, Kang J, Gligorich KM, Jones DA, Spangrude GJ, et al.: Transcription factor Oct1 Is a somatic and cancer stem cell determinant. PLoS Genet 2012, 8(11):e1003048.
Zhang M, Atkinson RL, Rosen JM: Selective targeting of radiation-resistant tumor-initiating cells. Proc Natl Acad Sci 2010, 107(8):3522–3527.
Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI: Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci 2003, 100(25):15178–15183.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN: Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444(7120):756–760.
Lendahl U, Zimmerman LB, McKay RDG: CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60(4):585–595.
Sládek N, Kollander R, Sreerama L, Kiang D: Cellular levels of aldehyde dehydrogenases (ALDH1A1 and ALDH3A1) as predictors of therapeutic responses to cyclophosphamide-based chemotherapy of breast cancer: a retrospective study. Cancer Chemother Pharmacol 2002, 49(4):309–321.
Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, Wilson C, Merchant M, Settleman J: The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res 2014, 74(13):3579–3590.
Klaus A, Birchmeier W: Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008, 8(5):387–398.
Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, Vernay B, Jacques TS, Taketo MM, Le Tissier P, et al.: Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A 2011, 108(28):11482–11487.
Takahashi-Yanaga F, Kahn M: Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res 2010, 16(12):3153–3162.
Pode-Shakked N, Harari-Steinberg O, Haberman-Ziv Y, Rom-Gross E, Bahar S, Omer D, Metsuyanim S, Buzhor E, Jacob-Hirsch J, Goldstein RS, et al.: Resistance or sensitivity of Wilms/' tumor to anti-FZD7 antibody highlights the Wnt pathway as a possible therapeutic target. Oncogene 2011, 30(14):1664–1680.
Agur Z, Kirnasovsky OU, Vasserman G, Tencer-Hershkowicz L, Kogan Y, Harrison H, Lamb R, Clarke RB: Dickkopf1 regulates fate decision and drives breast cancer stem cells to differentiation: an experimentally supported mathematical model. PLoS One 2011, 6(9):e24225.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES: Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138(4):645–659.
Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA: Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci 2011, 108(32):13253–13257.
Tang S-N, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK: Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer 2012, 131(1):30–40.
Pham P, Phan N, Nguyen N, Truong N, Duong T, Le D, Truong K, Phan N: Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med 2011, 9(1):209.
Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A: Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med 2009, 1(6–7):338–351.
Takezaki T, Hide T, Takanaga H, Nakamura H, Kuratsu J-I, Kondo T: Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci 2011, 102(7):1306–1312.
Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al.: Inhibition of the Hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009, 361(12):1164–1172.
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al.: Treatment of medulloblastoma with Hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009, 361(12):1173–1178.
Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, et al.: Smoothened mutation confers resistance to a hedgehog pathway inhibitor in Medulloblastoma. Science 2009, 326(5952):572–574.
Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, Koorstra J-BM, Habbe N, Karikari C, Mullendore M, et al.: An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther 2008, 7(9):2725–2735.
Mueller MT, Hermann PC, Witthauer J, Rubio–Viqueira B, Leicht SF, Huber S, Ellwart JW, Mustafa M, Bartenstein P, D'Haese JG, et al.: Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137(3):1102–1113.
Liu S, Dontu G, Mantle ID, Patel S, Ahn N-S, Jackson KW, Suri P, Wicha MS: Hedgehog signaling and bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006, 66(12):6063–6071.
Wicha MS: Targeting self-renewal, an Achilles' heel of cancer stem cells. Nat Med 2014, 20(1):14–15.
Kreso A, Van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W, Sydorenko N, et al.: Self-renewal as a therapeutic target in human colorectal cancer. Nat Med 2014, 20(1):29–36.
Wang Z, Li Y, Banerjee S, Sarkar FH: Emerging role of Notch in stem cells and cancer. Cancer Lett 2009, 279(1):8–12.
Bolós V, Grego-Bessa J, Pompa JL: Notch signaling in development and cancer. Endocr Rev 2007, 28(3):339–363.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306(5694):269–271.
Dickson BC, Mulligan AM, Zhang H, Lockwood G, O'Malley FP, Egan SE, Reedijk M: High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol 2007, 20(6):685–693.
Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE: High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 2005, 65(18):8530–8537.
Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, Sozzi G, Fontanella E, Menard S, Tagliabue E: Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin Cancer Res 2009, 15(6):2010–2021.
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li Y-M, Maciaczyk J, et al.: NOTCH Pathway blockade depletes CD133-Positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28(1):5–16.
Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS: Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res 2004, 6(6):R605–615.
Kondratyev M, Kreso A, Hallett RM, Girgis-Gabardo A, Barcelon ME, Ilieva D, Ware C, Majumder PK, Hassell JA: Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene 2012, 31(1):93–103.
Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB: Regulation of breast cancer stem cell activity by signaling through the notch4 receptor. Cancer Res 2010, 70(2):709–718.
Hill R, Wu H: PTEN, Stem cells, and cancer stem cells. J Biol Chem 2009, 284(18):11755–11759.
Li H, Gao Q, Guo L, Lu SH: The PTEN/PI3K/Akt pathway regulates stem-like cells in primary esophageal carcinoma cells. Cancer Biol Ther 2011, 11(11):950–958.
Moon SH, Kim DK, Cha Y, Jeon I, Song J, Park KS: PI3K/Akt and Stat3 signaling regulated by PTEN control of the cancer stem cell population, proliferation and senescence in a glioblastoma cell line. Int J Oncol 2013, 42(3):921–928.
Porta C, Paglino C, Mosca A: Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 2014, 4: 64.
Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC: PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008, 22(4):436–448.
Marinov M, Ziogas A, Pardo OE, Tan LT, Dhillon T, Mauri FA, Lane HA, Lemoine NR, Zangemeister-Wittke U, Seckl MJ, et al.: AKT/mTOR Pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res 2009, 15(4):1277–1287.
Pardoll D: Does the immune system see tumors as foreign or self? Annu Rev Immunol 2003, 21: 807–839.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD: Cancer immunoediting: from immunosurveillance to tumor escape. Nature Immunol 2002, 3(11):991–998.
Zou W: Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005, 5(4):263–274.
Scheller J, Ohnesorge N, Rose-John S: Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand J Immunol 2006, 63(5):321–329.
Bromberg J, Wang TC: Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009, 15(2):79–80.
Marotta LLC, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al.: The JAK2/STAT3 signaling pathway is required for growth of CD44 + CD24- stem cell-like breast cancer cells in human tumors. J Clin Invest 2011, 121(7):2723–2735.
Korkaya H, Liu S, Wicha MS: Regulation of cancer stem cells by cytokine networks: attacking cancer's inflammatory roots. Clin Cancer Res 2011, 17(19):6125–6129.
Korkaya H, Kim G-i, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi Ahmed A, Tawakkol N, D'Angelo R, Paulson AK: Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012, 47(4):570–584.
Sethi N, Dai X, Winter CG, Kang Y: Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 2011, 19(2):192–205.
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, et al.: CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest 2010, 120(2):485–497.
Lan K-H, Lu C-H, Yu DH: Mechanisms of trastuzumab resistance and their clinical implications. Ann N Y Acad Sci 2005, 1059(1):70–75.
Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al.: PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6(2):117–127.
Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, Malik F, Paulson AK, D'Angelo RC, Korkaya S, et al: SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene 2014,. Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, Malik F, Paulson AK, D'Angelo RC, Korkaya S, et al: SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model.Oncogene 2014,.
Brosnan CA, Voinnet O: The long and the short of noncoding RNAs. Curr Opin Cell Biol 2009, 21(3):416–425.
Calin GA, Croce CM: MicroRNA signatures in human cancers. Nat Rev Cancer 2006, 6(11):857–866.
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE: Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 2005, 102(10):3627–3632.
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al.: A microRNA polycistron as a potential human oncogene. Nature 2005, 435(7043):828–833.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al.: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci 2002, 99(24):15524–15529.
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, et al.: Reduced expression of the let-7 MicroRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004, 64(11):3753–3756.
Pichler M, Winter E, Stotz M, Eberhard K, Samonigg H, Lax S, Hoefler G: Down-regulation of KRAS-interacting miRNA-143 predicts poor prognosis but not response to EGFR-targeted agents in colorectal cancer. Br J Cancer 2012, 106(11):1826–1832.
Bach D, Fuereder J, Karbiener M, Scheideler M, Ress AL, Neureiter D, Kemmerling R, Dietze O, Wiederstein M, Berr F, et al.: Comprehensive analysis of alterations in the miRNome in response to photodynamic treatment. J Photochem Photobiol B 2013, 120: 74–81.
Van Roosbroeck K, Pollet J, Calin GA: miRNAs and long noncoding RNAs as biomarkers in human diseases. Expert Rev Mol Diagn 2013, 13(2):183–204.
Calin GA, Konopleva M: Small gene, big number, many effects. Blood 2012, 120(2):240–241.
Rinn JL, Chang HY: Genome regulation by long noncoding RNAs. Annu Rev Biochem 2012, 81: 145–166.
Wilusz JE, Sunwoo H, Spector DL: Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 2009, 23(13):1494–1504.
Korpal M, Lee ES, Hu G, Kang Y: The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of e-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008, 283(22):14910–14914.
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al.: Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138(3):592–603.
Chen J, Wang L, Matyunina LV, Hill CG, McDonald JF: Overexpression of miR-429 induces mesenchymal-to-epithelial transition (MET) in metastatic ovarian cancer cells. Gynecol Oncol 2011, 121(1):200–205.
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K: Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell 2010, 39(5):761–772.
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al.: Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138(3):592–603.
Lim Y, Wright JA, Attema JL, Gregory PA, Bert AG, Smith E, Thomas D, Drew PA, Khew-Goodall Y, Goodall GJ: Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem cell-like state. J Cell Sci 2013,. Lim Y, Wright JA, Attema JL, Gregory PA, Bert AG, Smith E, Thomas D, Drew PA, Khew-Goodall Y, Goodall GJ: Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem cell-like state.J Cell Sci 2013,.
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al.: let-7 Regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131(6):1109–1123.
Yu F, Deng H, Yao H, Liu Q, Su F, Song E: Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 2010, 29(29):4194–4204.
Wang Y, Yu Y, Tsuyada A, Ren X, Wu X, Stubblefield K, Rankin-Gee EK, Wang SE: Transforming growth factor-[beta] regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene 2011, 30(12):1470–1480.
Han M, Liu M, Wang Y, Chen X, Xu J, Sun Y, Zhao L, Qu H, Fan Y, Wu C: Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS One 2012, 7(6):e39520.
Liu S, Clouthier S, Wicha M: Role of microRNAs in the regulation of breast cancer stem cells. J Mammary Gland Biol Neoplasia 2012, 17(1):15–21.
Deng L, Shang L, Bai S, Chen J, He X, Trevino RM, Chen S, Li X, Meng X, Yu B, Wang X, Liu Y, McDermott SP, Ariazi AE, Ginestier C, Ibarra I, Ke J, Luther TK, Clouthier SG, Xu L, Shan G, Song E, Yao H, Hannon GJ, Weiss SJ, Wicha MS, Liu S: microRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res 2014,. Deng L, Shang L, Bai S, Chen J, He X, Trevino RM, Chen S, Li X, Meng X, Yu B, Wang X, Liu Y, McDermott SP, Ariazi AE, Ginestier C, Ibarra I, Ke J, Luther TK, Clouthier SG, Xu L, Shan G, Song E, Yao H, Hannon GJ, Weiss SJ, Wicha MS, Liu S: microRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development.Cancer Res 2014,.
Liu S, Patel SH, Ginestier C, Ibarra I, Martin-Trevino R, Bai S, McDermott SP, Shang L, Ke J, Ou SJ, et al.: MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet 2012, 8(6):e1002751.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai M-C, Hung T, Argani P, Rinn JL, et al.: Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464(7291):1071–1076.
Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, et al.: Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res 2011, 71(20):6320–6326.
Pádua Alves C, Fonseca AS, Muys BR, De Barros E Lima Bueno R, Bürger MC, De Souza JES, Valente V, Zago MA, Silva WA: Brief report: the lincrna hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells 2013, 31(12):2827–2832.
Yildirim E, Kirby James E, Brown Diane E, Mercier Francois E, Sadreyev Ruslan I, Scadden David T, Lee Jeannie T: Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152(4):727–742.
Agostini F, Cirillo D, Bolognesi B, Tartaglia GG: X-inactivation: quantitative predictions of protein interactions in the Xist network. Nucleic Acids Res 2012,. Agostini F, Cirillo D, Bolognesi B, Tartaglia GG: X-inactivation: quantitative predictions of protein interactions in the Xist network.Nucleic Acids Res 2012,.
Salvador MA, Wicinski J, Cabaud O, Toiron Y, Finetti P, Josselin E, Lelièvre H, Kraus-Berthier L, Depil S, Bertucci F, et al.: The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low xist expression. Clin Cancer Res 2013, 19(23):6520–6531.
Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, Merajver SD, Dontu G, Wicha MS: BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci 2008, 105(5):1680–1685.
Ganesan S, Silver DP, Greenberg RA, Avni D, Drapkin R, Miron A, Mok SC, Randrianarison V, Brodie S, Salstrom J, et al.: BRCA1 Supports XIST RNA concentration on the Inactive X chromosome. Cell 2002, 111(3):393–405.
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al.: Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143(1):46–58.
Gumireddy K, Li A, Yan J, Setoyama T, Johannes GJ, Orom UA, Tchou J, Liu Q, Zhang L, Speicher DW, et al: Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. 2013, 32:. Gumireddy K, Li A, Yan J, Setoyama T, Johannes GJ, Orom UA, Tchou J, Liu Q, Zhang L, Speicher DW, et al: Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. 2013, 32:.
Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et al.: MALAT-1, a novel noncoding RNA, and thymosin [beta]4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22(39):8031–8041.
Ying L, Chen Q, Wang Y, Zhou Z, Huang Y, Qiu F: Upregulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol Biosyst 2012, 8(9):2289–2294.
Matouk IJ, Raveh E, Abu-lail R, Mezan S, Gilon M, Gershtain E, Birman T, Gallula J, Schneider T, Barkali M, et al.: Oncofetal H19 RNA promotes tumor metastasis. Biochim Biophys Acta 2014, 1843(7):1414–1426.
Kallen Amanda N, Zhou X-B, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi J-S, Zhang H, et al.: The imprinted H19 LncRNA Antagonizes Let-7 MicroRNAs. Mol Cell 2013, 52(1):101–112.
Loewer S, Cabili MN, Guttman M, Loh Y-H, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, et al.: Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 2010, 42(12):1113–1117.
Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, Wu M, Xiong J, Guo X, Liu H: Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell 2013, 25(1):69–80.
Hou P, Zhao Y, Li Z, Yao R, Ma M, Gao Y, Zhao L, Zhang Y, Huang B, Lu J: LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis 2014, 5: e1287.
J-h Y, Yang F, Wang F, Ma J-z, Guo Y-j, Tao Q-f, Liu F, Pan W, Wang T-t, Zhou C-c, et al.: A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25(5):666–681.
Acknowledgements
We would like to recognize Dr. Suling Liu’s and Dr. Hasan Korkaya’s research contributions related to EMT/MET transitions and cytokine regulation of cancer stem cells which took place while they were members of Dr. Wicha’s laboratory. These research findings help us to conceive this review. Furthermore, we would like to acknowledge the invaluable assistance of Denise Poirier. This work was supported by NIH grants CA101860 and CA66233 (to MSW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
MSW holds equity in OncoMed Pharmaceuticals and currently receives research support from Verastem, MedImmune and Dompe Pharmaceuticals. MSW is a scientific advisor to MedImmune, Verastem, Cerulean and Paganini. All other authors have no competing interests to disclose.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Zhu, Y., Luo, M., Brooks, M. et al. Biological and clinical significance of cancer stem cell plasticity. Clin Trans Med 3, 32 (2014). https://doi.org/10.1186/s40169-014-0032-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-014-0032-3