
Abstract
Background
Dietary fiber is an integral part of a healthy diet, but questions remain about the mechanisms that underlie effects and the causal contributions of the gut microbiota. Here, we performed a 6-week exploratory trial in adults with excess weight (BMI: 25–35 kg/m2) to compare the effects of a high-dose (females: 25 g/day; males: 35 g/day) supplement of fermentable corn bran arabinoxylan (AX; n = 15) with that of microbiota-non-accessible microcrystalline cellulose (MCC; n = 16). Obesity-related surrogate endpoints and biomarkers of host-microbiome interactions implicated in the pathophysiology of obesity (trimethylamine N-oxide, gut hormones, cytokines, and measures of intestinal barrier integrity) were assessed. We then determined whether clinical outcomes could be predicted by fecal microbiota features or mechanistic biomarkers.
Results
AX enhanced satiety after a meal and decreased homeostatic model assessment of insulin resistance (HOMA-IR), while MCC reduced tumor necrosis factor-α and fecal calprotectin. Machine learning models determined that effects on satiety could be predicted by fecal bacterial taxa that utilized AX, as identified by bioorthogonal non-canonical amino acid tagging. Reductions in HOMA-IR and calprotectin were associated with shifts in fecal bile acids, but correlations were negative, suggesting that the benefits of fiber may not be mediated by their effects on bile acid pools. Biomarkers of host-microbiome interactions often linked to bacterial metabolites derived from fiber fermentation (short-chain fatty acids) were not affected by AX supplementation when compared to non-accessible MCC.
Conclusion
This study demonstrates the efficacy of purified dietary fibers when used as supplements and suggests that satietogenic effects of AX may be linked to bacterial taxa that ferment the fiber or utilize breakdown products. Other effects are likely microbiome independent. The findings provide a basis for fiber-type specific therapeutic applications and their personalization.
Trial registration
Clinicaltrials.gov, NCT02322112, registered on July 3, 2015.
Video Abstract
Similar content being viewed by others
Background
Obesity and its comorbidities such as type II diabetes have reached epidemic proportions worldwide [1]. Observational research has linked dietary fiber with reduced prevalence of chronic diseases [2, 3], and mechanisms by which fibers exert their benefits have been established in animal models [4, 5]. The health effects of fibers are dependent on their physicochemical properties [6, 7]. Viscous fibers show efficacy in improving lipid and glucose metabolism [8, 9], which is reflected in a health claim by the European Food Safety Authority for moderately viscous wheat endosperm arabinoxylans (AXs) and postprandial glycemic control [10]. Given that average fiber consumption remains low in industrialized societies [11], fiber supplementation could be an effective treatment or preventive strategy for obesity-related chronic diseases [12].
Clinical evidence for the health effects of fiber supplements remains highly inconsistent [13], and it has been questioned whether purified (isolated) forms of fiber maintain their physiological effects once removed from the three-dimensional plant cell wall matrix [2, 14]. In this context, the mechanistic foundations for the beneficial effects of purified fibers remain insufficiently understood in humans. The strongest evidence exists for viscous fibers, which likely prolong satiety and lower postprandial metabolic responses by delaying gastric emptying and intestinal nutrient absorption [7]. Fermentable fibers are hypothesized to favorably modulate the gut microbiota [4, 15] in a manner that mitigates obesity and related comorbidities [16, 17]. For example, selective fermentation of fibers can alter compositional features of the gut microbiota in a structure-dependent manner [18, 19], selecting for bacterial taxa associated with metabolic effects (e.g., Prevotella copri [20] and Eubacterium rectale [21]). Moreover, fiber fermentation generates metabolites such as short-chain fatty acids (SCFAs), which act as signaling molecules that may maintain intestinal barrier integrity and immune homeostasis, and induce hormones that regulate satiety and glucose metabolism [5, 22]. Some fiber structures can also bind bile acids, which display immunomodulatory and metabolic properties [23, 24]. The reconfiguration of the bile acid pool might, therefore, constitute another mechanism for the physiological effects of fiber [15].
Although the role of the gut microbiome in the health effects of dietary fiber has received tremendous attention [15, 25], studies in humans have so far only established correlations between physiological effects and shifts in specific bacterial taxa [26, 27] or metabolites [28, 29] that cannot assign causality [30]. A causal role of the microbiome has been established in animal models [31], but experiments in conventional and germ-free mice have demonstrated that the effects of fiber can also be completely independent of the gut microbiota [32]. Therefore, it remains unclear whether the gut microbiota is mechanistically implicated in the physiological effects of fiber, and if so, which effects are microbiome-dependent and which microbes are involved. Although causality is difficult to establish in humans [30], comparisons of physicochemically distinct fibers that differ in their degree of fermentability could be used to determine whether health outcomes are predictable through compositional and functional responses of the gut microbiota.
In a previous study [33], we compared the effects of high doses of two purified fibers—a moderately viscous [34] and fermentable [35] AX with an insoluble, non-accessible [33] large-particle microcrystalline cellulose (MCC)—on compositional and functional features of fecal microbiota in adults with excess weight (body mass index [BMI]: 25–35 kg/m2). This study showed that AX, but not MCC, induced global shifts in the fecal microbiota, and enriched, although with a substantial degree of inter-individual variation, bacterial taxa (e.g., P. copri) [20, 36] and metabolic functions (propionate) [36,37,38] implicated in host metabolic features linked to obesity and satiety. Here, we extended this research and assessed the effects of the purified fibers on perceived satiety and obesity-related surrogate endpoints in the same individuals. We hypothesized that administration of a fermentable AX would induce physiological effects that were linked to compositional and functional changes in fecal microbiota (e.g., fermentation), as well as molecular markers of biological processes induced through microbiome metabolites (e.g., SCFAs). To test this hypothesis, we compared the physiological effects of AX with non-accessible MCC and explored associations with fecal microbiota compositional (fiber-responsive taxa and ecological variables of the broader community [33]) and functional (SCFAs and bile acids) features. To gain insight into potential mechanisms, we assessed biomarkers of host-microbiome interactions implicated in the pathophysiology of obesity (i.e., trimethylamine N-oxide [TMAO], gut hormones, cytokines, and measures of intestinal barrier integrity). To determine the role of fiber fermentation, we analyzed the fecal microbiota from participants that consumed AX to identify bacterial taxa that were involved in the utilization of AX by employing an approach that combined ex vivo anaerobic fermentations with bioorthogonal non-canonical amino acid tagging (BONCAT) and fluorescence-activated cell sorting (FACS) [39]. We integrated the data using a machine learning approach to determine whether effects on satiety or surrogate endpoints could be predicted by fecal microbiota features or biomarkers of host-microbiome interactions.
Results
Baseline characteristics of study participants
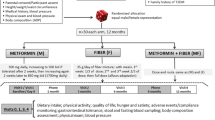
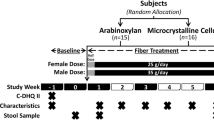
2To compare the effects of AX and MCC supplements at high doses (females: 25 g/day; males: 35 g/day) and high purity (> 80% fiber) on human health, we performed a single-blind, parallel-arm, 6-week, randomized controlled exploratory trial in adults with excess weight (Fig. 1). A total of 31 participants (AX: 10F and 5M; MCC: 11F and 5M) aged 32.9 ± 8.5 years with a BMI of 28.7 ± 2.3 kg/m2 completed the study protocol and were included in statistical analyses. Mean protocol adherence to AX and MCC supplementation was 94.7 ± 6.5% and 95.0 ± 5.6%, respectively (Additional file 1: Figure S1). No differences in age, sex, surrogate endpoints, or other study variables were detected between groups at baseline (see Additional file 2: Table S1 for baseline characteristics).

Study design of the randomized controlled trial. ASA24-Canada, Canadian version of the web-based Automated Self-Administered 24-hour Dietary Assessment Tool; stool characteristics, self-reported stool consistency and bowel movement frequency
Dietary intake
Dietary fiber intake increased by 142% and 171% during fiber supplementation of AX and MCC, respectively, as compared to baseline (p = 0.0002 for both AX and MCC, permutational t-test) with no difference between groups. This corresponded to an increase from 21 ± 6 and 19 ± 8 g/day to 46 ± 12 and 44 ± 8 g/day for AX and MCC, respectively (Additional file 3: Table S2). Interestingly, sugar consumption also increased by 35% during AX (p = 0.04) and 46% during MCC (p = 0.03) supplementation, likely due to participants incorporating the powdered supplements into foods and drinks that contained sugar, such as yogurt and fruit smoothies. No differences were detected between groups (p > 0.1), suggesting similar dietary changes were made by participants in both groups.
AX and MCC differ markedly in their physiological effects
Principal component analysis ordination of perceived satiety and surrogate endpoints revealed no differences at baseline between the AX and MCC groups (p = 0.77, permutational multivariate analysis of variance; Fig. 2A). In contrast, shifts in the variables from baseline to week 6 showed strong clustering by treatment group (p = 0.006; Fig. 2B), indicating that the two fibers differed in their overall physiological effects.

Effects of AX and MCC supplementation on satiety and surrogate endpoints. Principal component analysis plots show A perceived satiety and surrogate endpoints at baseline and B their percentage change from baseline per AX and MCC groups. Line graphs show weekly SLIM scale ratings C 30–60 min after consuming a meal with AX or MCC and D upon awakening. Bars (insets) represent the area under the SLIM score curve (AUCBL–W6). Scatter plots show E HOMA-IR, F QUICKI, G fecal calprotectin, and H TNF-α at baseline and week 6 of AX or MCC supplementation, respectively. Bars (insets) represent the percent change from baseline values per group. To assess within-group changes relative to baseline, data were analyzed for C and D using repeated measures one-way ANOVA with permutations and for E to H using paired permutational t-tests. To assess between-group differences, data were analyzed for A and B using permutational multivariate analysis of variance based on Manhattan distance and for C to H using unpaired permutational t-tests. Statistical significance was set for A to D at p < 0.05 and for E to H at p < 0.01. Data for C to H presented as mean ± SD; for E to H symbols represent individual samples. AX, arabinoxylan; HOMA-IR, homeostatic model assessment of insulin resistance; MCC, microcrystalline cellulose; QUICKI, quantitative insulin sensitivity check index; SLIM, Satiety Labeled Intensity Magnitude; TNF-α, tumor necrosis factor-α
Effects of AX and MCC on perceived satiety
AX resulted in higher satiety scores, assessed by the Satiety Labeled Intensity Magnitude (SLIM) scale, 30–60 min after consuming a meal (referred to as ‘satiety after a meal’) compared to MCC (p = 0.035, permutational t-test), with between-group differences detected during weeks 2 (p = 0.04), 4 (p = 0.007), and 6 (p = 0.03) (Fig. 2C). Further evaluation of satiety scores revealed that after AX consumption, individuals perceived feeling between “moderately full” and “very full”, while those consuming MCC remained between “slightly full” and “moderately full”. After participants awoke, AX also consistently increased satiety scores towards feeling “slightly full” over the entire treatment period, while MCC tended to reduce satiety scores towards feeling “slightly hungry,” with between-group differences reaching significance during week 5 (p = 0.03) (Fig. 2D).
Effects of AX and MCC on obesity-related surrogate endpoints and biomarkers of host-microbiome interactions
Among the surrogate endpoints, AX consumption reduced homeostatic model assessment of insulin resistance (HOMA-IR; insulin resistance index; p = 0.006; Fig. 2E) and increased quantitative insulin sensitivity check index (QUICKI; insulin sensitivity index, p = 0.008; Fig. 2F) compared to the MCC group. Effects of AX on HOMA-IR showed a 36% difference relative to MCC, which is comparable to those reported for other microbiome-targeted strategies, such as fecal microbiota transplantation (FMT) [40,41,42]. MCC reduced fecal calprotectin (a surrogate endpoint of intestinal inflammation) when compared to the AX group (p = 0.002), and by 39% relative to baseline (p = 0.004) (Fig. 2G). No effects of AX or MCC were detected for any other surrogate endpoint (Additional file 4: Table S3).
Evaluating biomarkers of host-microbiome interactions revealed a 7% reduction from baseline in tumor necrosis factor-α (TNF-α) by MCC (p = 0.004; Fig. 2H). No other effects of AX or MCC were detected for the remaining biomarkers of host-microbiome interactions (Additional file 4: Table S3), despite the fact that the production of gut hormones and intestinal barrier integrity have been previously linked to fiber fermentation [5, 16].
To confirm significant effects were independent of potential confounders, analysis of covariance (ANCOVA) models were performed using age, sex, and changes in total fiber and sugar consumption as covariates (Additional file 5: Table S4). Stool characteristic variables were also included since obesity has been associated with altered bowel habits [43] and, in our preceding study [33], AX and MCC promoted more frequent bowel movements (p < 0.05), while AX promoted softer stool consistencies compared to MCC (p < 0.05) (Additional file 4: Table S3). ANCOVA models showed that the observed effects were not confounded by host factors, dietary changes, or stool characteristics (p < 0.05).
Effects of AX and MCC on functional features of the fecal microbiota
As previously shown [33], AX consumption directed microbial output of SCFAs in favor of propionate, while MCC did not alter fecal SCFAs. Since bile acid derivatives also possess immunomodulatory and metabolic properties [23, 24], we applied targeted metabolomics to determine the effects of AX and MCC on fecal bile acids. This analysis showed that MCC decreased concentrations of apocholic acid (p = 0.009, permutational t-test) and hyodeoxycholic acid (p = 0.009) relative to baseline. Reductions in total fecal concentrations of bile acids and five secondary bile acids—deoxycholic acid, isolithocholic acid (ILCA), taurolithocholic acid (TLCA), taurodeoxycholic acid (TDCA), and glycodeoxycholic acid (GDCA)—also approached significance (0.01 < p < 0.05; Table 1 and Additional file 6: Table S5). By contrast, AX did not reduce bile acid concentrations relative to baseline but, when compared to MCC, increased concentrations of 7αOH-3-oxo-4-cholestenoic acid (p = 0.0096). Changes induced by both treatments also showed large standard deviations, indicating that bile acid shifts were highly individualized. Overall, our findings suggest that consumption of large-particle MCC alters the fecal bile acid profile by reducing secondary bile acid concentrations.
Identification of bacterial consortia involved in AX degradation
Considering that bacteria involved in fiber fermentation dictate the production of health-related metabolites [44], we aimed to identify the bacterial taxa within participants’ fecal microbiota that were involved in the fermentation of AX and utilization of breakdown products released during fermentation. We used BONCAT to fluorescently label metabolically active bacterial cells within the fecal microbiota [39], then sorted and profiled the active cells by FACS and 16S rRNA gene amplicon sequencing, respectively (Fig. 3 and Additional file 7: Figure S2). Initial comparison of fluorescent cells in incubations that contained the cellular activity marker L-azidohomoalanine (AHA) plus Cy5-dye, but differed by AX amendment, revealed no basal cellular activity in the absence of AX; thus, demonstrating that BONCAT was highly specific and only detected AX-induced cellular activity (Fig. 3).

Schematic representation of the ex vivo detection assay based on BONCAT. Stool samples stored frozen were thawed, filtered, and washed in PBS and then incubated in the presence of AX and the cellular activity marker L-azidohomoalanine (AHA) to detect AX-stimulated bacterial cells. A no-amendment control, containing only AHA, was incubated to detect possible basal activity in the absence of AX. Microscopic inspection showed no BONCAT signal for all controls; thus, no basal activity was detected. AX-incubated samples were then fixed in ethanol and active cells were stained using a Cu(I)-catalyzed click reaction using a Cy5 dye solution. A and B A representative picture of fecal microbiota incubated for 6 h A with AX and B without AX (BONCAT control). Stimulated cells, shown in pink as a Cy5-positive BONCAT signal, were sorted by FACS, with all microbial cells shown in blue (DAPI stained). DNA was extracted from both sorted cells and samples at 0-h and 6-h anaerobic incubations. The 16S rRNA gene was amplified by PCR and amplicons were sequenced using the Illumina Miseq platform. AX, arabinoxylan; BONCAT, bioorthogonal non-canonical amino acid tagging; FACS, fluorescence-activated cell sorting
Compared to the total bacterial community prior to incubation with AX, the active consortia had lower α-diversity (Shannon index: p = 0.0008, one-way ANOVA with permutations) and richness (Chao1 index: p < 0.0001) with, on average, 31% fewer amplicon sequence variant (ASV) numbers (p = 0.0001) (Additional file 8: Table S6). Several bacterial taxa previously shown to utilize AX were represented among the 31 dominant ASVs (mean relative abundance > 1%) in the active consortia (Additional file 8: Table S6), including ASVs related to Bifidobacterium longum, Blautia obeum, Bacteroides ovatus, Bacteroides cellulosilyticus, and E. rectale [33, 45]. Overall, these findings suggest that AX fermentation is not limited to a few cooperative species but extends to numerous members of the broader bacterial community, and involves several primary degraders as well as secondary fermenters.
To compare the abundance of individual taxa between the active consortia and fecal bacterial community, a differential abundance test (DESeq2) was applied. This analysis revealed the families Bacteroidaceae, Lactobacillaceae, and Enterobacteriaceae were more abundant in the active consortia, while Rikenellaceae, Ruminococcaceae, and Streptococcaceae were underrepresented. Fourteen ASVs were identified to differ between the active consortia and fecal bacterial communities prior to incubation with AX. Bacteroides koreensis, Bacteroides plebeius, Bacteroides xylanisolvens, Lactobacillus spp., and Escherichia/Shigella spp. were more abundant in the active consortia (p < 0.01). ASVs related to Coprococcus eutactus, Faecalibacterium prausnitzii, and Dialister invisus [46], which might be utilizing sugars and metabolic by-products (e.g., acetate and lactate) released during AX degradation [5, 33], were also metabolically active during incubation with AX but less abundant in the active consortia (p < 0.01).
Identification of microbiota-related predictors of satiety and surrogate endpoints
To gain insight into the role of the gut microbiota in the physiological effects of fiber, we used a machine learning approach to determine predictors for the physiological effects of the fibers on perceived satiety and surrogate endpoints. Variables related to microbiota compositional (fiber-responsive taxa in feces [33], BONCAT identified active taxa [differentially abundant bacterial ASVs and all metabolically active bacterial ASVs with average relative abundances ≥ 0.15%] and ecological characteristics [α-diversity and principal components] [33]) and functional (SCFAs and bile acids) features, biomarkers of host-microbiome interactions (TMAO, gut hormone, cytokine, and barrier function measures), and calorie-adjusted macronutrient intake data were included in the models. For each endpoint affected by fiber consumption, high- and low-responders were first identified according to the study cohort median (Additional file 9: Figures S3A and S3B), and datasets were used as predictor variables for the training of independent random forest classifiers (RFCs) to rank microbiota-related predictors that discriminate high-responders from low-responders (Additional file 9: Figures S3C and S3D).
For the effect of AX on satiety after a meal, only RFCs trained on the metabolically active taxa identified by BONCAT resulted in models with significant predictive ability. The best model was obtained with the 14 differentially abundant ASVs (receiver operating characteristic curves [AUC-ROC] = 0.95; Fig. 4A), but the model that was based on the 90 active ASVs still showed a high prediction accuracy (AUC-ROC = 0.84; Fig. 4B). A weak positive correlation that approached significance was detected between propionate-producing D. invisus (ASV6pygnt) and satiety (rs = 0.63, q = 0.08, Spearman’s correlation; Fig. 4A). Satiety was also negatively correlated with formate-producing Dorea formicigenerans (ASV2xmw96; rs = − 0.81, q = 0.007), with butyrate producers Eubacterium ramulus (ASV56kx74; rs = − 0.60, q = 0.08) and F. prausnitzii (ASV; rs = − 0.56, q = 0.098) also showing weak negative correlations (Fig. 4B). F. prausnitzii and D. formicigenerans further showed increases only in low-responders (p < 0.01). Although AX induced fecal propionate [33], which is the SCFA with the strongest evidence for satietogenic effects [22, 37], RFCs based on fecal SCFA shifts could not predict satiety after a meal (OOB error > 0.6) (Additional file 9: Figure S3C).

Identification of gut microbiota compositional features and biomarkers of host-microbiome interactions that predict satiety and surrogate endpoint responses by machine learning. (left) AUC-ROC curves show the performance accuracy of random forest classifiers trained to predict high-vs-low responders for: A and B perceived satiety after a meal with AX using the relative abundance of bacterial taxa activated during ex vivo incubation with AX; C HOMA-IR and D fecal calprotectin for AX and MCC, respectively, using fecal bile acid shifts; and E TNF-α for MCC using baseline intakes of calorie-adjusted macronutrients. (center) Horizontal bars represent Spearman’s correlation coefficients between endpoints and A and B metabolically active ASVs, C and D fecal bile acids, or E macronutrients shown to be important for predicting responses. Mean importance values were determined by random forest, which identifies factors that contribute the most to the model. (right) Scatter plots show the association between endpoints and the most discriminative microbiota-related factors that correlate with AX-induced A and B satiety after a meal and C HOMA-IR attenuation, and D MCC-induced fecal calprotectin attenuation. Vertical bar graphs show the most discriminative microbiota-related factors grouped by high- and low-responders. High-responders (black) and low-responders (gray) were defined according to the study cohort median. Statistical significance was set at p < 0.05 and FDR adjusted q values < 0.05. ∆, absolute change from baseline to week 6; %∆, percent change from baseline to week 6; 3√, cube root transformed before analysis; All ASVs, amplicon sequence variants with average relative abundances ≥ 0.15%; AX, arabinoxylan; AUC-ROC, area under the receiver operating characteristic curve; BL, baseline; Diff. Abundant ASVs, differentially abundant amplicon sequence variants among the bacterial consortia recovered by fluorescence-activated cell sorting; GDCA, glycodeoxycholic acid; HOMA-IR, homeostatic model assessment of insulin resistance; ILCA, isolithocholic acid; LCA, lithocholic acid; MCC, microcrystalline cellulose; TLCA, taurolithocholic acid; TNF-α, tumor necrosis factor-α
The only RFC that predicted HOMA-IR was AX-induced shifts in secondary bile acids (AUC-ROC = 0.70; Fig. 4C). MCC-induced fecal calprotectin responses were also predicted by MCC-induced shifts in secondary bile acids (AUC-ROC = 0.72; Fig. 4D), but different secondary bile acids were predictors. Improvements in HOMA-IR showed weak inverse correlations with reductions in the concentrations of lithocholic acid (LCA) (rs = − 0.62, q = 0.08) and its derivative ILCA (rs = − 0.60, q = 0.08) (Fig. 4C), while calprotectin responses were negatively correlated with reductions in conjugated bile acids TLCA (rs = − 0.68, q = 0.03), TDCA (rs = − 0.70, q = 0.03), and GDCA (rs = − 0.63, q = 0.05) (Fig. 4D). A comparison of fiber-induced bile acid shifts revealed significant differences between high-responders and low-responders, with shifts only being detected in low-responders (Fig. 4C and D). For the TNF-α response, only the RFC based on calorie-adjusted intakes of macronutrients at baseline could predict the effect of MCC (AUC-ROC = 0.73; Fig. 4E). While the most discriminative nutrient was saturated fat, consumption at baseline did not associate with TNF-α responses (rs = 0.46, q = 0.17).
To determine whether associations between surrogate endpoints and microbiome features were independent of confounders shown to impact microbiome composition [47], we used multivariate generalized linear models (GLMs) to control for age, sex, changes in total fiber and sugar intake, stool consistency, and bowel movement frequency as covariates (Additional file 10: Table S7). These analyses showed that the observed associations were not confounded by host or dietary factors (p < 0.1). As dietary fibers may indirectly influence the intestinal bile acid pool by altering environmental conditions such as pH and water availability [6, 48], links to fecal pH and moisture content were also evaluated (Additional file 11: Figure S4). Though values were not changed by either fiber (p > 0.1) [33], positive associations were detected between the shifts in fecal pH (q = 0.06) and moisture content (q < 0.05) and the bile acid shifts predictive of AX- and MCC-induced effects, respectively.
Our study was specifically designed to evaluate mechanistic associations between fiber-responsive bacterial taxa [33] and mechanistic biomarkers of host-microbiome interactions that relate to fiber fermentation. Although SCFAs, barrier function, and the dominant AX-responsive taxa (P. copri and B. longum) have been implicated in satiety, insulin resistance, and inflammation [16, 33], significant predictions were not detected (AUC-ROCs < 0.70). Additional univariate GLMs reaffirmed that the dominant fiber-induced shifts in fecal microbiota composition, propionate, and bile acids were not linked to the physiological benefits of fiber supplementation (q > 0.05; Additional file 12: Figure S5).
Discussion
This study established physiological effects for two isolated forms of dietary fiber, AX and MCC, that are therapeutically relevant for individuals with excess weight. By using BONCAT, we provide evidence that the ability of AX to enhance satiety was predicted by bacterial taxa involved in its utilization, suggesting that fermentation of fiber by the gut microbiota may influence its satietogenic effects. In contrast, the AX-induced attenuation of insulin resistance, although linked to fecal bile acid shifts, did not show positive correlations with microbiota features. Interestingly, MCC, which is non-accessible by gut microbiota, showed anti-inflammatory effects in the gut by reducing fecal calprotectin levels that were also predicted by bile acid shifts. Surprisingly, molecular markers of biological processes hypothesized to link metabolic activities of the gut microbiome with host metabolism and immunology (i.e., TMAO, gut hormones, cytokines, and intestinal barrier integrity) were not affected by fermentable AX and could not predict its effects. By exploring the role of the gut microbiota in the effects of physicochemically distinct fibers, our study provides insight as to how fibers may impact satiety and obesity-related surrogate endpoints in humans.
The ability of AX to induce satiety is in agreement with previous research [49] on both long-chain AX supplements [50, 51] and AX-rich whole grains [52,53,54]. Due to its viscous properties, AX may delay gastric emptying, thereby prolonging postprandial satiety [7]. Our results indicate that microbial fermentation of AX might also contribute to perceived satiety during AX consumption since satiety scores were predicted by the bacterial taxa involved in the breakdown of AX, as detected by BONCAT. We observed significant random forest models (Fig. 4A and B) and weak positive correlations between satiety and propionate producers (D. invisus and B. plebeius), as well as negative associations with butyrate producers, such as E. ramulus and F. prausnitzii. The findings are in overall agreement with the analysis of fecal SCFAs, which showed that AX shifts the propionate to butyrate ratio in favor of propionate [33]. Therefore, substrate competition between propionate and butyrate producers may have influenced satiety, as only propionate is shown to be satietogenic in humans [22, 37]. Although additional studies are needed to elucidate exact mechanisms and cross-feeding interactions, our findings provide evidence that the gut microbiota might contribute to the satiety-enhancing properties of AX in humans.
Although BONCAT has the potential to provide a direct assessment of bacterial taxa involved in fiber utilization, the approach is not without limitations. As BONCAT applies an in vitro fermentation, in vivo conditions are unlikely to be accurately replicated, and starting inoculums may have been affected by oxygen exposure during fecal collection, loss of viability during storage, and freeze-thawing. Microbes that do not import amino acids and incorporate them into newly synthesized proteins would also fail to be detected by BONCAT. Being a long-chain highly branched and viscous fiber, the AX is further likely to have behaved differently ex vivo than in the gastrointestinal tract. These limitations may provide an explanation for the failure of B. longum and P. copri to become metabolically active during the in vitro fermentation (Additional file 8: Table S6), even though these species expanded in vivo through AX [33]. However, the significant random forest models that resulted from including all bacterial ASVs identified by BONCAT support the value of this approach and its application in human nutritional studies. Future research is needed to further explore and refine the application of BONCAT to determine the interactions between dietary compounds and the microbiome, and to improve and validate experimental procedures.
The ability of different types of AX to improve glucose and insulin metabolism is well supported [49, 50, 55, 56] and has resulted in an European Food Safety Authority health claim [10]. The effect size detected in our study was comparable with that in studies that tested alternative strategies aimed to attenuate insulin resistance in individuals with obesity, such as a plant-based diet [40], Akkermansia muciniphila [41], or FMT [42]. Compared to these strategies, AX supplements would be more cost-effective and, since it is a food-grade dietary fiber, AX would constitute a promising opportunity for the development of functional foods and beverages. Interestingly, HOMA-IR increased in the MCC group, an effect likely attributable to the elevated sugar consumption during the treatment period. Given that sugar consumption increased equally in both groups (Additional file 3: Table S2), our findings indicate that AX may have counteracted the detrimental effects of sugar on insulin sensitivity.
Anti-inflammatory effects of MCC have, to our knowledge, not yet been reported in humans. These findings are, however, in agreement with research in mice, where very high cellulose diets mitigated chemically induced colitis [57, 58] and improved LPS-induced intestinal permeability [59]. Since the anti-inflammatory effects of MCC are novel in humans, larger studies are needed to confirm our findings and explore whether anti-inflammatory properties are specific to large-particle, highly crystalline cellulose.
Although the physiological effects of AX and MCC differed, both responses were predictable in random forest models through shifts in fecal secondary bile acids, albeit different derivatives. For AX, HOMA-IR responses were inversely associated with shifts in LCA and ILCA, two bile acids that can regulate glucose homeostasis via activating FXR- and TGR5-mediated signaling pathways [24]. Fecal calprotectin responses associated with MCC-induced reductions in TLCA, TDCA, and GDCA. While immunoregulatory properties of these conjugated bile acids remain poorly defined, TLCA may exert anti-inflammatory effects by inhibiting inflammasome signaling [60]. These findings are relevant given the emerging role of bile acids in the regulation of metabolism and immunology [23, 24]. However, despite good predictive power, our correlation analyses identified only negative correlations between physiological benefits and bile acids shifts, and significant shifts in bile acids were only observed in low-responders. Cause-and-effect relationships cannot be unraveled with our correlative data, and it is possible that the negative associations arose through indirect effects of the fibers, such as changes in pH (through AX fermentation) or moisture content (through MCC stimulating fluid secretions) [6, 7, 48], as suggested by our GLMs (Additional file 11: Figure S4). Nevertheless, the lack of positive associations between fiber-induced bile acid shifts and surrogate endpoints suggest that modulation of the bile acid pool does not provide a primary mechanism for the clinical benefits of AX and MCC. The specific effects of the two fibers as they relate to reductions in secondary bile acids are still therapeutically relevant as they provide information on how fibers can selectively manipulate bile acids. Secondary bile acids are considered to be cytotoxic [61]; therefore, the observed reductions might constitute a mechanism by which dietary fibers protect against the development of colon cancer [62].
Through a direct comparison of the effects of microbiota-accessible and non-accessible fibers in a human trial, this study was specifically designed to evaluate the role of the microbiome in the effects of fibers and test commonly hypothesized mechanistic links between microbial fermentation and physiological effects of fibers [16, 17]. For instance, the experimental design allowed us to determine whether fiber-induced shifts in fecal SCFAs were linked to effects on satiety and insulin sensitivity via gut hormones or systemic inflammation through improvements in barrier function [5, 16]. However, despite effects on satiety and insulin resistance through AX, none of the mechanistic biomarkers significantly changed in the AX and MCC groups. Even though this was surprising given that animal models provide convincing evidence for the importance of these mechanisms [4, 5], findings from human studies with fermentable fibers are, at best, inconclusive [63,64,65]. In addition, even though B. longum and P. copri were the numerically dominant AX-responders [33] and have been previously linked to improvements in inflammation [66], satiety [36], and insulin sensitivity [20] in humans, these taxa did not correlate with physiological outcomes. A microbiome independent effect is in agreement with the fact that MCC is not accessible to the microbiota, that non-accessible forms of AXs such as psyllium can improve insulin sensitivity [67], and that fibers can improve insulin sensitivity in germ-free mice [32]. Overall, our findings serve as a reminder that certain physiological effects of fiber might be microbiome independent and related to physicochemical attributes of fiber, such as viscosity or fecal bulking [6, 7]. Although we acknowledge the limitations of mechanistic studies in humans [30], as well as the challenges in accurately measuring biomarkers [68, 69] and the small sample size of our study, one has to also consider that some principles of the mechanistic actions of fibers detected in animal models, especially as they relate to the gut microbiota, might not apply to humans.
Conclusion
In summary, this study provides evidence for the physiological benefits of purified forms of dietary fibers, a notion that has been increasingly questioned in recent nutrition literature [2, 14]. Given the importance of satiety, insulin resistance, and systemic inflammation in the etiology of obesity and cardiometabolic diseases, our findings establish the potential role of purified fibers in the prevention and treatment of chronic diseases, while also warranting future studies to explore the anti-inflammatory effects of MCC. The distinct effects of the two fiber types can serve as a basis for a more targeted application of dietary fiber, such as AX for type II diabetes and MCC for inflammatory diseases, which could ultimately inform nutritional guidelines. Our findings also provide evidence for the role of fiber-microbiome interactions in inducing satiety, while the metabolic and immunological effects of the fibers may be primarily microbiota-independent. A better understanding of mechanisms by which fibers induce physiological effects in humans would contribute to a conceptual framework for the development of fiber structures or designer carbohydrates with improved clinical efficacy. In this respect, our findings that the effects of AX on satiety were linked to the ability of an individual’s microbiome to utilize the fiber provides a mechanistic basis to optimize fiber applications through a personalized approach.
Methods
Study design
Registration
As previously described [33], this randomized controlled exploratory trial was prospectively registered in July 2015 at ClinicalTrials.gov (NCT02322112) as part of a large clinical trial aimed at comparing the effects of four structurally distinct dietary fibers on the gut microbiota and human health. In response to requests by reviewers of a grant application, which advised against including a premarket ingredient in a large human trial, the AX arm was separated from the original trial in October 2016. Data from the participants that completed the AX protocol were analyzed independently and compared to data from the participants that completed the MCC protocol (microbiota-non-accessible controls) during the same period.
Dietary intervention
In brief, 38 individuals with excess weight (BMI: 25–35 kg/m2) were enrolled and randomly assigned to supplement their habitual diet with either AX or MCC at a daily dose of 25 g (females) or 35 g (males), an amount that resembles Health Canada recommendations for the intake of dietary fiber [70] (Fig. 1). Of the 19 individuals enrolled per arm, 15 participants completed the AX protocol, and 16 participants completed the MCC protocol [33]. The AX used was AGRIFIBER SFC (previously named BIOFIBER GUM), a fermentable, long-chain AX isolated from corn bran (AgriFiber Solutions LLC, USA), while the MCC was MICROCEL MC-12, a non-accessible, large-particle (160-micron average) wood-derived cellulose (Blanver Farmoquimica LTDA, Brazil) [33]. The rationale to focus on a BMI range of 25–35 kg/m2 was that these individuals would be more likely to have elevated levels of risk factors that predict cardiometabolic disease, but less likely to have these chronic diseases [21, 71]. Our overall goal was to explore if dietary supplements could attenuate these risk factors prior to disease onset.
Dietary intake
To assess whether dietary fiber supplementation influenced dietary intake, participants completed two non-consecutive 24-h dietary recalls at baseline and weeks 3 and 6 (Fig. 1) using the Canadian version of the web-based Automated Self-Administered 24-hour Dietary Assessment Tool [72]. Mean values of baseline (two recalls) and of weeks 3 and 6 (four recalls) were used in statistical analyses. Prior to assessing associations, diet data were first calorie-adjusted as previously described [73].
Perceived satiety
Perceived satiety was evaluated at baseline and weekly during the intervention using the validated SLIM questionnaire (Fig. 1), which is a 100 mm, bidirectional hunger-fullness scale anchored by “greatest imaginable fullness” (50 mm) and “greatest imaginable hunger” (− 50 mm), with “neither hungry nor full” in the center (0 mm) [74]. Each week, a SLIM scale was completed within (i) 30 mins of waking and (ii) 30–60 min of consuming a meal with AX or MCC added. For between-group comparisons, the area under the SLIM curve (AUCBL–W6) was calculated by using the linear trapezoidal method in GraphPad Prism. When applicable, missing data points were imputed using the mean of the participants’ known values, as previously described [75].
Obesity-related surrogate endpoints
Blood pressure was measured at baseline and week 6 with an automatic sphygmomanometer (Welch Allyn, Hill-Rom Inc., Indiana, USA). Blood samples were also collected at baseline and week 6 after a 12-h overnight fast using separation tubes, K2EDTA-coated tubes, and P800 tubes to obtain serum, plasma, and inhibitor-treated plasma, respectively. P800 tubes are pre-coated with K2EDTA plus a proprietary cocktail of protease, esterase, and DPP-IV inhibitors to prevent hormone degradation (BD Biosciences, USA). Total cholesterol, high-density lipoprotein cholesterol, triglycerides, and glucose were then quantified in serum on a Beckman Coulter DxC 800 (Beckman Coulter Inc., California, USA), with insulin measured in inhibitor-treated plasma by electrochemiluminescence immunoassay (ECLIA; K15174C, MesoScale Discovery®, Maryland, USA; intra-assay coefficient of variation [CV] 4.5%). Low-density lipoprotein cholesterol, HOMA-IR, and QUICKI values were then calculated as previously described [76,77,78]. To assess intestinal and systematic inflammation, fecal calprotectin and plasma high-sensitivity C-reactive protein were quantified by enzyme-linked immunosorbent assay (ELISA; K6927, Immundiagnostik AG, Bensheim, Germany) and ECLIA (K151STD; CV 2.1%), respectively. Finally, whole blood was collected for immediate quantification of complete blood count parameters using a Sysmex XN-10 analyzer (Sysmex Corporation, Kobe, Japan).
Biomarkers of host-microbiome interactions implicated in the pathophysiology of obesity
Appetite, glucose metabolism, and systemic inflammation
Several hormonal regulators of appetite and glucose metabolism were quantified in inhibitor-treated plasma by ECLIA (MesoScale Discovery®). This included total ghrelin and peptide YY by a U-PLEX® Assay (K151ACL; CV 3.2% and 2.4%), along with active glucagon-like peptide-1, glucagon, and leptin by a MULTI-SPOT® Assay (K15174C; CV 6.6%, 6.0%, and 5.1%). In addition, both adipocyte-derived hormone adiponectin, and the cytokines TNF-α, interleukin-6, interleukin-8, and interleukin-10 were measured in plasma by single-plex (K151BXC; CV 3.3%) and multiplex (K15049D; CV 3.0%, 4.3%, 3.7%, and 5.8%) assays, respectively.
Intestinal barrier integrity
Lipopolysaccharide-binding protein, albumin, and zonulin were quantified by ELISA as measures of gut barrier function recently validated by our group [79]. While lipopolysaccharide-binding protein was measured in plasma diluted 1:1300 in phosphate-buffered saline (PBS) (SEB406Hu, USCN Life Science and Technology, Texas, USA; CV 12.6%), albumin and zonulin were measured in fecal samples prepared by the Stool Sample Application System (K6330, K5600, and K6998SAS, Immundiagnostik AG), which diluted samples 1:100 in a proprietary extraction buffer.
TMAO
TMAO, a metabolite generated in the liver from the oxidation of bacterial-derived trimethylamine (TMA), was measured in serum by high-performance liquid chromatography-tandem mass spectrometry as previously described [80], with a CV of 5.0%. Briefly, for TMAO extraction, 50 μL of thawed serum was spiked with 50 μL of internal standard solution (TMAO-d9 and TMA-d9) and 150 μL of methanol with 0.1% formic acid. The mixture was then vortexed for 1 min and centrifuged at 10,000 rpm for 15 min at 15 °C. The supernatant was collected and stored frozen at − 20 °C until analysis. For sample derivatization, 25 μL of supernatant was reacted with 50 μL of ethyl bromoacetate (4 mg/ml in acetonitrile) in the presence of 3 μL concentrated ammonium hydroxide for 40 min at room temperature. Then, HPLC-grade water containing 0.5% formic acid was added to obtain the final volume of 500 μL and then stored at – 20 °C until analysis.
An Agilent 1200 series HPLC system (Agilent Technologies Inc., California, USA) coupled to a 3200 QTRAP mass spectrometer (AB SCIEX, Ontario, Canada) was used under turbospray positive mode to analyze standard and sample solutions. An Ascentis Express HILIC column (15 cm × 2.1 mm, 2.7 μm particle size; Sigma, Missouri, USA) was used at room temperature for LC separation. Composition of the mobile phase used for isocratic elution was (solvent A) 0.1% formic acid in acetonitrile and (solvent B) 10 mM ammonium formate (70:30, v/v). The run time was set as 6 min with a flow rate of 0.25 mL/min. Electrospray ionization was used under positive-ion detection with multiple-reaction monitoring scans. All other instrumental parameters used were as follows: curtain gas at 20 arbitrary units; gas 1 at 50; gas 2 at 60; ion spray voltage at 5200 V. The dwell time for each transition ion was 300 ms and the ion source temperature was 400 °C. Multiple-reaction monitoring transitions used were as follows: TMA derivative: 146.1 > 118.1; TMAO: 76.1 > 58.1; TMA-d9 derivative: 155.1 > 127.1; TMAO-d9: 85.1 > 68.1.
Fecal bile acids
Fecal bile acids were characterized by targeted metabolomics of 60 bile acids with ultrahigh performance liquid chromatography/multiple-reaction monitoring-mass spectrometry at the University of Victoria Genome British Columbia Proteomics Centre as previously described [81]. Briefly, bile acids were extracted from lyophilized and homogenized fecal samples by adding 1 mL of 75% acetonitrile to 10 mg of sample, followed by 20 sec of vortexing at 3,000 rpm, 5 min of sonication in an ice water bath, and 5 sec of additional vortexing. Samples were centrifuged at 15,000 rpm and 10 °C for 15 min, and then 20 μL of the supernatant was mixed with 60 μL of 50% methanol and 40 μL of internal standard. Finally, 10 μL of the mixture was injected onto an Agilent 1290 series HPLC system coupled to a 4000 QTRAP mass spectrometer. A Waters BEH C18 UPLC column (Waters Corp., Massachusetts, USA) was used for chromatographic separation. Composition of the mobile phase used for binary-solvent gradient elution was 0.01% formic acid in water (solvent A) and 0.01% formic acid in acetonitrile (solvent B). The flow rate was 0.35 mL/min with the column temperature maintained at 45 °C.
Ex vivo characterization of the bacterial consortia that utilize AX
Collection and processing of fecal samples
Fecal samples were collected at 6 weeks of AX supplementation as previously described [33]. Briefly, fecal material was collected in a stool specimen container and placed in an air-tight bag that contained a GasPak™ EZ Anaerobe Sachet (BD, Canada) to maintain anaerobicity. Samples were brought to the clinic within 4 h of defecation and processed immediately in a Bactron Anaerobic Chamber (Shel Lab, Oregon, USA) under anaerobic conditions (5% H2, 5% CO2, and 90% N2). Fecal material used for BONCAT was then diluted 1:10 in pre-reduced molecular grade PBS with 10% glycerol (a method previously shown to maintain microbiota viability [82]), aliquoted, and stored at − 80 °C until being shipped frozen on dry ice to the University of Vienna for further analysis.
BONCAT of AX-utilizing bacterial cells
To identify bacterial taxa involved in the degradation of AX and utilization of breakdown products released during degradation, we applied BONCAT [39], a fluorescence-based single-cell labeling of cellular activity, to week 6 fecal samples from the AX treatment group (Fig. 3). Briefly, BONCAT is based on incorporating the non-canonical amino acid AHA instead of L-methionine during protein synthesis, followed by fluorescent labelling of AHA-containing cellular proteins by azide-alkyne click chemistry. Using a Cu(I)-catalyzed reaction, a terminal alkyne coupled fluorescence dye, such as Cy5, can be linked to the azide group of the incorporated AHA; thus, marking microbial cells that have undergone protein synthesis during incubation with AHA and AX [83].
To apply BONCAT, fecal homogenates were first thawed and introduced into an anaerobic tent (85% N2, 10% CO2, 5% H2) upon arrival to the University of Vienna, with all reagents and laboratory consumables being introduced into the anaerobic tent two days prior to the experiment to ensure anaerobicity at the start of the experiment. Fecal homogenates were filtered (40 μm filter, Corning, Germany) to remove particles, washed twice in 1× PBS to remove residual glycerol, and diluted 1:10 in 1× PBS, as opposed to nutrient-rich media, to limit background noise, avoid autofluorescence in the Cy5 dye solution, and select for bacterial cells that preferentially utilize AX. Samples were then added to sterile Hungate tubes with 1 mM of cellular activity marker AHA (Baseclick GmbH, Germany) and 2 mg/mL of in vitro pre-digestion AX [84] (consistent with dietary intakes of AX [85, 86]), and then incubated in an anaerobic tent at 37 °C for 6 h. For each sample, a non-amended negative control, wherein only 1 mM of AHA was added, was also incubated to account for potential basal activity, of which there was no basal activity detected. After 6 h of incubation, biomasses were washed with 1× PBS, fixed in ethanol, and stored at – 20 °C in 1:1 ethanol/PBS.
To prepare the Cy5 dye solution, 1.25 μl of 20 mM CuSO4, 2.50 μl of 50 mM THPTA (Baseclick, Germany), and 0.30 μl of Cy5 alkyne dye (Jena Bioscience, Germany) were left in the dark for 3 min to react and then added to 221 μl of 1× PBS, 12.5 μl of 100 mM sodium ascorbate (Sigma-Aldrich, Austria), and 12.5 μl of 100mM aminoguanidine hydrochloride (Sigma-Aldrich, Austria). Next, 300–500 μl of the fixed biomasses were centrifuged at 10,000 rpm for 10 min and resuspended in 96% ethanol once the supernatants were removed. Finally, 60–100 μl of the dye solution was added to the fixed biomasses, incubated in the dark at room temperature for 30 min, washed three times with 1× PBS, and then filtered with 35 μm nylon mesh using 12 × 75 mm BD tubes (BD, Germany) immediately before being sorted by flow cytometry. Biomasses were also collected from the amended samples at 0- and 6-h incubations for additional DNA extractions.
FACS of AX-utilizing bacterial cells
Flow cytometry FACS of Cy5-labeled bacterial cells was done with an ultra-high-speed cell sorter MoFlo Astrios EQ (Beckman Coulter, California, USA) using the software Summit v6.2 (Beckman Coulter), as represented in Additional file 7: Figure S2. To standardize measurements and assess bacterial size, silica calibration beads (100, 500, and 1000 nm, Kisker Biotech, Germany) with refractive indexes close to that of biological material were recorded. The sorting of Cy5-labeled bacteria was performed as followed: background noise of the machine was first detected using the parameters forward scatter and side scatter. 488-nm FSC1-Height-Log vs 488-nm SSC2-Height-Log was then used to show the different sizes of silica beads in the first measurement and the scattering of the bacteria in subsequent measurements. Bacteria were pre-gated and displayed on a third scatter plot with 488 nm SSC-Area-Log vs 640 nm 671/30-Area-Log axes. Cy5-positive bacteria were then sorted out into tubes with a maximum event rate of 50,000 events/s. Reanalysis of the samples showed a purity of more than 99%.
DNA extraction, 16S rRNA gene amplicon sequencing, and inference of bacterial ASVs
Bacterial DNA from both FACS-sorted cells (6 h) and fecal incubations (0 and 6 h) were extracted using QIAamp DNA Mini Kit (Qiagen, Germany) following the protocol for bacteria according to the manufacturer’s instructions. Cell lysis was further performed enzymatically by first using Proteinase K and then by a second lysozyme step (Sigma-Aldrich, Austria). The V3–V4 region of the 16S rRNA gene was amplified and barcoded using a previously described 2-step PCR approach [87] with 16S rRNA gene primers S-D-Bact-0341-b-S-17 (5′-CCTACGGGNGGCWGCAG-3′) and S-D-Bact-0785-a-A-21 (5′-GACTACHVGGGTATCTAATCC-3′) [88]. Barcoded samples were then purified and normalized over a SequalPrep™ Normalization Plate Kit (Invitrogen) using a Biomek® NXP Span-8 pipetting robot (Beckman Coulter, California, USA), then pooled and concentrated on columns (Anlaytik Jena). Next, sequence libraries were prepared with the Illumina TruSeq Nano Kit as previously described [87] by sequencing in paired-end mode (2 × 300 nt; v3 chemistry) on an Illumina MiSeq. After sequencing, amplicon pools were extracted from the raw sequencing data using the FASTQ workflow in BaseSpace (Illumina) with default parameters, and then sequences were demultiplexed with the python package demultiplex [89] by permitting one mismatch each for barcodes, linkers, and primers. Contaminants, including mitochondria and chloroplast sequences, were removed using the R package decontam v1.6.0 [90] with the prevalence method and a threshold setting of 0.01. Sequence data were then analyzed using a bioinformatic approach based on ASVs as described previously [39, 91].
Statistical analyses
Statistical assessment of clinical effects
Statistical analyses were performed using R v3.5.1, Stata v15.0, and GraphPad Prism v9.1.2. Prior to statistical analyses, outliers were identified and removed based on a mean ± five standard deviation cutoff (≤ 2 participants per endpoint) [92]. To assess the overall effects of fiber supplementation on perceived satiety and surrogate endpoints, data were ordinated by principal component analysis using factoextra [93] and FactoMineR [94] packages. Then, between-group differences (AX-vs-MCC) were assessed by permutational multivariate analysis of variance based on Manhattan distances [95] using the Adonis function in the vegan [96] package. For perceived satiety, surrogate endpoints, biomarkers (apart from SCFAs [33]), and diet variables, repeated measures one-way ANOVA and paired t-tests with permutations (n = 1000) were applied to compare within-group differences relative to baseline using the permuco [97] package. Between-group differences were assessed by unpaired permutational t-tests (n = 1000) using the lmPerm [98] package. For BONCAT identified bacterial ASVs, the DEseq2 [99] package was used to identify ASVs whose abundance differed in the bacterial consortia recovered by FACS after a 6-h incubation in the presence of AX as compared to the total fecal bacterial communities after 0- and 6-h incubations. For surrogate endpoints, mechanistic biomarkers, and bacterial ASVs, a more stringent cutoff of p < 0.01 was considered significant to account for multiple comparisons and to detect only the robust effects of AX and MCC consumption, while p < 0.05 was considered significant for the remaining analyses where relatively few comparisons were made.
Machine learning models to predict clinical effects
To identify potential determinants of host-microbiome interactions that predicted the effects of fiber supplementation on perceived satiety and surrogate endpoints, separate RFCs were independently trained on changes in bacterial composition, ecological variables of the broader bacterial community (as previously determined [33]), mechanistic biomarkers, and macronutrient intake datasets (refer to Additional file 13: Table S8 for a description of each predictor dataset). Random forest uses supervised tree-based machine learning algorithms that are purported to be a robust approach for the discriminant analysis of high dimensional, low sample size data [100,101,102]. Prior to analysis, participants were classified as high- or low-responders for each endpoint according to the study cohort median, as in satiety after a meal (AUCBL–W6), HOMA-IR, fecal calprotectin, and TNF-α (percentage change). Since HOMA-IR and QUICKI indexes showed significant collinearity (rs = − 0.97 and p < 0.0001, Pearson’s correlation) with the same classifications, RFC analyses were only performed on HOMA-IR.
Independent RFCs were performed using the default settings in the randomForest [103] package, with the generalization error of each RFC estimated across 100 replicates using the leave-one-out cross-validation as previously described [104]. To evaluate the performance of each RFC, AUC-ROCs were generated from the true possible cross-validated results using the pROC [105] package and average out-of-bag error rates were estimated across 100 replicates. RFCs with AUC-ROC values ≥ 0.7 and out-of-bag error rates < 0.6 were considered to have good prediction accuracy [102]. A confusion matrix was further generated to evaluate subgroup prediction accuracy. To determine the importance of each individual variable for the classification of high-vs-low-responders, average mean importance scores were calculated by 100 replicates estimation.
To support the RFCs, Spearman’s correlations were performed between the endpoint and its best predictors, which showed the directionality of the associations. Univariate GLMs were further performed between the endpoint and the dominant fiber-induced shifts in fecal microbiota composition, propionate, and bile acids observed. To account for multiple comparisons, false discovery rate adjusted q values < 0.05 were considered significant. Data distributions were visually assessed by inspection of residual and histogram plots. Non-normally distributed data were cubed-root transformed prior to analysis by Gaussian-distribution GLMs with the identity link. Binominal-distribution GLMs with the logistic link were alternatively applied for HOMA-IR, as percentage change data were binomially distributed.
Adjusting for potential confounding effects
ANCOVA models were used to adjust for covariates that may have confounded the observed clinical effects. Multivariate GLMs were alternatively used to adjust for covariates that may have confounded the associations detected between surrogate endpoints and microbiome markers. Due to limitations in statistical power (small sample size), separate ANCOVA models and GLMs were performed for each covariate. Statistical significance was considered at p < 0.05 as relatively few comparisons were made.
Availability of data and materials
The 16S rRNA gene amplicon sequencing data were deposited in the NCBI Sequence Read Archive and are available for download under BioProjects PRJNA564636 (fecal) and PRJNA630848 (ex vivo). Remaining deidentified individual participant data, methods, and study materials will be made available from the corresponding authors upon reasonable request.
Abbreviations
- ANCOVA:
-
Analysis of covariance
- ASV:
-
Amplicon sequence variant
- AUC-ROC:
-
Area under the receiver operating characteristic curve
- AX:
-
Arabinoxylan
- BMI:
-
Body mass index
- BONCAT:
-
Bioorthogonal non-canonical amino acid tagging
- CV:
-
Intra-assay coefficient of variation
- ECLIA:
-
Electrochemiluminescence immunoassay
- ELISA:
-
Enzyme-linked immunosorbent assay
- FACS:
-
Fluorescence-activated cell sorting
- FMT:
-
Fecal microbiota transplantation
- GDCA:
-
Glycodeoxycholic acid
- GLM:
-
Generalized linear model
- HOMA-IR:
-
Homeostatic model assessment of insulin resistance
- ILCA:
-
Isolithocholic acid
- LCA:
-
Lithocholic acid
- MCC:
-
Microcrystalline cellulose
- PBS:
-
Phosphate-buffered saline
- QUICKI:
-
Quantitative insulin sensitivity check index
- RFC:
-
Random forest classifier
- SCFA:
-
Short-chain fatty acid
- SLIM:
-
Satiety Labeled Intensity Magnitude
- TDCA:
-
Taurodeoxycholic acid
- TLCA:
-
Taurolithocholic acid
- TMA:
-
Trimethylamine
- TMAO:
-
Trimethylamine N-oxide
- TNF-α:
-
Tumor necrosis factor-α
References
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–81.
Reynolds A, Mann J, Cummings J, Winter N, Mete E, Te Morenga L. Carbohydrate quality and human health: a series of systematic reviews and meta-analyses. Lancet. 2019;393:434–45.
AlEssa HB, Bhupathiraju SN, Malik VS, Wedick NM, Campos H, Rosner B, et al. Carbohydrate quality and quantity and risk of type 2 diabetes in US women. Am J Clin Nutr. 2015;102:1543–53.
Kaczmarczyk MM, Miller MJ, Freund GG. The health benefits of dietary fiber: beyond the usual suspects of type 2 diabetes mellitus, cardiovascular disease and colon cancer. Metabolism. 2012;61:1058–66.
Deehan EC, Duar RM, Armet AM, Perez-Muñoz ME, Jin M, Walter J. Modulation of the gastrointestinal microbiome with nondigestible fermentable carbohydrates to improve human health. Microbiol Spectr. 2017;5:1–24.
McRorie JWJ, McKeown NM. Understanding the physics of functional fibers in the gastrointestinal tract: an evidence-based approach to resolving enduring misconceptions about insoluble and soluble fiber. J Acad Nutr Diet. 2017;117:251–64.
Capuano E. The behavior of dietary fiber in the gastrointestinal tract determines its physiological effect. Crit Rev Food Sci Nutr. 2017;57:3543–64.
Jovanovski E, Khayyat R, Zurbau A, Komishon A, Mazhar N, Sievenpiper JL, et al. Should viscous fiber supplements be considered in diabetes control? Results from a systematic review and meta-analysis of randomized controlled trials. Diabetes Care. 2019;42:755–66.
Vuksan V, Jenkins AL, Rogovik AL, Fairgrieve CD, Jovanovski E, Leiter LA. Viscosity rather than quantity of dietary fibre predicts cholesterol-lowering effect in healthy individuals. Br J Nutr. 2011;106:1349–52.
EFSA Panel on Dietetic Products Nutrition and Allergies. Scientific Opinion on the substantiation of health claims related to arabinoxylan produced from wheat endosperm and reduction of post-prandial glycaemic responses (ID 830) pursuant to Article 13(1) of Regulation (EC) No 1924/2006. EFSA J. 2011;9:2205.
King DE, Mainous AG, Lambourne CA. Trends in dietary fiber intake in the United States, 1999-2008. J Acad Nutr Diet. 2012;112:642–68.
Deehan EC, Walter J. The fiber gap and the disappearing gut microbiome: implications for human nutrition. Trends Endocrinol Metab. 2016;27:239–42.
Armet AM, Deehan EC, Thöne JV, Hewko SJ, Walter J. The effect of isolated and synthetic dietary fibers on markers of metabolic diseases in human intervention studies: a systematic review. Adv Nutr. 2020;11:420–38.
Augustin LS, Aas A-M, Astrup A, Atkinson FS, Baer-Sinnott S, Barclay AW, et al. Dietary fibre consensus from the International Carbohydrate Quality Consortium (ICQC). Nutrients. 2020;12:2553.
Makki K, Deehan EC, Walter J, Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. 2018;23:705–15.
Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19:55–71.
Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020;20:40–54.
Tuncil YE, Thakkar RD, Arioglu-Tuncil S, Hamaker BR, Lindemann SR. Subtle variations in dietary-fiber fine structure differentially influence the composition and metabolic function of gut microbiota. mSphere. 2020;5:e00180–20.
Deehan EC, Yang C, Perez-Muñoz ME, Nguyen NK, Cheng CC, Triador L, et al. Precision microbiome modulation with discrete dietary fiber structures directs short-chain fatty acid production. Cell Host Microbe. 2020;27:389–404.e6.
Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, et al. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 2015;22:971–82.
Martínez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013;7:269–80.
Blaak EE, Canfora EE, Theis S, Frost G, Groen AK, Mithieux G, et al. Short chain fatty acids in human gut and metabolic health. Benef Microbes. 2020;11:411–55.
Winston JA, Theriot CM. Diversification of host bile acids by members of the gut microbiota. Gut Microbes. 2020;11:158–71.
Chiang JY, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr. 2019;39:175–200.
Holscher HD. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes. 2017;8:172–84.
Dewulf EM, Cani PD, Claus SP, Fuentes S, Puylaert PGB, Neyrinck AM, et al. Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut. 2013;62:1112–21.
Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359:1151–6.
Fechner A, Kiehntopf M, Jahreis G. The formation of short-chain fatty acids is positively associated with the blood lipid-lowering effect of lupin kernel fiber in moderately hypercholesterolemic adults. J Nutr. 2014;144:599–607.
Nicolucci AC, Hume MP, Martinez I, Mayengbam S, Walter J, Reimer RA. Prebiotic reduces body fat and alters intestinal microbiota in children with overweight or obesity. Gastroenterology. 2017;153:711–22.
Walter J, Armet AM, Finlay BB, Shanahan F. Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell. 2020;180:221–32.
Murga-Garrido SM, Hong Q, Cross T-WL, Hutchison ER, Han J, Thomas SP, et al. Gut microbiome variation modulates the effects of dietary fiber on host metabolism. Microbiome. 2021;9:117.
Bindels LB, Segura Munoz RR, Gomes-Neto JC, Mutemberezi V, Martinez I, Salazar N, et al. Resistant starch can improve insulin sensitivity independently of the gut microbiota. Microbiome. 2017;5:12.
Nguyen NK, Deehan EC, Zhang Z, Jin M, Baskota N, Perez-Muñoz ME, et al. Gut microbiota modulation with long-chain corn bran arabinoxylan in adults with overweight and obesity is linked to an individualized temporal increase in fecal propionate. Microbiome. 2020;8:118.
Kale MS, Yadav MP, Hicks KB, Hanah K. Concentration and shear rate dependence of solution viscosity for arabinoxylans from different sources. Food Hydrocoll. 2015;47:178–83.
Rumpagaporn P, Reuhs BL, Kaur A, Patterson JA, Keshavarzian A, Hamaker BR. Structural features of soluble cereal arabinoxylan fibers associated with a slow rate of in vitro fermentation by human fecal microbiota. Carbohydr Polym. 2015;130:191–7.
Sandberg J, Kovatcheva-Datchary P, Bjorck I, Bäckhed F, Nilsson A. Abundance of gut Prevotella at baseline and metabolic response to barley prebiotics. Eur J Nutr. 2018;58:2365–76.
Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SEK, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut. 2015;64:1744–54.
Chambers ES, Byrne CS, Morrison DJ, Murphy KG, Preston T, Tedford C, et al. Dietary supplementation with inulin-propionate ester or inulin improves insulin sensitivity in adults with overweight and obesity with distinct effects on the gut microbiota, plasma metabolome and systemic inflammatory responses: a randomised cross-over trial. Gut. 2019;68:1430–8.
Riva A, Kolimár D, Spittler A, Wisgrill L, Herbold CW, Abrankó L, et al. Conversion of rutin, a prevalent dietary flavonol, by the human gut microbiota. Front Microbiol. 2020;11:585428.
Kahleova H, Tura A, Hill M, Holubkov R, Barnard ND. A plant-based dietary intervention improves beta-cell function and insulin resistance in overweight adults: a 16-week randomized clinical trial. Nutrients. 2018;10:189.
Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. 2019;25:1096–103.
Mocanu V, Zhang Z, Deehan EC, Kao DH, Hotte N, Karmali S, et al. Fecal microbial transplantation and fiber supplementation in patients with severe obesity and metabolic syndrome: a randomized double-blind, placebo-controlled phase 2 trial. Nat Med. 2021;27:1272–9.
Ballou S, Singh P, Rangan V, Iturrino J, Nee J, Lembo AJ. Obesity is associated with significantly increased risk for diarrhoea after controlling for demographic, dietary and medical factors: a cross-sectional analysis of the 2009-2010 National Health and Nutrition Examination Survey. Aliment Pharmacol Ther. 2019;50:1019–24.
Tannock GW, Liu Y. Guided dietary fibre intake as a means of directing short-chain fatty acid production by the gut microbiota. J R Soc N Z. 2020;50:434–55.
Rivière A, Gagnon M, Weckx S, Roy D, De Vuyst L. Mutual cross-feeding interactions between Bifidobacterium longum subsp. longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan oligosaccharides. Appl Environ Microbiol. 2015;81:7767–81.
Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41.
Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–4.
Hofmann AF, Mysels KJ. Bile acid solubility and precipitation in vitro and in vivo: the role of conjugation, pH, and Ca2+ ions. J Lipid Res. 1992;33:617–26.
Delzenne NM, Olivares M, Neyrinck AM, Beaumont M, Kjolbaek L, Larsen TM, et al. Nutritional interest of dietary fiber and prebiotics in obesity: Lessons from the MyNewGut consortium. Clin Nutr. 2020;39:414–24.
Hartvigsen ML, Gregersen S, Laerke HN, Holst JJ, Bach Knudsen KE, Hermansen K. Effects of concentrated arabinoxylan and β-glucan compared with refined wheat and whole grain rye on glucose and appetite in subjects with the metabolic syndrome: a randomized study. Eur J Clin Nutr. 2014;68:84–90.
Brum JM, Gibb RD, Peters JC, Mattes RD. Satiety effects of psyllium in healthy volunteers. Appetite. 2016;105:27–36.
Isaksson H, Sundberg B, Åman P, Fredriksson H, Olsson J. Whole grain rye porridge breakfast improves satiety compared to refined wheat bread breakfast. Food Nutr Res. 2008;52:1809.
Cioffi I, Ibrugger S, Bache J, Thomassen MT, Contaldo F, Pasanisi F, et al. Effects on satiation, satiety and food intake of wholegrain and refined grain pasta. Appetite. 2016;107:152–8.
Hartvigsen ML, Laerke HN, Overgaard A, Holst JJ, Bach Knudsen KE, Hermansen K. Postprandial effects of test meals including concentrated arabinoxylan and whole grain rye in subjects with the metabolic syndrome: a randomised study. Eur J Clin Nutr. 2014;68:567–74.
Lu ZX, Walker KZ, Muir JG, Mascara T, O'Dea K. Arabinoxylan fiber, a byproduct of wheat flour processing, reduces the postprandial glucose response in normoglycemic subjects. Am J Clin Nutr. 2000;71:1123–8.
Garcia AL, Otto B, Reich S-C, Weickert MO, Steiniger J, Machowetz A, et al. Arabinoxylan consumption decreases postprandial serum glucose, serum insulin and plasma total ghrelin response in subjects with impaired glucose tolerance. Eur J Clin Nutr. 2007;61:334–41.
Nagy-Szakal D, Hollister EB, Luna RA, Szigeti R, Tatevian N, Smith CW, et al. Cellulose supplementation early in life ameliorates colitis in adult mice. PLoS One. 2013;8:e56685.
Kim Y, Hwang SW, Kim S, Lee Y-S, Kim T-Y, Lee S-H, et al. Dietary cellulose prevents gut inflammation by modulating lipid metabolism and gut microbiota. Gut Microbes. 2020;11:944–61.
Di Caro V, Alcamo AM, Cummings JL, Clark RSB, Novak EA, Mollen KP, et al. Effect of dietary cellulose supplementation on gut barrier function and apoptosis in a murine model of endotoxemia. PLoS One. 2019;14:e0224838.
Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–16.
Zeng H, Umar S, Rust B, Lazarova D, Bordonaro M. Secondary bile acids and short chain fatty acids in the colon: a focus on colonic microbiome, cell proliferation, inflammation, and cancer. Int J Mol Sci. 2019;20:1214.
Ocvirk S, O'Keefe SJ. Influence of bile acids on colorectal cancer risk: potential mechanisms mediated by diet - gut microbiota interactions. Curr Nutr Rep. 2017;6:315–22.
Müller M, Hermes GD, Canfora EE, Holst JJ, Zoetendal EG, Smidt H, et al. Effect of wheat bran derived prebiotic supplementation on gastrointestinal transit, gut microbiota, and metabolic health: a randomized controlled trial in healthy adults with a slow gut transit. Gut Microbes. 2020;12:1704141.
Liu F, Li P, Chen M, Luo Y, Prabhakar M, Zheng H, et al. Fructooligosaccharide (FOS) and galactooligosaccharide (GOS) increase Bifidobacterium but reduce butyrate producing bacteria with adverse glycemic metabolism in healthy young population. Sci Rep. 2017;7:11789.
Canfora EE, van der Beek CM, Hermes GDA, Goossens GH, Jocken JWE, Holst JJ, et al. Supplementation of diet with galacto-oligosaccharides increases Bifidobacteria, but not insulin sensitivity, in obese prediabetic individuals. Gastroenterology. 2017;153:87–97.e3.
Wong CB, Odamaki T, Xiao J-z. Beneficial effects of Bifidobacterium longum subsp. longum BB536 on human health: modulation of gut microbiome as the principal action. J Funct Foods. 2019;54:506–19.
McRorie J. Clinical data support that psyllium is not fermented in the gut. Am J Gastroenterol. 2013;108:1541.
Müller M, Hernández MAG, Goossens GH, Reijnders D, Holst JJ, Jocken JWE, et al. Circulating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and GLP-1 concentrations in humans. Sci Rep. 2019;9:12515.
Gurry T, Nguyen LTT, Yu X, Alm EJ. Functional heterogeneity in the fermentation capabilities of the healthy human gut microbiota. PLoS One. 2021;16:e0254004.
Bélanger M, Poirier M, Jbilou J, Scarborough P. Modelling the impact of compliance with dietary recommendations on cancer and cardiovascular disease mortality in Canada. Public Health. 2014;128:222–30.
Kivimäki M, Kuosma E, Ferrie JE, Luukkonen R, Nyberg ST, Alfredsson L, et al. Overweight, obesity, and risk of cardiometabolic multimorbidity: pooled analysis of individual-level data for 120 813 adults from 16 cohort studies from the USA and Europe. Lancet Public Health. 2017;2:e277–e85.
Kirkpatrick SI, Subar AF, Douglass D, Zimmerman TP, Thompson FE, Kahle LL, et al. Performance of the automated self-administered 24-hour recall relative to a measure of true intakes and to an interviewer-administered 24-h recall. Am J Clin Nutr. 2014;100:233–40.
Willett W, Stampfer MJ. Total energy intake: implications for epidemiologic analyses. Am J Epidemiol. 1986;124:17–27.
Cardello AV, Schutz HG, Lesher LL, Merrill E. Development and testing of a labeled magnitude scale of perceived satiety. Appetite. 2005;44:1–13.
Engels JM, Diehr P. Imputation of missing longitudinal data: a comparison of methods. J Clin Epidemiol. 2003;56:968–76.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402–10.
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502.
Seethaler B, Basrai M, Neyrinck AM, Nazare J-A, Walter J, Delzenne NM, et al. Biomarkers for assessment of intestinal permeability in clinical practice. Am J Physiol Gastrointest Liver Physiol. 2021;321:G11–7.
Mi S, Zhao Y-Y, Jacobs RL, Curtis JM. Simultaneous determination of trimethylamine and trimethylamine N-oxide in mouse plasma samples by hydrophilic interaction liquid chromatography coupled to tandem mass spectrometry. J Sep Sci. 2017;40:688–96.
Han J, Liu Y, Wang R, Yang J, Ling V, Borchers CH. Metabolic profiling of bile acids in human and mouse blood by LC-MS/MS in combination with phospholipid-depletion solid-phase extraction. Anal Chem. 2015;87:1127–36.
de Carvalho NM, Oliveira DL, Dib Saleh MA, Pintado M, Madureira AR. Preservation of human gut microbiota inoculums for in vitro fermentations studies. Fermentation. 2021;7:14.
Hatzenpichler R, Scheller S, Tavormina PL, Babin BM, Tirrell DA, Orphan VJ. In situ visualization of newly synthesized proteins in environmental microbes using amino acid tagging and click chemistry. Environ Microbiol. 2014;16:2568–90.
Jin M, Kalainy S, Baskota N, Chiang D, Deehan EC, McDougall C, et al. Faecal microbiota from patients with cirrhosis has a low capacity to ferment non-digestible carbohydrates into short-chain fatty acids. Liver Int. 2019;39:1437–47.
Slavin JL. Fiber and prebiotics: mechanisms and health benefits. Nutrients. 2013;5:1417–35.
Lockyer S, Stanner S. Prebiotics – an added benefit of some fibre types. Nutr Bull. 2019;44:74–91.
Herbold CW, Pelikan C, Kuzyk O, Hausmann B, Angel R, Berry D, et al. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front Microbiol. 2015;6:731.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Laros JF. demultiplex. 2020. version 1.1.0. https://github.com/jfjlaros/demultiplex.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Wolever TMS, van Klinken BJ-W, Spruill SE, Jenkins AL, Chu Y, Harkness L. Effect of serving size and addition of sugar on the glycemic response elicited by oatmeal: a randomized, cross-over study. Clin Nutr ESPEN. 2016;16:48–54.
Kassambara A, Mundt F. factoextra: extract and visualize the results of multivariate data analyses. 2017. R-package version 1.0.5. https://CRAN.R-project.org/package=factoextra.
Lê S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Stat Softw. 2008;25:1–18.
Brito IL, Gurry T, Zhao S, Huang K, Young SK, Shea TP, et al. Transmission of human-associated microbiota along family and social networks. Nat Microbiol. 2019;4:964–71.
Oksanen J, Blanchet GF, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara R, Simpson GL, Solymos P et al. vegan: community ecology package. 2019. R-package version 2.5-5. https://CRAN.R-project.org/package=vegan.
Frossard J, Renaud O. permuco: Permutation tests for regression, (repeated measures) ANOVA/ANCOVA and comparison of signals. 2019. R-package version 1.1.0. https://CRAN.R-project.org/package=permuco.
Wheeler B, Torchiano M. lmPerm: permutation tests for linear models. 2016. R-package version 2.1.0. https://CRAN.R-project.org/package=lmPerm.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Chen X, Ishwaran H. Random forests for genomic data analysis. Genomics. 2012;99:323–9.
Knights D, Costello EK, Knight R. Supervised classification of human microbiota. FEMS Microbiol Rev. 2011;35:343–59.
Xia J, Broadhurst DI, Wilson M, Wishart DS. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics. 2013;9:280–99.
Liaw A, Wiener M. Classification and regression by randomforest. R News. 2002;2:18–22.
Maldonado-Gómez MX, Martínez I, Bottacini F, O'Callaghan A, Ventura M, van Sinderen D, et al. Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell Host Microbe. 2016;20:515–26.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77.
Acknowledgements
We thank AgriFiber Solutions LLC (Illinois, USA) for providing AGRIFIBER SFC, and Blanver Farmoquimica LTDA (São Paulo, Brazil) for providing MICROCEL MC-12. We also thank Petra Pjevac, Jasmin Schwarz, and Gudrun Kohl from the Joint Microbiome Facility (Vienna, Austria) for supporting the ex vivo microbiota analysis.
Funding
This study was co-funded by a joint European Research Area and Joint Programming Initiative-A Healthy Diet for a Healthy Life (ERA-HDHL) grant (Biomarkers for Nutrition and Health; The FiberTAG Project; www.healthydietforhealthylife.eu/) with specific contributions from the Canadian Institutes of Health Research (RN-298871-372173). The ex vivo microbiota analysis was supported by the European Research Council (Starting Grant: FunKeyGut 741623). ECD acknowledges support from the Queen Elizabeth II Graduate Scholarship, Elizabeth Russell MacEachran Scholarship, and Anthony Fellowship in Human Nutrition. JW is funded in part by a Science Foundation Ireland Centre grant to APC Microbiome Ireland (APC/SFI/12/RC/2273_P2) and a Science Foundation Ireland Professorship (19/RP/6853). The funding sources had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
Conceptualization: ECD and JW; Methodology: ECD, ZZ, JC, CMP, and JW; Funding Acquisition: ECD, JC, JAN, NMD, SCB, CMP, and JW; Clinical Investigation: ECD, JC, and CMP; Perceived Satiety Assessment: ECD, JC, and WVW; Surrogate Endpoint Assessment: ECD, MEPM, JAK, JC, SDP, and CJF; Cytokine and Hormonal Measures: ECD, MEPM, and CJF; Barrier Function Measures: ECD, ZZ, BS, and SCB; TMAO Quantification: YZ and JMC; Fecal Microbiota Analysis: NKN and MEPM; ex vivo Microbiota Analysis: AR, BH, AS, and DB; Statistical Analysis: ECD, ZZ, FL, JAB, DK, and JW; Machine Learning: ZZ and DK; Visualization: ECD and ZZ; Project Administration: ECD; Supervision: CMP and JW; Writing—Original Draft: ECD, ZZ, AMA, and JW; All authors commented on and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from each participant in the study prior to enrollment. The study protocol, including the separation of the AX arm from the original clinical trial, was approved by the University of Alberta Health Research Ethics Board (Pro00050274).
Consent for publication
Not applicable.
Competing interests
Since January 2021, ECD has been an employee of AgriFiber Solutions LLC, although all statistical analyses and manuscript preparations were completed before employment. JW has received research funding and consulting fees from industry sources involved in the manufacture and marketing of dietary fibers, including AgriFiber Solutions LLC. JW is further a co-owner of Synbiotics Health, a developer of synbiotic products. These interests did not influence his judgement or presentation of study findings. All other authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Adherence to the study protocol as estimated by the average amount (weight) of dietary fiber consumed during the intervention period.
Additional file 2: Table S1.
Baseline clinical measurements. Table provides the anthropometric measurements, surrogate endpoints, and biomarkers of host-microbiota interactions assessed at baseline, with participant grouped by arabinoxylan or microcrystalline cellulose supplementation. Data provided as mean ± SD.
Additional file 3: Table S2.
Macronutrient consumption at baseline and during arabinoxylan or microcrystalline cellulose supplementation. Table provides total dietary calories and macronutrients as assessed by the Canadian version of the web-based Automated Self-Administered 24-hour Dietary Assessment Tool, with participant grouped by arabinoxylan or microcrystalline cellulose supplementation. Data provided as mean ± SD.
Additional file 4: Table S3.
Clinical measurements at baseline and six weeks of arabinoxylan or microcrystalline cellulose supplementation. Table provides the anthropometric measurements, surrogate endpoints, biomarkers of host-microbiota interactions, and stool characteristics assessed, with participant grouped by arabinoxylan or microcrystalline cellulose supplementation. Data provided as mean ± SD.
Additional file 5: Table S4.
Covariate-adjustment of dietary fiber treatment effects by analysis of covariance (ANCOVA). Table provides p values from separate ANCOVA models used to adjust for the following covariates: age, sex, total dietary fiber intake, total dietary sugar intake, stool consistency, and bowel movement frequency.
Additional file 6: Table S5.
Fecal concentrations of remaining bile acids detected at baseline and six weeks of arabinoxylan or microcrystalline cellulose supplementation. Table provides the remaining 29 bile acid compounds that were detected in < 90% of fecal samples, with participant grouped by arabinoxylan or microcrystalline cellulose supplementation.
Additional file 7: Figure S2.
Sorting of AX-stimulated bacterial cells by FACS on a MoFlow Astrios EQ cell sorter. As shown in the dot plots, A background noise of the machine was detected using FSC and SSC parameters. B Bacterial cells were measured in the same setting and pre-gated. C An example of Cy5-negative cells is presented in the dot plot showing the Cy5 channel via the SSC channel. D An example of Cy5-positive fluorescent cells (activated by AX) that were gated and sorted out by FACS. AX, arabinoxylan; FACS, fluorescence-activated cell sorting; FSC, forward scatter; SSC, side scatter.
Additional file 8: Table S6.
Relative abundance of bacterial taxa activated through ex vivo incubation with arabinoxylan and recovered by fluorescence-activated cell sorting (FACS). Table provides α-diversity and the bacterial taxa that were activated at 6-h incubation with arabinoxylan, recovered by FACS, showed a mean relative abundance > 1%, and/or showed a differential abundance in the recovered consortia relative to the total fecal communities. Data provided as mean ± SD.
Additional file 9: Figure S3.
Confirmation of gut microbiota compositional features and mechanistic biomarkers that predict clinical responses. Line graphs show differences in the effects of A AX on perceived satiety after a meal and HOMA-IR and B MCC on fecal calprotectin and TNF-α for high and low responders, as defined according to the study cohort median. AUC-ROC values show the performance accuracy of random forest classifiers for predicting high-vs-low responders in C AX-induced perceived satiety after a meal and HOMA-IR attenuation, D MCC-induced fecal calprotectin and TNF-α attenuation, and E AX and MCC induced changes in HOMA-IR, fecal calprotectin, and TNF-α. High and low responders were defined according to the study cohort median. Black cells denote OOB error rates ≥ 0.6. Prediction performance of random forest classifiers trained to predict high-vs-low responders in AX-induced F and G satiety after a meal and H HOMA-IR attenuation, and MCC-induced I fecal calprotectin and J TNF-α attenuation. OOB shows the mean prediction error of the random forests model with boosted decision trees (n = 500). The confusion matrix shows subgroup prediction accuracy, where row i and column j indicates the number of subjects predicted as i but were actually classified as j. Error rates indicate the percentage of incorrect classifications. ∆, absolute change from baseline to week 6; %∆, percent change from baseline to week 6; ASV, amplicon sequence variant; AX, arabinoxylan; AUC-ROC, area under the receiver operating characteristic curve; HOMA-IR, homeostatic model assessment of insulin resistance; MCC, microcrystalline cellulose; OTU, operational taxonomic unit; OOB: out-of-bag; SLIM, Satiety Labeled Intensity Magnitude; TNF-α, tumor necrosis factor-α.
Additional file 10: Table S7.
Univariate and covariate-adjusted generalized linear models (GLMs) assessing fecal microbiota-related factors that associate with surrogate endpoints of dietary fiber supplementation. Table provides β-coefficient directionality and p values from separate GLMs used to adjust for the following covariates: age, sex, total dietary fiber intake, total dietary sugar intake, stool consistency, and bowel movement frequency.
Additional file 11: Figure S4.
Scatter plots show Spearman’s correlations between shifts in A and B fecal pH and C and D fecal moisture content and changes in the fecal concentrations of bile acids shown to predict the arabinoxylan and microcrystalline cellulose induced reductions in HOMA-IR and fecal calprotectin, respectively. Statistical significance was set at FDR adjusted q values < 0.05. GDCA, glycodeoxycholic acid; HOMA-IR, homeostatic model assessment of insulin resistance; ILCA, isolithocholic acid; LCA, lithocholic acid; TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid.
Additional file 12: Figure S5.
Associations between the effects on perceived satiety and surrogate endpoints and the dominant fecal microbiota features affected by fiber supplementation. Heatmap shows cubed-root transformed β-coefficients of univariate generalized linear models performed on the compositional (dominant AX-responsive taxa at baseline, shifts, and ex vivo) and functional (fecal propionate and bile acid shifts) features of the gut microbiota. Statistical significance was considered at FDR corrected q values < 0.05. ∆, absolute change from baseline to week 6; %∆, percent change from baseline to week 6; 7αOHCA; 7αOH-3-oxo-4-cholestenoic acid; ApoCA; apocholic acid; ASV, amplicon sequence variant; AUC, area under the curve; AX, arabinoxylan; BL, baseline; HDCA, hyodeoxycholic acid; HOMA-IR, homeostatic model assessment of insulin resistance; MCC, microcrystalline cellulose; OTU, operational taxonomic unit; TNF-α, tumor necrosis factor-α.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Deehan, E.C., Zhang, Z., Riva, A. et al. Elucidating the role of the gut microbiota in the physiological effects of dietary fiber. Microbiome 10, 77 (2022). https://doi.org/10.1186/s40168-022-01248-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-022-01248-5