
Abstract
Background
Feed efficiency is a crucial parameter in swine production, given both its economic and environmental impact. The gut microbiota plays an essential role in nutrient digestibility and is, therefore, likely to affect feed efficiency. This study aimed to characterize feed efficiency, fatness traits, and gut microbiome composition in three major breeds of domesticated swine and investigate a possible link between feed efficiency and gut microbiota composition.
Results
Average daily feed intake (ADFI), average daily gain (ADG), feed conversion ratio (FCR), residual feed intake (RFI), backfat, loin depth, and intramuscular fat of 615 pigs belonging to the Duroc (DR), Landrace (LR), and Large White (LW) breeds were measured. Gut microbiota composition was characterized by 16S rRNA gene sequencing. Orthogonal contrasts between paternal line (DR) and maternal lines (LR+LW) and between the two maternal lines (LR versus LW) were performed. Average daily feed intake and ADG were statistically different with DR having lower ADFI and ADG compared to LR and LW. Landrace and LW had a similar ADG and RFI, with higher ADFI and FCR for LW. Alpha diversity was higher in the fecal microbial communities of LR pigs than in those of DR and LW pigs for all time points considered. Duroc communities had significantly higher proportional representation of the Catenibacterium and Clostridium genera compared to LR and LW, while LR pigs had significantly higher proportions of Bacteroides than LW for all time points considered. Amplicon sequence variants from multiple genera (including Anaerovibrio, Bacteroides, Blautia, Clostridium, Dorea, Eubacterium, Faecalibacterium, Lactobacillus, Oscillibacter, and Ruminococcus) were found to be significantly associated with feed efficiency, regardless of the time point considered.
Conclusions
In this study, we characterized differences in the composition of the fecal microbiota of three commercially relevant breeds of swine, both over time and between breeds. Correlations between different microbiome compositions and feed efficiency were established. This suggests that the microbial community may contribute to shaping host productive parameters. Moreover, our study provides important insights into how the intestinal microbial community might influence host energy harvesting capacity. A deeper understanding of this process may allow us to modulate the gut microbiome in order to raise more efficient animals.
Video Abstract
Similar content being viewed by others


Background
Feed efficiency is an essential trait in pig production since feed accounts for 50 to 85% of pork producers’ costs [1, 2]. Feed conversion ratio and residual feed intake are two traits that have been routinely used to evaluate feed efficiency [3]. An animal with a low feed conversion ratio and low residual feed intake consumes less feed per unit of body weight than expected and is considered efficient, whereas an animal with a high feed conversion ratio and residual feed intake consumes more feed than expected and is considered inefficient [3]. Increasing a pig’s feed efficiency can thus decrease the total feed it consumes, reducing a farm’s costs and energy use [4]. Most efforts to optimize feed efficiency in pigs to date have focused on host genetics, management practices, and diet [5, 6]. Despite these factors that influence feed efficiency, little is known about the relationship between feed efficiency and different breeds of pigs. Do et al. (2013) [7] discovered breed differences in heritability estimates of feed efficiency as well as variations in their phenotypic and genetic correlations among Duroc, Landrace, and Large White.
The gut microbiota is a complex system that plays an important role in health and immunity in all mammals [8]. It is comprised of diverse populations of bacteria and other microorganisms whose abundances are impacted by both environmental and host genetic factors [9]. Recently, studies have demonstrated associations of microbial profiles with nutrition and productivity parameters [10,11,12]. Notably, the gut microbiota metabolizes various food components, providing nutrients to the host in the form of fermentation end-products and other by-products, amino acids, vitamins, and indole derivatives [13]. In the context of swine feed efficiency, the gut microbiota plays important roles in nutrient uptake, energy harvest, and carbohydrate metabolism, particularly in processing indigestible polysaccharides [14, 15]. Recent studies have reported that the composition and alpha diversity of the pig gut microbiota are correlated with nutrient digestibility, average daily gain, and body weight [11, 16]. Variation in the gut microbiome has also been associated with life stage [9, 17]. However, to the best of our knowledge, only a few studies have reported the effect of different microbial populations on feeding efficiency of different breeds. Singh et al. [18] reported a correlation between gut microbiota diversity and feed efficiency, while Tan et al. [19] identified differences in the microbiomes of pigs with high and low feed efficiencies. The genera Bacteroides, Cellulosilyticum, and Prevotella, were more abundant in low feed efficiency pigs, and Oscillibacter and Rhodococcus were found in animals that were more feed efficient [19, 20].
Duroc pigs are often used in breeding programs as the terminal sires in three-way crosses with Landrace × Large White or Large White × Landrace sows. The greater relative selection emphasis on feed efficiency and growth in Duroc compared with Landrace and Large White might have inadvertently selecting for more efficient microbiomes. Previous studies have reported gut microbiome differences when comparing hosts with different genetic backgrounds [21, 22]. This may partially explain why the association between the gut microbiome and feed efficiency shows low repeatability among studies [18,19,20]. In addition, diet has been reported to be a principal factor affecting gut microbiota composition [23]. The objectives of this study were (i) to characterize differences in feed efficiency, growth, and fatness traits between three different commercial breeds of pigs; (ii) to study how the composition of the gut microbiome within and between breeds changes as animals grow; and (iii) to investigate whether there is an association between gut microbiome composition and feed efficiency in swine.
Results
Duroc pigs are currently used in breeding programs as paternal lines, while crosses between Landrace and Large White pigs are used as maternal lines. The different aims of selection for paternal and maternal lines might have significantly shaped feed efficiency and gut microbiome composition. For this reason, to investigate the differences between paternal and maternal lines and between the two maternal lines with the minimum number of orthogonal contrasts, we performed the comparison between Duroc and the average of Landrace and Large White [DR vs. (LR and LW)/2] and Landrace versus Large White (LR vs. LW). In this study, the “Results” section was divided into four parts. In the first part, we reported differences between breeds in terms of feed efficiency and fatness traits. In the second part, gut microbiome differences between breeds at each time point were showed. In the third part, the effects of amplicon sequence variants (ASV) and the interaction between ASV and breed on feed efficiency were reported separately at the three time points. Lastly, the associations between gut microbiome and feed efficiency were presented.
Feed efficiency and fatness traits in the Duroc, Landrace, and Large White breeds
Table 1 and Additional file 1 summarizes descriptive statistics and breed difference estimates for average daily feed intake (ADFI), average daily gain (ADG), feed efficiency, and fatness traits. Average daily feed intake, ADG, back fat, and loin depth were all lower in Duroc (DR) pigs than the average of Landrace (LR) and Large White (LW) animals. Landrace and LW pigs were similar in terms of ADG, residuals calculated by regressing ADFI on ADG (RF1), residuals calculated by regressing ADFI on ADG and body weight (RF2), and intramuscular fat (IMF); however, LW pigs had significantly higher ADFI, feed conversion ratio (FCR), and backfat.
Least squares means for ADFI, ADG, feed conversion ratio, residual feed intake 1 (RF1), and residual feed intake 2 (RF2) in each of the three breeds across three time points (73, 123, and 158 days) are reported in Fig. 1. Significant (P < 0.05) differences were observed. Across time points, ADG for the three breeds rang

Average daily gain and feed efficiency (least squares mean ± confidence interval) of Duroc (DR), Landrace (LR), and Large White (LW) at three time points 73 days (T1), 123 days (T2), and 158 days (T3) in the growth trial
ed from 0.48 to 0.77 kg/d. At 123 days, ADG was significantly higher in LW (0.68 ± 0.058 kg/d) than DR (0.64 ± 0.057 kg/d) and LR (0.65 ± 0.055 kg/d), while no statistically significant differences were observed at 73 and 158 days. Average daily feed intake ranged from 1503 to 2795 g/d across time points. Average daily feed intake was significantly higher in LW at 123 and 158 days than in DR and LR, while no significant differences were observed at 73 days. The FCR of the three breeds on average ranged from 3.21 to 3.80 across time points. Feed conversion ratio was significantly lower in LR at 73 and 123 days compared to DR and LW. With regard to the residual feed intake, RF1 ranged from − 205 to 95, while RF2 ranged from − 226 to 136 across time points. These significant differences between breeds were confirmed by linear discriminant analysis (LDA) (Fig. 2). The first component explained 93.4% and the second 6.6% of the total variance. The first component differentiated DR and LR from LW based on FCR (loading 10.4) and IMF (loading − 0.18), while the second component discriminated DR and LR based on FCR (loading 8.4) and backfat (loading − 0.25).

Linear discriminant analysis (LDA) of feed efficiency and fatness trait data from three breeds of pig. Scores [a] and loadings [b] are reported
Gut microbiome composition in the Duroc, Landrace, and Large White breeds
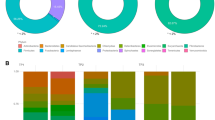
Figure 3 illustrates the relative abundance of microbial ASV when aggregated at the family level for the three breeds and the three time points surveyed in this study. Over the three time points, about 80% of ASV were classified into just 7 families: Lactobacillaceae, Clostridiaceae, Streptococcaceae, Prevotellaceae, Ruminococcaceae, Eubacteriaceae, and Lachnospiraceae.

Relative abundance of microbiome taxa at family level of Duroc (DR), Landrace (LR), and Large White (LW) in three time points 73 days (T1), 123 days (T2), and 158 days (T3) of the feeding trial
Alpha diversity of pig gut microbiome was measured using the Shannon, Simpson, and Inverse Simpson indices (Fig. 4). On average, the alpha diversity across time points ranged from 4.05 to 4.43, from 0.93 to 0.95, and from 18.7 to 27.2 for the Shannon, Simpson, and Inverse Simpson indices, respectively. Comparing the breeds across time points, Shannon index values were significantly (P < 0.05) higher in LR than in DR and LW at 123 days. Duroc pigs had lower Shannon index values than LR and LW at 73 days, while no statistically differences were observed at 123 and 158 days. Simpson index values were lower in DR than in LR and LW at 73 and 123 days. The Inverse Simpson index was significantly lower in DR than in LR and LW at 73 days. Landrace had a higher Inverse Simpson index than DR and LW at 123 days, while no statistically significant differences were observed at 158 days.

Measurements of fecal microbiome alpha diversity at ASV level using the Shannon, Simpson, and Inverse Simpson indices (least squares means ± confidence interval) in the Duroc (DR), Landrace (LR), and Large White (LW) breeds for three time points 73 days (T1), 123 days (T2), and 158 days (T3) in the growth trial.
Clustering analyses were focused on identifying clusters (enterotypes) among the fecal samples of pigs collected at each time point. Each of these enterotypes may be driven by specific genera that contribute to microbial compositions. The clusters of samples and genera that significantly separate the enterotypes according to breed and time points are shown in Fig. 5 and Supplementary Figure S1. The optimal number of clusters based on Calinski–Harabasz index maximization was 2, with the exception of 73 and 123 days for DR and LW samples where the optimal number of clusters was more than 2. In order to identify specific bacterial genera that were characteristic to the three breeds within each time point, we performed an LDA analysis coupled with LDA Effect Size LEfSe. Figure 5 shows the genera that were differentially represented among the three breeds and time points. Anaerostipes and Turicibacter genera had very high LDA scores across all breeds and enterotypes. At 73 days, the three enterotypes in DR pigs were distinguished by Dorea, Faecalibacterium, and Anaerovibrio, while the enterotypes for LR and LW pigs were distinguished by Anaerostipes and Turicibacter. At 123 days, the four enterotypes in DR pigs were distinguished by a total of 12 genera. Of these, Turicibacter, Faecalibacterium, Anaerostipes, and Dorea were most abundant in enterotypes A, B, C, and D, respectively. At the same time point, the two enterotypes for LR were significantly (P < 0.05) distinguished for either Turicibacter or Anaerostipes, while the three enterotypes for LW were significantly (P < 0.05) distinguished by Turicibacter, Anaerostipes, and Clostridium. Enterotypes A and B for DR and LR at 158 days were significantly (P < 0.05) distinguished by Turicibacter and Anaerostipes, while the two enterotypes for LW were distinguished by 11 genera. Of these, Anaerostipes and Sporobacterium were significantly (P < 0.05) more abundant. This analysis revealed a different grouping of the samples across time points for DR and LW, while the LR samples were mainly clustered into Anaerostipes and Turicibacter enterotypes.

Clustering and LDA analyses of gut microbiome data collected from Duroc [a], Landrace [b], and Large White [c] pigs at three time points 73 days (T1), 123 days (T2), and 158 days (T3) of the feeding trial. (Above) Genera that differentiate enterotypes at each time point. Bar length represents a log10-transformed linear discriminant score. Color corresponds to the enterotype in which a genus was found to be most abundant. (Below) Enterotype clusters identified using Calinski–Harabasz (CH) index maximization
ASV differentially represented in the Duroc, Landrace, and Large White breeds
Analyses of ASV representation performed at each time point revealed the gut microbiomes of DR, LR, and LW pigs to be distinct (Supplementary Table S1). When ASV abundances in DR pigs were compared to the average of those in LR and LW pigs at 73, 123, and 158 days, a total of 441, 401, and 324 ASV were found to be significantly different in terms of their representation (false discovery rate (FDR), 5%). Of these, 261 ASV classified as Firmicutes (177), Bacteroidetes (55), Proteobacteria (12), Spirochaetes (11), Actinobacteria (3), Chlamydiae (1), Fusobacteria (1), and Tenericutes (1) were shared between 73 and 123 days; 267 ASV classified as Firmicutes (180), Bacteroidetes (59), Proteobacteria (11), Spirochaetes (10), Actinobacteria (5), Fusobacteria (1), and Tenericutes (1) were shared between 123 and 158 days, and 191 ASV classified as phylum Firmicutes (128), Bacteroidetes (41), Proteobacteria (9), Spirochaetes (8), Actinobacteria (3), Fusobacteria (1), and Tenericutes (1) were shared between 73 and 158 days.
The relative abundances of 184, 153, and 123 ASV were significantly different between LR versus LW pigs at 73, 123, and 158 days, respectively. Of these, 56 consistently separated LR from LW pigs at 73 and 123 days, specifically, Firmicutes (24), Bacteroidetes (16), Proteobacteria (9), Spirochaetes (3), Actinobacteria (3), and Fusobacteria (1). There were 38 ASV that were systematically different between LR and LW at 123 and 158 days. These belonged to different phyla such as Firmicutes (24), Bacteroidetes (5), Proteobacteria (5), Actinobacteria (3), and Spirochaetes (1). Thirteen ASV classified as Firmicutes (5), Bacteroidetes (4), Proteobacteria (3), and Actinobacteria (1) were shared between 73 and 158 days.
The significant ASV in the orthogonal contrasts of DR versus the average of LR and LW and LR versus LW at all three time points are presented as volcano plots (Fig. 6a–c), while the relative abundance is reported in Additional file 2. A comparison of the Duroc sire line with the combined LR and LW maternal lines at 73 days revealed a higher abundance of 6 Firmicutes. The most significant of these were classified as Ruminococcus and Clostridium. In a comparison of the two maternal lines with one another at 73 days, LR had higher representation of Bacteroidetes and Spirochaetes than LW. Within these phyla, the most significant genera were Prevotella, Bacteroides, and Treponema. Landrace also had a lower proportion of several ASV than LW at 73 days, many of which were classified as Clostridium, Campylobacter, Blautia, Eubacterium, Lactobacillus, and Roseburia (Fig. 6a).

ASV with significantly higher or lower relative abundance when comparing the Duroc (DR) sire line vs the average of the Landrace and Large White maternal lines (DR vs (LR+LW)/2) (left panel) or the two maternal lines (LR vs LW) (right panel) at three time points (73 days (T1) [a], 123 days (T2) [b], and 158 days (T3) [c]). Each volcano plot simultaneously indicates the log2 fold change (x-axis) and the –log10(p value) (y-axis) for each ASV plotted. Dashed lines indicate arbitrary thresholds of logFC and p value. Each point represents an ASV, annotation highlight the genus of the ASV, while color represents the phylum.
Significant differences were also observed at 123 days when comparing ASV abundances in DR pigs with the average of LR and LW. Duroc pigs had higher proportion of 8 Firmicutes, 1 Actinobacteria, and 1 Bacteroidetes than the two maternal lines. The most abundant genera within these phyla were Catenibacterium, Prevotella, and Collinsella. On the other hand, DR, compared to the average of LR and LW at 123 days had a lower proportion of 8 Firmicutes, 4 Bacteroidetes, and 2 Spirochaetes. Within these phyla, the most significant genera were classified as Clostridium, Prevotella, and Treponema. Remarkable differences in ASV abundances were also observed when comparing LR to LW at 123 days. Specifically, LR had a higher proportion of 6 Bacteroidetes, 6 Firmicutes, 2 Proteobacteria, and 1 Spirochaetes. Within these phyla, the predominant genera were Anaerococcus, Barnesiella, Clostridium, and Prevotella. Interestingly, the gut microbiome of LR at 123 days also had a lower proportion of ASV belonging to Clostridium and Eubacterium than LW (Fig. 6b).
The orthogonal contrast between paternal and maternal lines at 158 days revealed a higher abundance of 13 Firmicutes, 2 Bacteroidetes, and 1 Actinobacteria. Within the Firmicutes phylum, as observed in 73 and 123 days, the most significant genera were Catenibacterium and Clostridium. The paternal line had a lower abundance of 5 Firmicutes and 1 Spirochaetes. Of these, one of the most significant genera was Turicibacter as observed in 73 and 123 days. We also discovered differences between the two maternal lines at 158 days. LR compared to LW had a particularly higher abundance of Bacteroides, Campylobacter, Coprococcus, Enterococcus, and Fusobacterium, as well as a lower abundance of Lactobacillus, Ruminococcus, and Desulfovibrio (Fig. 6c). Of these, the genus Bacteroides was the most significant discriminant of LR and LW across the three time points.
Association between pig phenotypes and gut microbiota
We identified several ASV whose relative abundances correlated significantly with feed efficiency and fatness traits (Supplementary Table S2). A total of 16, 33, and 93 ASV were significantly associated with feed efficiency and fatness traits at 73, 123, and 158 days, respectively. They belonged mainly to 4 phyla: Firmicutes, Bacteroidetes, Proteobacteria, and Spirochaetes. At the genus level, 14 were classified as Ruminococcus, 12 as Clostridium, 10 as Eubacterium, 6 as Lactobacillus, 5 as Bacteroides, and 4 as Prevotella. Twelve ASV were classified as belonging to 7 other minor genera, and 79 were unassigned. We identified 20, 1, 20, 26, 61, 6, and 8 taxa that were significantly associated with ADFI, ADG, RF1, RF2, FCR, backfat, and loin depth, respectively, at the three different time points.
The interaction effects between ASV and breed were significant for 2 and 48 ASV at 73 and 158 days (Supplementary Table S3), respectively, while no significant interaction effects were observed at 123 days. The significant ASV belonged to the Firmicutes and Bacteroidetes. Eight of these belonged to the Faecalibacterium genus, 4 to the Eubacterium, 4 to the Bacteroides, 4 to the Oscillibacter, 4 to the Ruminococcus, 2 to the Anaerovibrio, and 24 were unassigned. Of the ASV identified, 22, 22, and 4 were significantly associated with ADFI, RF1, and FCR, respectively, at 73 and 158 days.
We found across time points that the genus Peptococcus and Turicibacter were positively correlated (rs ~ 0.27) with backfat at 123 and 158 days. The genera Faecalibacterium and Oscillibacter were negatively correlated with feed efficiency (rs = − 0.22) at 73 days. A negative correlation (rs ~ − 0.20) was also found between Anaerovibrio, Catenibacterium, Dorea, and Roseburia and fatness traits at 73, 123, and 158 days (Additional file 3).
We discovered within breed and across time points that the genus Oscillibacter was negatively correlated (rs ~ − 0.30) with feed efficiency (RF1, RF2, and FCR), backfat, and loin depth in DR at 73 days. The genera Blautia, Dorea, Eubacterium, Faecalibacterium, Lactobacillus, and Ruminococcus, on the other hand, were positively correlated with feed efficiency (rs ~ 0.15) and fatness traits (rs ~ 0.22) in Duroc pigs at 123 and 158 days. Similarly, a negative correlation was obtained between the genus Sarcina and growth (rs = − 0.38) as well as IMF (rs = − 0.23) at 158 days (Fig. 7a–c).

Heat maps showing Spearman correlations and level of significance between genera, feed efficiency, and fatness traits of Duroc, Landrace, and Large White in three time points of the growth trial (73 days (T1) [a], 123 days (T2) [b], and 158 days (T3) [c]). Correlations were analyzed between ASV at the genus level. The color represents the correlation while the saturation represents the level of significance
The genus Oscillibacter was negatively correlated (rs ~ − 0.17) with ADFI and feed efficiency in LR at 73 and 123 days. This maternal line also had a positive correlation (rs ~ 0.17) at 123 and 158 days between ASV classified as genera Clostridium and feed efficiency.
In addition, we found a positive correlation (rs ~ 0.20) between four genera (Corynebacterium, Lactobacillus, Finegoldia, and Psychrobacter) and both ADFI and feed efficiency in LW pigs at 123 days, while the genus Desulfovibrio was negatively correlated (rs ~ − 0.33) with ADFI, feed efficiency, and backfat at 73 and 158 days.
We found within time points and across breeds that the genus Oscillibacter was negatively correlated with ADFI and feed efficiency for DR, LR, and LW pigs at 73 days. The genera Anaerovibrio, Clostridium, Faecalibacterium, Eubacterium, and Ruminococcus, showed negative correlations with feed efficiency and backfat in both LR and LW at 73 days. ASV classified as Dorea, Eubacterium, and Lactobacillus were positively correlated with feed efficiency in DR and LW at 123 days. Additionally, the genus Blautia was positively correlated with feed efficiency in DR and LR at 123 days. ASVs classified as Dorea and Lactobacillus were positively correlated with ADFI and feed efficiency in DR and LW at 158 days.
Discussion
In the past decade, many studies have investigated the potential impact of the swine microbiome on different phenotypes [9, 11]. However, few studies have compared the gut microbiomes of different pig breeds [21, 24]. To the best of our knowledge, this is the first large-scale study that explores the relationship between breed, gut microbiome composition, and feed efficiency. The data used in this study were collected from DR, LR, and LW, which are the most common commercial breeds worldwide due to their favorable lean growth, maternal behavior, feed efficiency, and production traits [25, 26].
The gut microbiome of all three breeds was dominated by the phyla Firmicutes and Bacteroidetes, consistent with previous findings [9, 19, 27]. However, the most abundant bacterial family in the breeds characterized here was Lactobacillaceae, rather than Clostridiaceae and Prevotellaceae, as reported by Lu et al. [9]. Several factors potentially contributed to gut microbiota variation, among them breed, age, body weight, and diet [17]. Furthermore, previous studies revealed a significant effect of host genetics and environmental factors (e.g., pen, kinship) on gut microbiome composition [14, 28]. Evidence of host genome influence on microbiome composition has been reported in humans by Goodrich et al. [29], and studies of the pig gut microbiome have reported non-zero heritability estimates of certain microbial taxa [30]. A genome-wide association study has also recently been conducted to explore potential links between single-nucleotide polymorphisms within the pig genome, and the abundance of taxa was shown to be significantly associated with growth and fatness parameters [31]. A significant association between the pig genome and the relative abundance of six relevant genera was also found by Crespo-Piazuelo et al. [28].
Landrace pigs had a more diverse gut microbiome compared with the other two breeds, in agreement with Pajarillo et al. [21]. Some researchers have suggested that very low diversity in a microbiome is a good predictor of poor health status [32, 33]. Landrace pigs were also characterized by higher feed efficiency compared to DR and LW, which is consistent with findings reported by Quan et al. [34] who suggested a higher Shannon index in high feed efficiency crossbred pigs.
The gut microbiome composition of DR pigs was significantly different from that of LR and LW animals, while the LR and LW microbiomes were more similar. At the genus level, Catenibacterium, Clostridium, and Turicibacter significantly influenced the separation between sire and maternal lines across the time points considered. The association between the genus Catenibacterium and DR compared with LR and LW was also previously reported by Pajarillo et al. [21]. In mice, it has been demonstrated that prevalent bacterial genera with a higher abundance in the same breed usually have a phylogenetic or functional association with their host [35]. Comparing the two maternal lines, LR pigs had a significantly higher proportion of Bacteroides than LW pigs across time points, instead of Lactobacillus as reported by Pajarillo et al. [21]. Large White had higher ADFI and FCR compared to LR. It is known in pigs that the type and quantity of feed consumed can affect both host biology and the relative abundance of different bacterial species in the gastrointestinal tract. For example, Frese et al. [36] indicated that the diet composition can widely modify the gut microbiome composition (e.g., increase of Prevotella), which reflected the different functional capacities of the microbial community in different time points.
A variation across time point was observed with an increase in abundance of some taxa associated with Firmicutes and Bacteroidetes in the gut of DR pigs compared to LR and LW. A significant change in the ratio of Firmicutes to Bacteroidetes in the gut of developing pigs over time and after cohabitation was observed [37, 38]. Changes in the abundance of Firmicutes and Bacteroidetes have been associated with a modification in the carcass fat deposition [39]. Bacteroidetes had a negative correlation with fat mass and had the ability to utilize various sugar derivatives from vegetables in pigs [40].
At the genus level, the microbiome shift over time was mainly related to Oscillibacter, Prevotella, Campylobacter, and Treponema. The relative abundance of Oscillibacter and Prevotella increased as the pigs aged in DR compared to LR and LW. The proportion of Campylobacter increased over time in LR compared to LW, while Treponema decreased. An increase in the average proportion of Prevotella in the 15 weeks after weaning was also observed by Lu et al. [9]. Regarding a possible biological function of these genera in the gastrointestinal tract, Oscillibacter was reported as a probiotic and producer of anti-inflammatory metabolites [41], while Prevotella is known to be involved in the degradation of both plant-based dietary polysaccharides [42]. A higher relative amount of Prevotella was also reported in both high and low feed efficiency Landrace pigs by Tan et al. [19]. Lastly, taxa classified as Treponema were the main driver of the enterotype-like group within the pig gut microbiota associated with growth traits [43], which was involved in cellulose and lignin degradation [16].
It is expected that host genetics has the potential to meaningfully influence the gut microbiota [9, 12, 35], and consequently feed efficiency, by favoring or disfavoring microbes that significantly contribute to nutrient digestion and energy harvest. Therefore, the gut microbiome composition could be associated with intestinal morphology and physiology that can impact the production traits such as growth and feed intake [44].
Characterizing the relationship between gut microbial composition and feed efficiency revealed a positive association between four genera (Lactobacillus, Blautia, Dorea, and Eubacterium) and feed efficiency. In agreement with our results, Verschuren et al. [45] found a positive effect of Lactobacillus on the feed efficiency of three-way cross pigs. Wang et al. [46] reported an effect of Lactobacillus on growth and fat deposition in broiler chickens. Some genera of the family Lactobacillaceae were involved in the production of antimicrobial bacteriocins, which are related to gut microbiome composition [47]. A species of the genus Lactobacillus with the ability to promote intestinal metabolism was identified in a cluster of highly feed-efficient DR and LR pigs [14, 19]. According to this study, Yang et al. [14] and Quan et al. [34] reported positive associations between taxa classified as Ruminococcaceae and feed efficiency in DR and crossbred pigs. Interestingly, taxa belonging to Ruminococcaceae are able to produce short-chain fatty acids fermenting dietary polymers, such as polysaccharides, that are not degradable by the host [48]. Fermentation products may influence several aspects of the gastrointestinal tract, such as transit time and nutrient digestion [49]. Moreover, previous studies in swine demonstrated that an increase in the production of short-chain fatty acids could improve the absorptive capacity of the intestine, promoting the growth of beneficial bacteria [49], thereby increasing feed efficiency [14, 34].
Conclusions
Here, an effect of breeds characterized by different feed efficiency on gut microbiome was discovered. Microbiome differences between breeds were found mainly associated with the genera Catenibacterium, Clostridium, and Bacteroides. These results suggest that host genetics has an essential effect on the structure and composition of the pig gut microbiome. Association analyses between the gut microbiome and feed efficiency revealed a positive association between Blautia, Dorea, Eubacterium, Lactobacillus, and feed efficiency in pigs. We provide evidence that the gut microbiome was correlated with feed efficiency and fatness traits, which might be relevant to understand how the intestinal microbial community influences the host production traits. Therefore, these results suggest that the intestinal microbial composition can provide important knowledge in order to improve the feed efficiency of pigs in pork industry.
Methods
Experimental design and sample collection
Animal use approval was not needed for this study because the data analyzed were from an existing database provided by Smithfield Premium Genetics (Rose Hill, NC, USA). Data points from Duroc (DR) (n = 190), Landrace (LR) (n = 221), and Large White (LW) (n = 204) boars were used. The animals were the progeny of 27, 27, and 44 sires crossed with 119, 153, and 158 dams for DR, LR, and LW, respectively. The growth trial ran concurrently for the three lines from May to December 2017. During the growth trial, all pigs were provided the same pelleted feed and received standard vaccinations and medications on a nucleus farm composed of 8 rooms (Supplementary Table S4 and S5). In this period, animals were kept in single-breed groups with an average count of 11.3 ± 1.3 animals per group (8 pens/room). Each group had access to one single-space Feed Intake Recording Equipment (FIRE) feeder (Osborne Industries, Inc., Osborne, KS, USA). Each FIRE feeder was equipped with a weighing scale (ACCU-ARM Weigh Race; Osborne Industries, Inc., Osborne, KS, USA) to record the body weight of a pig accessing the feeder (Supplementary Figure S2). In addition, a pig’s identifier and animal consumption were recorded every time a pig visits the feeder. Average daily feed intake (ADFI) was calculated as the average amount of feed consumed daily during the growth trail. Average daily gain (ADG) for each tested boar was measured as the ratio between body weight and age across the testing period (Supplementary Figure S3). One estimate of residual feed intake (RF1) was calculated as the residual from a regression model of ADFI on ADG. A second estimate of residual feed intake (RF2) was calculated as for RF1 but also included a correction with respect to body weight as reported by [5]. The average feed conversion ratio (FCR) was calculated as the ratio between ADFI and ADG. At the end of the growth trial, ultrasound backfat, loin eye area, and intramuscular fat (IMF) were measured. Ultrasound images of all animals were recorded with an Aloka 500 ultrasound machine (Corometrics Medical Systems, Wallingford, CT, USA) and analyzed for IMF using the Swine Image Analysis Software (Designer Genes Technologies Inc., Harrison, AR, USA). Descriptive statistics of all the traits are presented in Table 1. Fecal samples were collected by swabbing the rectum of each animal at 73.2 ± 2.9 days, 123.4 ± 3.6 days, and 158.5 ± 4.4 days after birth and were subjected to microbiome sequencing (Supplementary Figure S4). A schematic illustration depicting the experimental design and sample collection is reported in Supplementary Figure S5.
DNA extraction and 16S rRNA gene amplicon sequencing of fecal
DNA was extracted from each rectal swab using the method reported in detail by Lu et al. [9]. Phased, bi-directional amplification of the V4 region (515–806) of the 16S rRNA gene was employed to generate indexed libraries for Illumina sequencing in the manner previously described [9]. All sequencing was performed at the DNA Sequencing Innovation Lab at the Center for Genome Sciences and Systems Biology at Washington University in St. Louis (USA). The raw sequence data from the Illumina platform were converted into read files using MiSeq Reporter. Pairs of V4 16S rRNA gene sequences were first merged into a single sequence using FLASH v1.2.11 [50] with a required overlap of at least 100 and no more than 250 base pairs in order to provide a confident overlap. Sequences were oriented in the forward direction, and any primer sequences were trimmed; during primer matching, up to 1 mismatch was allowed. Sequences were imported into Quantitative Insights Into Microbial Ecology (QIIME2 version 2017.12, https://qiime2.org/) for demultiplexing and the construction of an amplicon sequence variant (ASV) feature table using the Divisive Amplicon Denoising Algorithm 2 (DADA2) [51] with default settings and no truncation or length filtering (--p-trunc-len 0). Features present in only 1 sample were removed from the table. Finally, the Ribosomal Database Project (RDP) classifier (v2.4) was retrained in the manner previously described [52] and used to predict taxonomic assignments for each ASV sequence using a confidence cutoff of 0.8. Results were then exported for further analysis in the R environment [53].
Data editing
Feeding efficiency traits and body weight were obtained from the FIRE system records. To obtain an accurate prediction of individual feed intake and body weight, data editing was required [5]. In this study, feed intake records were edited on the basis of the procedure proposed by [5, 54], while body weights were edited following the procedure reported by [26, 55]. We fit a robust regression model using the “mass” [56] package in R [53], with age and squared age as covariates. Each data point was assigned a weight (from 0 to 1) to minimize the influence of extreme values. Data with weights of less than 0.5 units were treated as outliers and removed before any statistical analysis [55]. The predicted body weight from robust regression was used to calculate ADG for each boar. Average daily gain values that were outside the mean plus or minus three standard deviations were considered outliers and removed. The final data set comprised of 336,959 data points. We focused our analysis using ASV with a total count ≥ 1000 across all samples within a time point. In total, 765, 729, and 820 ASV met these criteria in the final data set for 73, 123, and 158 days, respectively.
Statistical analyses
Phenotypic differences
To investigate the impact of breed and to control the environmental factors on performance, growth and efficiency traits were analyzed using the following model with the MIXED Procedure in SAS 9.4 [57].
where yijklm was the overall mean of the observed trait (ADFI, ADG, RF1, RF2, FCR, backfat, loin depth, and IMF); μ was the overall intercept of the model; Bri was the fixed effect of the ith Breed (i = 1 to 3); Roj was the fixed effect of the jth Room (j = 1 to 8); Si(Br)k:i was the random effect of the kth sire (k = 1 to 100) within Breed which was assumed to be N(0,I\( {\sigma}_{Si}^2 \)); Pe(Ro)l:j was the random effect of the lth pen (l = 1 to 64) within Ro which was assumed to be N(0,I\( {\sigma}_{Pe}^2 \)); and eijklm was the residual error which was assumed to be N(0,I\( {\sigma}_e^2 \)), where σe2 was the residual variance.
Moreover, to assess the relationships between different breeds, a linear discriminant analysis (LDA) was performed using the “lda” function in the “mass” package [56] in R and overall mean of feed efficiency and fatness traits as variables.
Microbiome composition differences across breeds
The ASV for 73, 123, and 158 days were analyzed using a negative binomial model with the “edgeR” [58] package in R, including the fixed effects described in the model (1). Orthogonal contrasts were fitted as reported above to obtain ASV significance among breeds. The Benjamini–Hochberg method was used for multiple-testing correction [59]. A false discovery rate of 5% was used to declare whether or not an ASV was significantly different between breeds. Jensen-Shannon Divergence [60] was calculated for each breed at the three separate time points according to the relative abundance of each genus in each sample using the “dist.JSD” function from the phyloseq package in R [61, 62] to identify enterotypes among the samples. Based on the obtained distance matrix, the samples at each time point were clustered via partitioning around medoids by using the “pam” function in the R package “cluster” [63]. The optimal number of clusters was chosen by maximizing the Calinski–Harabasz index [64], using the “index.G1” function in the R package “clusterSim” [65], and the Silhouette index [66], using the “silhouette” function in the R package “cluster” [63]. To identify genera that had a significant effect on the division of the enterotypes at each time point, the LDA Effect Size based on the non-parametric Kruskal–Wallis sum-rank test was performed [67] with an alpha value of 0.05 for the factorial Kruskal–Wallis test among classes and a threshold of 2 on the logarithmic LDA score for discriminative features using the galaxy/hutlab website (http://huttenhower.sph.harvard.edu/galaxy/).
ASV–phenotype association
We investigated the effect of ASV at 73, 123, and 158 days on performance traits and residual feed intake using the following model with the MIXED Procedure in SAS 9.4 [57]:
where yijklmn was the phenotype within each time points (ADFI, ADG, RF1, RF2, FCR, backfat, loin depth, and IMF); μ was the overall intercept of the model; Bri and Roj were the fixed effects described in the model (1); Ask was the covariate of the kth ASV; As × Brik was the fixed effect of the ikth As × Brik interaction; Si(Br)l:i, Pe(Ro)m:j, and eijklmn were as in the model (1). Orthogonal contrasts among breeds were fitted as reported above. An FDR of 5% was used to declare whether or not an ASV was significantly associated with traits and/or statistically different between breeds. Spearman correlations were used to correlate feed efficiency, fatness traits, and ASVs with a genus assignment using the “Hmisc” (https://cran.r-project.org/web/packages/Hmisc/Hmisc.pdf) package in R.
Availability of data and materials
The phenotypes generated and/or analyzed during the current study are not publicly available due to third-party ownership but are available from the corresponding authors upon reasonable request. 16S rRNA gene sequences will be updated to a public repository upon acceptance of the manuscript for publication.
Abbreviations
- ASV:
-
Amplicon sequence variant
- ADG:
-
Average daily gain
- ADFI:
-
Average daily feed intake
- DR:
-
Duroc
- FCR:
-
Feed conversion ratio
- FIRE:
-
Feed Intake Recording Equipment
- FDR:
-
False discovery rate
- IMF:
-
Intramuscular fat
- LR:
-
Landrace
- LDA:
-
Linear discriminant analysis
- LW:
-
Large White
- RF1:
-
Residual feed intake 1 (calculated by regressing ADFI on ADG)
- RF2:
-
Residuals feed intake 2 (calculated by regressing ADFI on ADG and body weight)
References
Cadéro A, Aubry A, Brossard L, Dourmad JY, Salaün Y, Garcia-Launay F. Modelling interactions between farmer practices and fattening pig performances with an individual-based model. Animal. 2018;12:1277–86.
Young JM, Cai W, Dekkers JCM. Effect of selection for residual feed intake on feeding behavior and daily feed intake patterns in Yorkshire swine1. J Anim Sci. 2011;89:639–47.
Patience JF, Rossoni-Serão MC, Gutiérrez NA. A review of feed efficiency in swine: biology and application. Journal of Animal Science and Biotechnology. 2015;6. https://doi.org/10.1186/s40104-015-0031-2.
O’Shea CJ, Sweeney T, Bahar B, Ryan MT, Thornton K, O’Doherty JV. Indices of gastrointestinal fermentation and manure emissions of growing-finishing pigs as influenced through singular or combined consumption of Lactobacillus plantarum and inulin. J Anim Sci. 2012;90:3848–57.
Lu D, Jiao S, Tiezzi F, Knauer M, Huang Y, Gray KA, et al. The relationship between different measures of feed efficiency and feeding behavior traits in Duroc pigs. J Anim Sci. 2017;95:3370.
Ding R, Yang M, Wang X, Quan J, Zhuang Z, Zhou S, et al. Genetic architecture of feeding behavior and feed efficiency in a Duroc pig population. Front Genet. 2018;9. https://doi.org/10.3389/fgene.2018.00220.
Do DN, Strathe AB, Jensen J, Mark T, Kadarmideen HN. Genetic parameters for different measures of feed efficiency and related traits in boars of three pig breeds1. J Anim Sci. 2013;91:4069–79.
Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–70.
Lu D, Tiezzi F, Schillebeeckx C, McNulty NP, Schwab C, Shull C, et al. Host contributes to longitudinal diversity of fecal microbiota in swine selected for lean growth. Microbiome. 2018;6. https://doi.org/10.1186/s40168-017-0384-1.
Canibe N, Højberg O, Højsgaard S, Jensen BB. Feed physical form and formic acid addition to the feed affect the gastrointestinal ecology and growth performance of growing pigs. J Anim Sci. 2005;83:1287–302.
Mach N, Berri M, Estellé J, Levenez F, Lemonnier G, Denis C, et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes: role of early-life gut microbiome on pigs’ health. Environ Microbiol Rep. 2015;7:554–69.
Maltecca C, Bergamaschi M, Tiezzi F. The interaction between microbiome and pig efficiency: a review. J Anim Breed Genet. 2019. https://doi.org/10.1111/jbg.12443.
Feng W, Ao H, Peng C. Gut microbiota, short-chain fatty acids, and herbal medicines. Front Pharmacol. 2018;9. https://doi.org/10.3389/fphar.2018.01354.
Yang H, Huang X, Fang S, He M, Zhao Y, Wu Z, et al. Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficiency in pigs. Front Microbiol. 2017;8. https://doi.org/10.3389/fmicb.2017.01555.
Quan J, Cai G, Yang M, Zeng Z, Ding R, Wang X, et al. Exploring the fecal microbial composition and metagenomic functional capacities associated with feed efficiency in commercial DLY pigs. Front Microbiol. 2019;10:52.
Niu Q, Li P, Hao S, Zhang Y, Kim SW, Li H, et al. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci Rep. 2015;5. https://doi.org/10.1038/srep09938.
Wang X, Tsai T, Deng F, Wei X, Chai J, Knapp J, et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome. 2019;7. https://doi.org/10.1186/s40168-019-0721-7.
Singh KM, Shah TM, Reddy B, Deshpande S, Rank DN, Joshi CG. Taxonomic and gene-centric metagenomics of the fecal microbiome of low and high feed conversion ratio (FCR) broilers. J Appl Genet. 2014;55:145–54.
Tan Z, Yang T, Wang Y, Xing K, Zhang F, Zhao X, et al. Metagenomic analysis of cecal microbiome identified microbiota and functional capacities associated with feed efficiency in Landrace finishing pigs. Front Microbiol. 2017;8. https://doi.org/10.3389/fmicb.2017.01546.
McCormack UM, Curião T, Buzoianu SG, Prieto ML, Ryan T, Varley P, et al. Exploring a possible link between the intestinal microbiota and feed efficiency in pigs. Appl Environ Microbiol. 2017;83. https://doi.org/10.1128/AEM.00380-17.
Pajarillo EAB, Chae JP, Balolong MP, Kim HB, Seo K-S, Kang D-K. Pyrosequencing-based analysis of fecal microbial communities in three purebred pig lines. J Microbiol. 2014;52:646–51.
López-García A, Benítez R, Núňez Y, Gómez Izquierdo E, De Mercado E, García-Casco J, et al. Influence of breed and dietary energy source on gut microbiota composition in Iberian and Duroc pigs. 2019.
Isaacson R, Kim HB. The intestinal microbiome of the pig. Anim Health Res Rev. 2012;13:100–9.
Xiao Y, Kong F, Xiang Y, Zhou W, Wang J, Yang H, et al. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci Rep. 2018;8. https://doi.org/10.1038/s41598-018-24289-z.
Rauw WM, Soler J, Tibau J, Reixach J, Gomez RL. Feeding time and feeding rate and its relationship with feed intake, feed efficiency, growth rate, and rate of fat deposition in growing Duroc barrows1. J Anim Sci. 2006;84:3404–9.
Jiao S, Maltecca C, Gray KA, Cassady JP. Feed intake, average daily gain, feed efficiency, and real-time ultrasound traits in Duroc pigs: II. Genome wide association. J Anim Sci. 2014;92:2846–60.
Pedersen R, Ingerslev H-C, Sturek M, Alloosh M, Cirera S, Christoffersen BØ, et al. Characterisation of gut microbiota in Ossabaw and Göttingen minipigs as models of obesity and metabolic syndrome. PLoS ONE. 2013;8:e56612.
Crespo-Piazuelo D, Migura-Garcia L, Estellé J, Criado-Mesas L, Revilla M, Castelló A, et al. Association between the pig genome and its gut microbiota composition. Sci Rep. 2019;9. https://doi.org/10.1038/s41598-019-45066-6.
Goodrich JK, Davenport ER, Clark AG, Ley RE. The relationship between the human genome and microbiome comes into view. Annu Rev Genet. 2017;51:413–33.
Chen C, Huang X, Fang S, Yang H, He M, Zhao Y, et al. Contribution of host genetics to the variation of microbial composition of cecum lumen and feces in pigs. Front Microbiol. 2018;9. https://doi.org/10.3389/fmicb.2018.02626.
Bergamaschi M, Maltecca C, Schillebeeckx C, McNulty N, Schwab C, Shull C, et al. Heritability and genome-wide association of swine gut microbiome features with growth and fatness parameters. Scientific Reports. Submitted. 2020.
Bergamaschi, M., Maltecca, C., Schillebeeckx, C. et al. Heritability and genome-wide association of swine gut microbiome features with growth and fatness parameters. Sci Rep 2020;10:10134. https://doi.org/10.1038/s41598-020-66791-3.
Heiman ML, Greenway FL. A healthy gastrointestinal microbiome is dependent on dietary diversity. Molecular Metabolism. 2016;5:317–20.
Quan J, Cai G, Ye J, Yang M, Ding R, Wang X, et al. A global comparison of the microbiome compositions of three gut locations in commercial pigs with extreme feed conversion ratios. Sci Rep. 2018;8. https://doi.org/10.1038/s41598-018-22692-0.
Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, Wymore A, et al. Host genetic and environmental effects on mouse intestinal microbiota. The ISME Journal. 2012;6:2033–44.
Frese SA, Parker K, Calvert CC, Mills DA. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome. 2015;3. https://doi.org/10.1186/s40168-015-0091-8.
Pajarillo EAB, Chae JP, Kim HB, Kim IH, Kang D-K. Barcoded pyrosequencing-based metagenomic analysis of the faecal microbiome of three purebred pig lines after cohabitation. Appl Microbiol Biotechnol. 2015;99:5647–56.
Kim HB, Borewicz K, White BA, Singer RS, Sreevatsan S, Tu ZJ, et al. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet Microbiol. 2011;153:124–33.
Fan P, Liu P, Song P, Chen X, Ma X. Moderate dietary protein restriction alters the composition of gut microbiota and improves ileal barrier function in adult pig model. Sci Rep. 2017;7:43412.
Yang H, Huang X, Fang S, Xin W, Huang L, Chen C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci Rep. 2016;6. https://doi.org/10.1038/srep27427.
Li J, Sung CYJ, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci. 2016;113:E1306–15.
Patel DD, Patel AK, Parmar NR, Shah TM, Patel JB, Pandya PR, et al. Microbial and carbohydrate active enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet. Gene. 2014;545:88–94.
Ramayo-Caldas Y, Mach N, Lepage P, Levenez F, Denis C, Lemonnier G, et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. The ISME Journal. 2016;10:2973–7.
Campbell JM, Crenshaw JD, Polo J. The biological stress of early weaned piglets. J Animal Sci Biotechnol. 2013;4:19.
Verschuren LMG, Calus MPL, Jansman AJM, Bergsma R, Knol EF, Gilbert H, et al. Fecal microbial composition associated with variation in feed efficiency in pigs depends on diet and sex1. J Anim Sci. 2018;96:1405–18.
Wang H, Ni X, Qing X, Zeng D, Luo M, Liu L, et al. Live probiotic Lactobacillus johnsonii BS15 promotes growth performance and lowers fat deposition by improving lipid metabolism, intestinal development, and gut microflora in broilers. Front Microbiol. 2017;8. https://doi.org/10.3389/fmicb.2017.01073.
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–7.
Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–31.
Metzler BU, Mosenthin R. A review of interactions between dietary fiber and the gastrointestinal microbiota and their consequences on intestinal phosphorus metabolism in growing pigs. Asian Australas J Anim Sci. 2008;21:603–15.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. The ISME Journal. 2017;11:2639–43.
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214–4.
Team, R. Core. R: a language and environment for statistical computing. 2017.
Casey DS, Stern HS, Dekkers JCM. Identification of errors and factors associated with errors in data from electronic swine feeders1. J Anim Sci. 2005;83:969–82.
Zumbach B, Misztal I, Chen CY, Tsuruta S, Łukaszewicz M, Herring WO, et al. Use of serial pig body weights for genetic evaluation of daily gain. J Anim Breed Genet. 2010;127:93–9.
Venables WN, Ripley BD, Venables WN. Modern applied statistics with S. 4th ed. New York: Springer; 2002.
SAS Institute Inc. SAS User’s Guide: Statistics. Version 9.4. 2011.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300.
Endres DM, Schindelin JE. A new metric for probability distributions. IEEE Trans Inf Theory. 2003;49:1858–60.
MetaHIT Consortium (additional members), Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K, Studer M, et al. Package “cluster”. https://cran.r-project.org/web/packages/cluster/cluster.pdf.
Calinski T, Harabasz J. A dendrite method for cluster analysis. Communications in Statistics - Theory and Methods. 1974;3:1–27.
Walesiak M, Dudek A. ClusterSim: searching for optimal clustering procedure for a data set. https://cran.r-project.org/web/packages/clusterSim/index.html.
Rousseeuw PJ. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math. 1987;20:53–65.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Acknowledgements
The authors would like to thank Jessica Hoisington-Lopez and MariaLynn Crosby from the DNA Sequencing Innovation Lab at the Center for Genome Sciences and Systems Biology at Washington University in St. Louis for their sequencing expertise.
Funding
This study was supported by The NC Pork Council 2017-1929 grant: “The Role of Feeding Behavior on Gut Microbiome Composition and its Implications for Lean Growth Selection in Pigs.”
Author information
Authors and Affiliations
Contributions
MB performed the statistical analyses, as well as interpreted the results and drafted the manuscript. CM and FT designed the experiment and helped draft the manuscript. JH, YH, and KG supervised and performed phenotypic data collection and editing. JH, YH, KG, and NM provided comments for the manuscript. NM and CS were responsible for sequencing analysis. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Phenotypic records presented in this study came from nucleus farms belonging to Smithfield Premium Genetics. Procedures for fecal sample collection adhered to the guidelines of the Institutional Animal Care and Use Committee, North Carolina State University, and National Pork Board.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Descriptive statistics of feed efficiency and fatness traits of pigs.
Additional file 2.
Relative abundance of amplicon sequence variant (ASV) at genus level in the gut microbiome of pigs.
Additional file 3.
Spearman correlations between gut microbiome, feed efficiency, and fatness traits of pigs.
Additional file 4: Supplementary Table S1.
Differences between the gut microbiome of three breeds of pigs.
Additional file 5: Supplementary Table S2.
Results of ASV effect on feed efficiency and fatness traits of three purebred lines across three time points of the growth trial.
Additional file 6: Supplementary Table S3.
Results of the interaction between ASV and breed effect on feed efficiency and fatness traits of three purebred lines across three time points of the growth trial.
Additional file 7: Supplementary Table S4.
Composition of the diets fed to pigs in this study.
Additional file 8: Supplementary Table S5.
Medical treatments administered to pigs in this study.
Additional file 9: Supplementary Figure S1.
Calinski-Harabasz indexes (CH) for number of potential clusters of samples at 73 days (T1), 123 days (T2), and 158 days (T3) for Duroc (red), Landrace (blue), and Large White (green). The highest CH value at each time point indicates optimal number of cluster/enterotypes.
Additional file 10: Supplementary Figure S2.
Variation of body weight of Duroc (DR), Landrace (LR) and Large White (LW) during the feeding trial.
Additional file 11: Supplementary Figure S3.
Average daily gain of Duroc (DR), Landrace (LR) and Large White (LW) during the feeding trial.
Additional file 12: Supplementary Figure S4.
Average age of Duroc (DR), Landrace (LR) and Large White (LW) during the feeding trial.
Additional file 13: Supplementary Figure S5.
Illustration of the experimental design.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bergamaschi, M., Tiezzi, F., Howard, J. et al. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome 8, 110 (2020). https://doi.org/10.1186/s40168-020-00888-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-020-00888-9