
Abstract
Background
A vaginal microbiota dominated by lactobacilli (particularly Lactobacillus crispatus) is associated with vaginal health, whereas a vaginal microbiota not dominated by lactobacilli is considered dysbiotic. Here we investigated whether L. crispatus strains isolated from the vaginal tract of women with Lactobacillus-dominated vaginal microbiota (LVM) are pheno- or genotypically distinct from L. crispatus strains isolated from vaginal samples with dysbiotic vaginal microbiota (DVM).
Results
We studied 33 L. crispatus strains (n = 16 from LVM; n = 17 from DVM). Comparison of these two groups of strains showed that, although strain differences existed, both groups degraded various carbohydrates, produced similar amounts of organic acids, inhibited Neisseria gonorrhoeae growth, and did not produce biofilms. Comparative genomics analyses of 28 strains (n = 12 LVM; n = 16 DVM) revealed a novel, 3-fragmented glycosyltransferase gene that was more prevalent among strains isolated from DVM. Most L. crispatus strains showed growth on glycogen-supplemented growth media. Strains that showed less-efficient (n = 6) or no (n = 1) growth on glycogen all carried N-terminal deletions (respectively, 29 and 37 amino acid deletions) in a putative pullulanase type I protein.
Discussion
L. crispatus strains isolated from LVM were not phenotypically distinct from L. crispatus strains isolated from DVM; however, the finding that the latter were more likely to carry a 3-fragmented glycosyltransferase gene may indicate a role for cell surface glycoconjugates, which may shape vaginal microbiota-host interactions. Furthermore, the observation that variation in the pullulanase type I gene is associated with growth on glycogen discourages previous claims that L. crispatus cannot directly utilize glycogen.
Similar content being viewed by others


Introduction
The vaginal mucosa hosts a community of commensal, symbiotic, and sometimes pathogenic micro-organisms. Increasing evidence has shown that the bacteria within this community, referred to here as the vaginal microbiota (VM), play an important role in protecting the vaginal tract from pathogenic infection, which can have far-reaching effects on a woman’s sexual and reproductive health [1, 2]. Several VM compositions have been described, including VM dominated by (1) Lactobacillus iners, (2) L. crispatus, (3) L. gasseri, (4) L. jensenii, and (5) VM that are not dominated by a single bacterial species but rather consist of diverse anaerobic bacteria, including Gardnerella vaginalis and members of Lachnospiraceae, Leptotrichiaceae and Prevotellaceae [3,4,5]. Particularly, VM that are dominated by L. crispatus are associated with vaginal health, whereas a VM consisting of diverse anaerobes—commonly referred to as vaginal dysbiosis—has been shown to increase a woman’s odds for developing bacterial vaginosis (BV), acquiring STIs, including HIV, and having an adverse pregnancy outcome [1, 2, 4, 6].
The application of human vaginal L. crispatus isolates as therapeutic agents to treat dysbiosis may have much potential [7, 8], but currently, there are still many gaps in our knowledge concerning the importance of specific physiological properties of L. crispatus for a sustained domination on the mucosal surface of the vagina. Comparative genomics approaches offer a powerful tool to identify novel important functional properties of bacterial strains. The genomes of nine human L. crispatus isolates have previously been studied, also in the context of vaginal dysbiosis [9, 10]. Comparative genomics of these strains showed that about 60% of orthologous groups (genes derived from the same ancestral gene) were conserved among all strains, i.e., comprising a “core” genome [10]. The accessory genome was defined as genes shared by at least two strains, while unique genes are specific to a single strain. Currently, it is unclear whether traits pertaining to in vivo dominance are shared by all strains (core genome) or only by a subset of strains (accessory genome). For example, both women with and without vaginal dysbiosis can be colonized with L. crispatus (see, e.g., [11]), and we do not yet fully understand why in some women L. crispatus dominates and in others it does not.
The following bacterial traits may be of importance for L. crispatus to successfully dominate the vaginal mucosa: (1) the formation of an extracellular matrix (biofilm) on the vaginal mucosal surface, (2) the production of antimicrobials such as lactic acid, bacteriocins, and H2O2 that inhibit the growth and/or adhesion of urogenital pathogens, (3) efficient utilization of available nutrients—particularly glycogen, as this is the main carbon source in the vaginal lumen, and (4) the modulation of host-immunogenic responses. Considering these points, firstly, Ojala et al. [10] observed genomic islands encoding enzymes involved in exopolysacharide (EPS) biosynthesis in the accessory genome of L. crispatus and postulated that strain differences in this trait could contribute to differences in biofilm formation, adhesion, and competitive exclusion of pathogens. Secondly, experiments have shown that L. crispatus effectively inhibits urogenital pathogens through lactic acid production, but these studies included only strains originating from healthy women [12,13,14,15,16]. Abdelmaksoud et al. [9] compared L. crispatus strains isolated from Lactobacillus-dominated VM (LVM) with strains isolated from dysbiotic VM (DVM) and indeed observed decreased lactic acid production in one of the strains isolated from DVM, providing an explanation for its low abundance. However, no significant conclusion could be made as their study included only eight strains. Thirdly, there is a general consensus that vaginal lactobacilli (including L. crispatus) ferment glycogen thus producing lactic acid, but no actual evidence exists that L. crispatus produces the enzymes to directly degrade glycogen [10, 17]. Lastly, L. crispatus-dominated VM are associated with an anti-inflammatory vaginal cytokine profile [18, 19] and immune evasion is likely a crucial (but poorly studied) factor that allows L. crispatus to dominate the vaginal niche. A proposed underlying mechanism is that L. crispatus produces immunomodulatory molecules [20], but L. crispatus may also accomplish immune modulation by alternating its cell surface glycosylation, as has been suggested for gut commensals [21]. Taken together, there is a clear need to study the properties of more human (clinical) L. crispatus isolates to fully appreciate the diversity within this species.
Here we investigated whether L. crispatus strains isolated from the vaginal tract of women with LVM are pheno- or genotypically distinct from L. crispatus strains isolated from vaginal samples with DVM, with the aim of identifying bacterial traits pertaining to a successful domination of lactobacilli of the vaginal mucosa.
Results
Lactobacillus crispatus strain selection and whole genome sequencing
For this study, 40 nurse-collected vaginal swabs were obtained from the Sexually Transmitted Infections clinic in Amsterdam, The Netherlands, from June to August 2012, as described previously by Dols et al. [4]. In total, 33 L. crispatus strains were isolated from these samples (n = 16 from LVM samples; n = 17 L. crispatus strains from DVM samples). Following whole genome sequencing, four contigs (n = 3 strains from LVM; n = 1 strain from DVM) were discarded as they had less than 50% coverage with other assemblies or with the reference genome (125-2-CHN), suggesting that these isolates belonged to a different Lactobacillus species. One contig (from a strain isolated from LVM) aligned to the reference genome, but its genome size was above the expected range, suggestive of contamination with a second strain and was therefore also discarded. The remaining 28 isolates (n = 12 LVM and n = 16 DVM) were assembled and used for comparative genomics. These genomes have been deposited at DDBJ/ENA/GenBank under the accession numbers NKKQ00000000-NKLR00000000. The versions described in this paper are versions NKKQ01000000-NKLR01000000 (Table 1).
Lactobacillus crispatus pan-genome
The 28 L. crispatus genomes had an average length of 2.31 Mbp (range 2.16–2.56 MB) (Table 1), which was slightly larger than the reference genome (125-2-CHN; 2.04 Mbp). The GC content of the genomes was on average 36.8%, similar to other lactobacilli [10]. An average of 2099 genes were annotated per strain (Table 1; Fig. 1). This set of 28 L. crispatus genomes comprised 4261 different gene families. The core genome consisted of 1429 genes (which correspond to ~ 68% of a given genome) and the accessory genome averaged at 618 genes (~ 30%) per strain. Each strain had on average 54 unique genes (~ 2.0%). The number of accessory and unique genes did not significantly differ between strains isolated from LVM or from DVM, with respectively an average of 621 (range 481–855) and 55 (range 5–243) genes for LVM strains and 615 (range 488–837) and 53 (range 1–250) genes for DVM strains. The distribution of cluster of ortholog groups (COG) also did not differ between strains from Lactobacillus-dominated and DVM. The gene accumulation model [22] describes the expansion of the pan-genome as a function of the number of genomes assessed and estimated that this species has access to a larger gene pool than described here; the model estimated the L. crispatus pan-genome to include 4384 genes.

Whole genome alignments of the coding sequences from the Lactobacillus crispatus clinical isolates described in this study. The outermost ring represents COG annotated genes on the forward strand (color coded according to the respective COG). The positions of the genes discussed in this article are indicated. The third ring represents COG annotated genes on the reverse strand (color coded according to the respective COG). The next 12 rings each represent one genome of the LVM strains, followed by a separator ring and 16 rings each representing a genome of the DVM strains. The height of the bar and the saturation of the color in these rings indicate a BLAST hit of either > 90% identity (darker colored) or > 70% identity (lightly colored). Hits below 70% identity score are not shown and appear as white bars in the plots. The two inner most rings represent the GC content of that area and the GC-skew respectively. Abbreviations: COG: cluster ortholog genes; LVM: Lactobacillus-dominated vaginal microbiota; DVM: dysbiotic vaginal microbiota; WT: wild type
A fragmented glycosyltransferase gene was abundant among strains isolated from DVM
In a comparative genomics analysis, we aimed to identify genes that were specific to strains isolated from either LVM or DVM. We observed that three transposases, one of which was further classified as an IS30 family transposase, were more abundant among strains isolated from DVM than among strains from LVM. IS30 transposases are associated with genomic instability and have previously been found to flank genomic deletions in commercial Lactobacillus rhamnosus GG probiotic strains [23]. Most notably, we observed that strains from DVM were more likely to carry three gene fragments of a single glycosyltransferase (GT) than strains isolated from LVM. GTs are enzymes that are involved in the transfer of a sugar moiety to a substrate and are thus essential in synthesis of glycoconjugates like exopolysaccharides, glycoproteins, and glycosylated teichoic acids [24, 25].
The three differentially abundant GT gene fragments all align to different regions of a single family 2 A-fold GT gene of the 125-2-CHN L. crispatus strain (CGA_000165885.1) and are flanked by other genes potentially encoding GTs (Fig. 2). Fragment 1 aligns with 472 bp of the original unfragmented GT, while fragment 2 overlaps with the last 3 bp of fragment 1 and fragment 3 overlaps 7 bp with fragment 2. Given that all these fragments align to the non-fragmented GT gene in L. crispatus 125-2-CHN, we hypothesize that the three fragments belong to the same GT. The L. crispatus genomes however contained one or more of the three GT fragments, while the surrounding genes were conserved among the strains. The first fragment of 510 bp contains the true GT fold domain and is thus likely responsible for the catalytic activity of the GT. The second and third fragments are considerably shorter, respectively 228 and 328 bp, and do not harbor any significant relation to a known GT fold (Fig. 3). Four different combinations of GT fragments were observed in the studied genomes, namely a variant with (1) no fragments, (2) all three fragments, (3) fragments 1 and 3, and (4) fragments 1 and 2 (Fig. 2; Table 2).

Schematic overview of the organization of the glycosyltransferase fragments in the Lactobacillus crispatus genomes. The orientation of the fragments is dependent on the assembly and can therefore be different than depicted here. Also, the distance between the fragments is undetermined and can be of any length (depicted with diagonal lines). Abbreviations: GT: Glycosyltransferase; GTA, GTB: GT super families; GT1, GT2, GT3: GT fragments 1, 2, 3; UDP-GALAC: UDP-Galactopyranose mutase; GTF: GT family 1; TRAN: transposase; LVM: Lactobacillus-dominated vaginal microbiota; DVM: dysbiotic vaginal microbiota

Schematic overview of how the glycosyltransferase fragments align to the Lactobacillus crispatus 125-2-CHN reference genome. The first fragment comprises the conserved glycosyltransferase family 2 domain with catalytic activity. The shorter second and third fragments most probably do not harbor any catalytic GT activity. We hypothesize that these two fragments play a role in steering the specific activity of the GT (e.g., towards donor or substrate specificity). Abbreviation: GT: glycosyltransferase
Strains isolated from LVM were not phenotypically distinct from strains isolated from DVM
Phenotypic studies on the L. crispatus strains did not reveal any biofilm formation—as assessed by crystal violet assays, except for one strain (RL19) which produced a weak biofilm. In line with this, very low levels of autoaggregation (on average 5%) were observed and this also did not differ between the two groups of strains. Strain-specific carbohydrate fermentation profiles were observed, as assessed by a commercial API CH50 test, but the distribution of these profiles did not relate to whether the strains were isolated from LVM or from DVM. Strains isolated from LVM produced similar amounts of organic acids compared with strains isolated from DVM when grown on chemically defined medium mimicking vaginal fluids [26]. The strains mainly produced lactic acid. Other acids such as succinic acid, butyric acid, glutamic acid, phenylalanine, isoleucine, and tyrosine were also produced, but fourfold lower compared to lactic acid. Very small acidic molecules, such as acetic and propionic acid, were out of the detection range and could thus not be measured. We also assessed antimicrobial activity against a common urogenital pathogen Neisseria gonorrhoeae. Inhibition was similar for strains isolated from LVM and from DVM: N. gonorrhoeae growth was inhibited (i.e., lower OD600nm in stationary phase compared to the control), in a dose-dependent way, by on average 27.9 ± 15.8% for undiluted L. crispatus supernatants compared to the N. gonorrhoeae control. Undiluted neutralized L. crispatus supernatants inhibited N. gonorrhoeae growth by on average 15.7 ± 16.3% (Additional file 1).
Strain-specific glycogen growth among both LVM and DVM isolates
Of the 28 strains for which full genomes were available, we tested 25 strains (n = 12 LVM and n = 13 DVM) for growth on glycogen. We compared growth on glucose-free NYCIII medium supplemented with glycogen as carbon source to growth on NYCIII medium supplemented with glucose (positive control) and NYCIII medium supplemented with water (negative control). All except one strain (RL05) showed growth on glycogen; however, six strains showed substantially less-efficient growth on glycogen. One strain showed a longer lag time (RL19; on average 4.5 h, compared to an average of 1.5 h for other strains), and five strains (RL02, RL06, RL07, RL09, and RL26) showed a lower OD after 36 h of growth compared to other strains (Fig. 4). Growth on glycogen did not correlate to whether the strain was isolated from LVM or DVM.

Correlation between growth on glycogen and N-terminal mutation in a putative type I pullulanase protein. Growth curves for individual Lactobacillus crispatus strains on minimal medium supplemented with a glycogen, b glucose, and c water. Strains that showed less-efficient or no growth on glycogen carried a mutation in the N-terminal sequence of a putative type I pullulanase protein and are indicated with a dashed line
Growth on glycogen corresponded with variation in a putative pullulanase type I gene
We followed up on the glycogen growth experiments with a gene-trait analysis as glycogen is considered to be a key, although disputed, nutrient (directly) available to L. crispatus. We searched the L. crispatus genomes for the presence/absence of enzymes that can potentially be involved in glycogen metabolism. We thus searched for orthologs of the (1) glycogen debranching enzyme (encoded by glgX) in Escherichia coli [27, 28], (2) Streptococcus agalactiae pullulanase [29], (3) SusB of Bacteroides thetaiotaomicron [30], and (4) the amylase (encoded by amyE) of Bacillus subtilis [31]. This search revealed a gene that was similar to the glgX gene; this gene was annotated as a pullulanase type I gene. In other species, this pullulanase is bound to the outer S-layer of the cell wall, suggesting that this enzyme utilizes extracellular glycogen [32]. All except two strains (RL31, RL32) carried a copy of this gene. The genes are conserved except for variation in the sequence that encodes a putative N-terminal signal peptide that may be involved in extracellular localization of the enzyme. All strains with less-efficient growth on glycogen had a 29-amino acid deletion in the N-terminal sequence (strains: RL02, RL06, RL07, RL09, RL19, and RL26) and the strain that showed no growth (RL05) had an 8-amino acid deletion in the same region as the other strains in addition to 37-amino acid deletion further downstream (Table 3).
Discussion
Key findings of this paper
Here we report the full genomes of 28 L. crispatus clinical isolates, the largest contribution of L. crispatus clinical isolates to date. These strains were isolated from women with LVM and from women with DVM. A comparative genomics analysis revealed that a glycosyltransferase gene was more frequently found in the genomes of strains isolated from DVM as compared with strains isolated from LVM, suggesting a fitness advantage for carrying this gene in L. crispatus under dysbiotic conditions and a role for surface glycoconjugates in microbiota-host interactions. Comparative experiments pertaining to biofilm formation, antimicrobial activity, and nutrient utilization showed that these two groups of strains did not phenotypically differ from each other. Of particular novelty value, we found that these clinical L. crispatus isolates were capable of growth on glycogen and that variation in a pullulanase type I gene correlates to the level of this activity.
Vaginal dysbiotic conditions may pressurize Lactobacillus crispatus to vary its glycome
Several studies have shown that vaginal dysbiosis is associated with an increased pro-inflammatory response, including an increase in pro-inflammatory chemokines and cytokines, but also elevated numbers of activated CD4+ T cells [3, 19], although no clinical signs of inflammation are present and vaginal dysbiosis is seen as a condition rather than as a disease [33]. Nonetheless, it indicates that the vaginal niche in a dysbiotic state is indeed under some immune pressure and that immune evasion could be a key (but poorly studied) trait for probiotic bacterial survival and dominance on the vaginal mucosa.
Our comparative genomics analysis revealed a glycosyltransferase (GT) gene that was more common in strains isolated from DVM compared with strains isolated from LVM. The identified GT consists of three fragments, which all align to a single GT in the reference L. crispatus genome (125-2-CHN). Sequence analyses showed that the first and longest fragment exhibits close homology to a known GT-A fold and most probably harbors the active site of the GT (Fig. 3). The latter two fragments do not harbor any structural motifs resembling known GTs and most probably do not harbor any catalytic GT activity. We hypothesize that these two fragments play a role in steering the specific activity of the GT (e.g., towards donor or substrate specificity). This might point towards L. crispatus harnessing its genetic potential to change its surface glycome. Such a process is termed phase variation and allows bacteria to rapidly adapt and diversify their surface glycans, resulting in an evolutionary advantage in the arms race between the immune system and invading bacteria. Modulation of the surface glycome by phase variation of the GT coding sequence is a common immune evasion strategy, which has been extensively studied in pathogenic bacteria like Campylobacter jejuni [25], but could be utilized by commensals as well [21]. We hypothesize that L. crispatus in DVM exploits this genetic variation to allow for (a higher) variation in cell wall glycoconjugates providing a mechanism for L. crispatus to persist at low levels in DVM and remain stealth from the immune system (Fig. 5). Of note, evidence for expression of all of the three GT fragments comes from a recent transcriptomics study that studied the effect of metronidazole treatment on the VM of women with (recurring) BV [11]. Personal communication with Dr. Zhi-Luo Deng revealed that high levels of expression for the three putative GT peptides were present in the vaginal samples of two women who were responsive to treatment (i.e., their VM was fully restored to a L. crispatus-dominated VM following treatment with metronidazole). See Additional file 2. This finding is in line with our hypothesis that the presence of the fragmented GT gene has a selective advantage for L. crispatus under dysbiotic conditions. Further functional experiments are needed to test this hypothesized host-microbe interaction and determine if and how the variation of glycoconjugates is affected by this GT. Additionally, the immunological response of the host must be further studied in reference to these hypothesized microbial adaptations. The bacterial surface glycome and related variability events are currently overlooked features in probiotic strain selection, though they might be crucial to a strain’s survival and in vivo dominance [21].

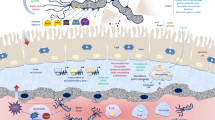
Model for enzymatic activity in glycosylation and glycogen degradation in Lactobacillus crispatus. Schematic representation of the vaginal environment with either LVM or DVM. Our comparative genomics analysis revealed a glycosyltransferase gene that was more common in Lactobacillus crispatus strains isolated from LVM (red bacteria) and DVM (low abundance of red lactobacilli, diverse bacterial population in multiple colors and forms, thinner mucus layer). We hypothesize that L. crispatus in DVM exploits this genetic variation to allow for (a higher) variation in cell wall glycoconjugates providing a mechanism for L. crispatus to persist at low levels in DVM and remain stealth from the immune system. Another finding of this work describes the ability of L. crispatus strains to utilize glycogen as a food source, which is associated with the presence of a full-length pullulanase gene (red dots on cell wall of L. crispatus). Abbreviations: LVM: Lactobacillus-dominated vaginal microbiota; DVM: dysbiotic vaginal microbiota, LC: Langerhans cell, CK: cytokines
No distinct phenotypes pertaining to dominance in vivo were observed
It has previously been postulated, relying merely on genomics data, that the accessory genome of L. crispatus could lead to strain differences relating to biofilm formation, adhesion, and competitive exclusion of pathogens [9, 10]; all of which could influence whether a strain dominates the vaginal mucosa or not. Our comparative experimental work, however, showed that L. crispatus—irrespective of whether the strain was isolated from a woman with LVM or with DVM—all formed little to no biofilm and demonstrated effective lactic acid production and effective antimicrobial activity against N. gonorrhoeae. The previous genomic analyses also suggested that L. crispatus has enzymes that can degrade various carbohydrates [10]. Indeed, we observed that L. crispatus ferments a broad range of carbohydrates, as assessed by a commercial API test, but these profiles did not differ between strains isolated from LVM or from DVM.
First evidence showing that Lactobacillus crispatus grows on glycogen
The vaginal environment of healthy reproductive-age women is distinct from other mammals in that it has low microbial diversity, a high abundance of lactobacilli, and high levels of lactic acid and luminal glycogen [34]. It has been postulated that proliferation of vaginal lactobacilli is supported by estrogen-driven glycogen production [35]; however, the “fly in the ointment”—as finely formulated by Nunn et al. [17]—is that evidence for direct utilization of glycogen by vaginal lactobacilli is absent. Moreover, previous reports have stated that the core genome of L. crispatus does not contain the necessary genes to break down glycogen [10, 36]. It has even been suggested that L. crispatus relies on amylase secretion by the host or other microbes for glycogen breakdown [17, 37], as L. crispatus does contain all the appropriate enzymes to consume glycogen breakdown products such as glucose and maltose [36]. Here we provide the first evidence suggesting that L. crispatus human isolates are capable of growing on extracellular glycogen and we identified variation in a gene which correlated with this activity. The identified gene putatively encodes a pullulanase type I enzyme belonging to the glycoside hydrolase family 13 [38]. Its closest ortholog is an extracellular cell-attached pullulanase found in Lactobacillus acidophilus [32]. The L. crispatus pullulanase gene described here carries three conserved domains, comprising an N-terminal carbohydrate-binding module family 41, a catalytic module belonging to the pullulanase super family, and a C-terminal bacterial surface layer protein (SLAP) [39] (Fig. 6). We observed that all except two of the strains in our study carry a copy of this gene. These two strains (RL31 and RL32) were no longer cultivable after their initial isolation. The six strains that showed less-efficient or no growth on glycogen all showed variation in the N-terminal part of the putative pullulanase protein. All of these deletions are upstream of the carbohydrate-binding module in a sequence encoding a putative signal peptide. This may explain the relatively slow and linear growth on glycogen of most of the mutants (Fig. 4a). The enzyme may be synthesized and not secreted in these mutants, but over time as some cells dye and lyse, the enzyme is released into the media allowing slow conversion of glycogen into sugars that can be imported to fuel growth. The presence of a SLAP domain suggests that the enzyme with a functional signal peptide is assigned to the outermost S-layer of the cell wall and is hence expected to be capable of degrading extracellular glycogen [32]. Further experiments are needed to fully characterize this pullulanase enzyme, to demonstrate its ability to metabolize glycogen in vitro, and to assess whether it degrades intra- or extracellular glycogen. Importantly, this pullulanase is likely part of a larger cluster of glycoproteins involved in glycogen metabolism in L. crispatus, which should be considered in future research.

Schematic overview of the organization of the putative pullulanase type I encoding gene in Lactobacillus crispatus. The enzyme comprises three conserved domains including an N-terminal carbohydrate-binding module family 41 with specific carbohydrate-binding sites, a catalytic module belonging to the pullulanase super family and a C-terminal bacterial surface layer protein (SLAP). The mutations (indicated by arrows) were located in an unconserved area that encodes a putative signal peptide (SP) that may be involved in subcellular localization. Abbreviations: SP: signal peptide; CBM41: carbohydrate-binding module family 41; PulA: pullulanase; SLAP: surface layer protein
Of note, we analyzed just one L. crispatus strain per vaginal sample, while it is plausible that multiple strain types co-exist in the vagina. So strain variability in growth on glycogen (and other carbohydrates) might actually benefit the L. crispatus population as a whole and explain the variation in growth on glycogen that we observed, especially considering that glycogen availability may fluctuate along with oscillating estrogen levels during the menstrual cycle. When developing probiotics, it could thus be beneficial to select for L. crispatus strains that ferment different carbohydrates (in addition to glycogen) [8] and also to supplement the probiotic with a prebiotic [40, 41].
Conclusion
Here we report whole genome sequences of 28 L. crispatus human isolates. Our comparative study led to three novel insights: (1) gene fragments encoding for a glycosyltransferase were disproportionally higher abundant among strains isolated from DVM, suggesting a role for cell surface glycoconjugates that shape vaginal microbiota-host interactions, (2) L. crispatus strains isolated from LVM do not differ from those isolated from DVM regarding the phenotypic traits studied here, including biofilm formation, pathogen inhibitory activity, and carbohydrate utilization, and (3) L. crispatus is able to grow on glycogen and this correlates with the presence of a full-length pullulanase type I gene.
Methods
L. crispatus strain selection
For this study, nurse-collected vaginal swabs were obtained from the Sexually Transmitted Infections clinic in Amsterdam, The Netherlands, from June to August 2012, as described previously by Dols et al. [4]. These vaginal samples came from women with LVM (Nugent score 0–3) and from women with DVM (Nugent score 7–10). LVM and DVM vaginal swabs were plated on tryptic soy agar supplemented with 5% sheep serum and 0.25% lactic acid and pH set to 5.5 with acetic acid and incubated under a microaerobic atmosphere (using an Anoxomat; Mart Microbiology B.V., The Netherlands) at 37 °C for 48–72 h. Candidate Lactobacillus spp. strains were selected based on colony morphology (white, small, smooth, circular, opaque colonies), and single colonies were subjected to 16S rRNA sequencing. One L. crispatus isolate per vaginal sample was taken forward for whole genome sequencing. A DNA library was prepared for these isolates using the Nextera XT DNA Library preparation kit, and the genome was sequenced using the Illumina Miseq generate FASTQ workflow.
Genome assembly and quality control
All analyses were run on a virtual machine running Ubuntu version 16.02. Contigs were assembled using the Spades assembly pipeline [42]. Contigs were discarded if they had less than 50% coverage with other assemblies or with the reference genome (N50 and NG50 values deviated more than 3 standard deviations from the mean as determined using QUAST [43]). The genomes were assembled with Spades 3.5.0 using default settings. The Spades pipeline integrates read-error correction, iterative k-mer (nucleotide sequences of length k)-based short read assembling, and mismatch correction. The quality of the assemblies was determined with Quast (History 2013) using default settings and the Lactobacillus crispatus 125-2-CHN strain as reference genome (Genbank FN692037).
Genome annotation and comparative genome analysis
After assembly, the generated contigs were sorted with Mauve contig mover [44], using the L. crispatus 125-2-CHN strain as reference genome. Contaminating sequences of human origin and adaptor sequences were identified using BLAST and manually removed. The reordered genomes were annotated using the Prokka automated annotation pipeline [45] using default settings. Additionally, the genomes were uploaded to Genbank and annotated using the NCBI integrated Prokaryotic Genome Annotation Pipeline [46]. The annotated genomes were analyzed using the sequence element enrichment analysis (SEER), which looks for an association between enriched k-mers and a certain phenotype [47]. Following the developer’s instructions, the genomes were split into k-mers using fsm-lite on standard settings and a minimum k-mer frequency of 2 and a maximum frequency of 28. The usage of k-mers enables the software to look for both SNPs as well as gene variation at the same time. After k-mer counting, the resulting file was split into 16 equal parts and g-zipped for parallelization purposes. In order to correct for the clonal population structure of bacteria, the population structure was estimated using Mash with default settings [48]. Using SEER, we looked for k-mers of various lengths that associated with whether the L. crispatus strains came from LVM or DVM. The results were filtered for k-mers with a chi-square test of association of < 0.01 and a likelihood-ratio test p value (a statistical test for the goodness of fit for two models) of < 0.0001. The resulting list of k-mers was sorted by likelihood-ratio p, and the top 50 hits were manually evaluated using BLASTx and BLASTn.
Pan and accessory genome analysis
We used the bacterial pan-genome analysis tool developed by Chaudhari et al. [49] using default settings. The circular image was created using CGview Comparison Tool [50] by running the build_blast_atlas_all_vs_all.sh script included in the package.
Comparative phenotype experiments
Not all strains were (consistently) cultivable after their initial isolation, so experimental data was collected for a subset of the strains and could differ per experiment. The ratio of cultivable LVM and DVM strains was however similar for each experiment. For a full overview of experimental procedures, we refer to the Additional file 1. In short, carbohydrate metabolism profiles were assessed using commercial API CH50 carbohydrate fermentation tests (bioMérieux, Inc., Marcy l’Etoile, France) according to the manufacturer’s protocol. To assess organic acid production, strains were grown on medium that mimicked vaginal secretions [26]. Total metabolite extracts from spent medium were assessed as previously described by Collins et al. [41]. Biofilm formation was assessed using the crystal violet assay as described by Santos et al. [51] and autoaggregation as described by Younes et al. [52]. Antimicrobial activity against Neisseria gonorrhoeae was assessed by challenging N. gonorrhoeae (WHO-L strain) with varying (neutralized with NaOH to pH 7.0) dilutions of L. crispatus supernatants. Inhibitory effect was assessed as percentile difference in OD600nm in a conditional stationary phase as compared to the control.
Glycogen degradation assay
Starter cultures were grown in regular NYCIII glucose medium for 72 h. For this assay, 1.1× carbohydrate-deprived NYCIII medium was supplemented with water (negative control), 5% glucose (positive control), or 5% glycogen (Sigma-Aldrich, Saint Louis, USA) and subsequently inoculated with 10% (v/v) bacterial culture (OD ~ 0.5; 109 CFU/ml). Growth on glycogen was compared to growth on NYCII without supplemented carbon source and to NYCIII with glucose. Growth curves were followed in a BioScreen (Labsystems, Helsinki, Finland). At least two independent experiments per strain were performed in triplicate.
Abbreviations
- COG:
-
Cluster ortholog groups
- DVM:
-
Dysbiotic vaginal microbiota
- GT:
-
Glycosyltransferase
- LVM:
-
Lactobacillus-dominated vaginal microbiota
- VM:
-
Vaginal microbiota
References
DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, Sun CL, Goltsman DS, Wong RJ, Shaw G, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci U S A. 2015;112(35):11060–5.
Tamarelle J, Thiébaut ACM, de Barbeyrac B, Bébéar C, Ravel J, Delarocque-Astagneau E. The vaginal microbiota and its association with human papillomavirus, Chlamydia trachomatis, Neisseria gonorrhoeae and Mycoplasma genitalium infections: a systematic review and meta-analysis. Clin Microbiol Infect. 2019 Jan;25(1):35-47.
Borgdorff H, van der Veer C, van Houdt R, Alberts CJ, de Vries HJ, Bruisten SM, Snijder MB, Prins M, Geerlings SE, van der Loeff MF S, et al. The association between ethnicity and vaginal microbiota composition in Amsterdam, the Netherlands. PLoS One. 2017;12(7):e0181135.
Dols JA, Molenaar D, van der Helm JJ, Caspers MP, de Kat A-BA, Schuren FH, Speksnijder AG, Westerhoff HV, Richardus JH, Boon ME, et al. Molecular assessment of bacterial vaginosis by Lactobacillus abundance and species diversity. BMC Infect Dis. 2016;16:180.
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–7.
van der Veer C, Bruisten SM, van der Helm JJ, de Vries HJ, van Houdt R. The cervicovaginal microbiota in women notified for chlamydia trachomatis infection: a case-control study at the Sexually Transmitted Infection Outpatient Clinic in Amsterdam, The Netherlands. Clin Infect Dis. 2017;64(1):24–31.
Kort R. Personalized therapy with probiotics from the host by TripleA. Trends Biotechnol. 2014;32(6):291–3.
Kort R, van der Veer C. A new probiotic composition for the prevention of bacterial vaginosis. 2017. European Patent 17181005.
Abdelmaksoud AA, Koparde VN, Sheth NU, Serrano MG, Glascock AL, Fettweis JM, Strauss JF 3rd, Buck GA, Jefferson KK. Comparison of Lactobacillus crispatus isolates from Lactobacillus-dominated vaginal microbiomes with isolates from microbiomes containing bacterial vaginosis-associated bacteria. Microbiology. 2016;162(3):466–75.
Ojala T, Kankainen M, Castro J, Cerca N, Edelman S, Westerlund-Wikstrom B, Paulin L, Holm L, Auvinen P. Comparative genomics of Lactobacillus crispatus suggests novel mechanisms for the competitive exclusion of Gardnerella vaginalis. BMC Genomics. 2014;15:1070.
Deng ZL, Gottschick C, Bhuju S, Masur C, Abels C, Wagner-Dobler I. Metatranscriptome analysis of the vaginal microbiota reveals potential mechanisms for protection against metronidazole in bacterial vaginosis. mSphere. 2018;3(3):e00262-18.
Atassi F, Brassart D, Grob P, Graf F, Servin AL. Lactobacillus strains isolated from the vaginal microbiota of healthy women inhibit Prevotella bivia and Gardnerella vaginalis in coculture and cell culture. FEMS Immunol Med Microbiol. 2006;48(3):424–32.
Foschi C, Salvo M, Cevenini R, Parolin C, Vitali B, Marangoni A. Vaginal lactobacilli reduce Neisseria gonorrhoeae viability through multiple strategies: an in vitro study. Front Cell Infect Microbiol. 2017;7:502.
Gong Z, Luna Y, Yu P, Fan H. Lactobacilli inactivate chlamydia trachomatis through lactic acid but not H2O2. PLoS One. 2014;9(9):e107758.
Graver MA, Wade JJ. The role of acidification in the inhibition of Neisseria gonorrhoeae by vaginal lactobacilli during anaerobic growth. Ann Clin Microbiol Antimicrob. 2011;10:8.
Nardini P, Nahui Palomino RA, Parolin C, Laghi L, Foschi C, Cevenini R, Vitali B, Marangoni A. Lactobacillus crispatus inhibits the infectivity of Chlamydia trachomatis elementary bodies, in vitro study. Sci Rep. 2016;6:29024.
Nunn KL, Forney LJ. Unraveling the dynamics of the human vaginal microbiome. Yale J Biol Med. 2016;89(3):331–7.
Borgdorff H, Gautam R, Armstrong SD, Xia D, Ndayisaba GF, van Teijlingen NH, Geijtenbeek TB, Wastling JM, van de Wijgert JH. Cervicovaginal microbiome dysbiosis is associated with proteome changes related to alterations of the cervicovaginal mucosal barrier. Mucosal Immunol. 2016;9(3):621–33.
Gosmann C, Anahtar MN, Handley SA, Farcasanu M, Abu-Ali G, Bowman BA, Padavattan N, Desai C, Droit L, Moodley A, et al. Lactobacillus-deficient Cervicovaginal bacterial communities are associated with increased HIV Acquisition in Young South African Women. Immunity. 2017;46(1):29–37.
Witkin SS, Mendes-Soares H, Linhares IM, Jayaram A, Ledger WJ, Forney LJ. Influence of vaginal bacteria and D- and L-lactic acid isomers on vaginal extracellular matrix metalloproteinase inducer: implications for protection against upper genital tract infections. MBio. 2013;4(4):e00460-13.
Tytgat HLP, de Vos WM. Sugar coating the envelope: glycoconjugates for microbe-host crosstalk. Trends Microbiol. 2016;24(11):853–61.
Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11(5):472–7.
Sybesma W, Molenaar D, van IW VK, Kort R. Genome instability in Lactobacillus rhamnosus GG. Appl Environ Microbiol. 2013;79(7):2233–9.
Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–55.
Tytgat HL, Lebeer S. The sweet tooth of bacteria: common themes in bacterial glycoconjugates. Microbiol Mol Biol Rev. 2014;78(3):372–417.
Geshnizgani AM, Onderdonk AB. Defined medium simulating genital tract secretions for growth of vaginal microflora. J Clin Microbiol. 1992;30(5):1323–6.
Strydom L, Jewell J, Meier MA, George GM, Pfister B, Zeeman S, Kossmann J, Lloyd JR. Analysis of genes involved in glycogen degradation in Escherichia coli. FEMS Microbiol Lett. 2017;364(3):1–7.
Dauvillee D, Kinderf IS, Li Z, Kosar-Hashemi B, Samuel MS, Rampling L, Ball S, Morell MK. Role of the Escherichia coli glgX gene in glycogen metabolism. J Bacteriol. 2005;187(4):1465–73.
Santi I, Pezzicoli A, Bosello M, Berti F, Mariani M, Telford JL, Grandi G, Soriani M. Functional characterization of a newly identified group B Streptococcus pullulanase eliciting antibodies able to prevent alpha-glucans degradation. PLoS One. 2008;3(11):e3787.
Kitamura M, Okuyama M, Tanzawa F, Mori H, Kitago Y, Watanabe N, Kimura A, Tanaka I, Yao M. Structural and functional analysis of a glycoside hydrolase family 97 enzyme from Bacteroides thetaiotaomicron. J Biol Chem. 2008;283(52):36328–37.
Yamazaki H, Ohmura K, Nakayama A, Takeichi Y, Otozai K, Yamasaki M, Tamura G, Yamane K. Alpha-amylase genes (amyR2 and amyE+) from an alpha-amylase-hyperproducing Bacillus subtilis strain: molecular cloning and nucleotide sequences. J Bacteriol. 1983;156(1):327–37.
Moller MS, Goh YJ, Rasmussen KB, Cypryk W, Celebioglu HU, Klaenhammer TR, Svensson B, Abou Hachem M. An extracellular cell-attached pullulanase confers branched alpha-glucan utilization in human gut Lactobacillus acidophilus. Appl Environ Microbiol. 2017;83(12):e00402-17.
Reid G. Is bacterial vaginosis a disease? Appl Microbiol Biotechnol. 2018;102(2):553–8.
Petrova MI, van den Broek M, Balzarini J, Vanderleyden J, Lebeer S. Vaginal microbiota and its role in HIV transmission and infection. FEMS Microbiol Rev. 2013;37(5):762–92.
Mirmonsef P, Hotton AL, Gilbert D, Burgad D, Landay A, Weber KM, Cohen M, Ravel J, Spear GT. Free glycogen in vaginal fluids is associated with Lactobacillus colonization and low vaginal pH. PLoS One. 2014;9(7):e102467.
France MT, Mendes-Soares H, Forney LJ. Genomic comparisons of Lactobacillus crispatus and Lactobacillus iners reveal potential ecological drivers of community composition in the vagina. Appl Environ Microbiol. 2016;82(24):7063–73.
Spear GT, French AL, Gilbert D, Zariffard MR, Mirmonsef P, Sullivan TH, Spear WW, Landay A, Micci S, Lee BH, et al. Human alpha-amylase present in lower-genital-tract mucosal fluid processes glycogen to support vaginal colonization by lactobacillus. J Infect Dis. 2014;210(7):1019–28.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database issue):D490–5.
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(Database issue):D222–6.
Gibson GR, Hutkins R, Sanders ME, Prescott SL, Reimer RA, Salminen SJ, Scott K, Stanton C, Swanson KS, Cani PD, et al. Expert consensus document: the International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol. 2017;14(8):491–502.
Collins SL, McMillan A, Seney S, van der Veer C, Kort R, Sumarah MW, Reid G. Promising prebiotic candidate established by evaluation of lactitol, lactulose, raffinose, and oligofructose for maintenance of a lactobacillus-dominated vaginal microbiota. Appl Environ Microbiol. 2018;84(5).
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5.
Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics. 2009;25(16):2071–3.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44(14):6614–24.
Lees JA, Vehkala M, Valimaki N, Harris SR, Chewapreecha C, Croucher NJ, Marttinen P, Davies MR, Steer AC, Tong SY, et al. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat Commun. 2016;7:12797.
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17(1):132.
Chaudhari NM, Gupta VK, Dutta C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373.
Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView comparison tool. BMC Genomics. 2012;13:202.
Santos CM, Pires MC, Leao TL, Hernandez ZP, Rodriguez ML, Martins AK, Miranda LS, Martins FS, Nicoli JR. Selection of Lactobacillus strains as potential probiotics for vaginitis treatment. Microbiology. 2016;162(7):1195–207.
Younes JA, van der Mei HC, van den Heuvel E, Busscher HJ, Reid G. Adhesion forces and coaggregation between vaginal staphylococci and lactobacilli. PLoS One. 2012;7(5):e36917.
Acknowledgements
We thank Dr. Titia Heijman of the Sexually Transmitted Infections clinic in Amsterdam, The Netherlands, for organizing the collection of the clinical vaginal samples. We acknowledge Liesbeth Hoekman (TNO) for isolation and initial characterization of Lactobacillus crispatus strains. We thank Mark Sumarah and Justin Renaud for facilitating the metabolomics analysis. We also thank Dr. Zhi-Luo Deng for mining his transcriptomics data for the GT gene fragments and pullulanase gene transcripts.
Funding
This research was funded by Public Health Service Amsterdam (GGD), the VU University of Amsterdam (VU) and the Netherlands Organization for Applied Scientific Research (TNO). HT holds a Marie Sklodowska-Curie fellowship of the European Union’s Horizon 2020 research and innovation program under agreement No 703577 (Glycoli) to support her work at ETH Zurich.
Availability of data and materials
The 28 Lactobacillus crispatus sequenced genomes described in this paper have been deposited at DDBJ/ENA/GenBank BioProject PRJNA390079 under the accessions NKKQ00000000-NKLR00000000.
Author information
Authors and Affiliations
Contributions
RK, SB, HdV, and FS conceptualized the study. CV and JS performed the experimental work, supervised by AdKA, SB, and RK. JS performed the bio-informatic analyses, supervised by DM and RK. RH did the initial glycogen finding and provided further expertise. HT provided expertise for the glycosyltransferase finding and GR for the potential of probiotic applications. CV drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research proposed in this study was evaluated by the ethics review board of the Academic Medical Center (AMC), University of Amsterdam, The Netherlands. According to the review board no additional ethical approval was required for this study, as the vaginal samples used here were collected as part of routine procedure for cervical examinations at the STI clinic in Amsterdam (document reference number W12_086 # 12.17.0104).
Consent for publication
Clients of the STI clinic were notified that remainders of their samples could be used for scientific research, after anonymization of client clinical data and samples. If the clients objected, their data and samples were discarded. This procedure has been approved by the AMC ethics review board (reference number W15_159 # 15.0193).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Biofilm formation, auto-aggregation, carbohydrate degradation, organic acid production, and antimicrobial activity of Lactobacillus crispatus strains. (DOCX 2146 kb)
Additional file 2:
Transcripts of Lactobacillus crispatus glycosyltransferase gene fragments in vaginal samples of women treated for bacterial vaginosis. (XLSX 11 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
van der Veer, C., Hertzberger, R.Y., Bruisten, S.M. et al. Comparative genomics of human Lactobacillus crispatus isolates reveals genes for glycosylation and glycogen degradation: implications for in vivo dominance of the vaginal microbiota. Microbiome 7, 49 (2019). https://doi.org/10.1186/s40168-019-0667-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-019-0667-9