
Abstract
Background
Increasing evidence suggests that gut microbiota play a role in the pathogenesis of breast cancer. The composition and functional capacity of gut microbiota associated with breast cancer have not been studied systematically.
Methods
We performed a comprehensive shotgun metagenomic analysis of 18 premenopausal breast cancer patients, 25 premenopausal healthy controls, 44 postmenopausal breast cancer patients, and 46 postmenopausal healthy controls.
Results
Microbial diversity was higher in breast cancer patients than in controls. Relative species abundance in gut microbiota did not differ significantly between premenopausal breast cancer patients and premenopausal controls. In contrast, relative abundance of 45 species differed significantly between postmenopausal patients and postmenopausal controls: 38 species were enriched in postmenopausal patients, including Escherichia coli, Klebsiella sp_1_1_55, Prevotella amnii, Enterococcus gallinarum, Actinomyces sp. HPA0247, Shewanella putrefaciens, and Erwinia amylovora, and 7 species were less abundant in postmenopausal patients, including Eubacterium eligens and Lactobacillus vaginalis. Acinetobacter radioresistens and Enterococcus gallinarum were positively but weakly associated with expression of high-sensitivity C-reactive protein; Shewanella putrefaciens and Erwinia amylovora were positively but weakly associated with estradiol levels. Actinomyces sp. HPA0247 negatively but weakly correlated with CD3+CD8+ T cell numbers. Further characterization of metagenome functional capacity indicated that the gut metagenomes of postmenopausal breast cancer patients were enriched in genes encoding lipopolysaccharide biosynthesis, iron complex transport system, PTS system, secretion system, and beta-oxidation.
Conclusion
The composition and functions of the gut microbial community differ between postmenopausal breast cancer patients and healthy controls. The gut microbiota may regulate or respond to host immunity and metabolic balance. Thus, while cause and effect cannot be determined, there is a reproducible change in the microbiota of treatment-naive patients relative to matched controls.
Similar content being viewed by others

Background
The human gut harbors thousands of bacterial species, together making up a population as large as 1013–14 microbes, which encode 150-fold more genes than the human genome [1,2,3]. The gut microbiota is composed of a large number of anaerobic microorganisms, predominantly Bacteroidetes and Firmicutes [4, 5], which are affected by a multitude of factors including host genetics, lifestyle, and environment. The microbiota plays important roles in maintaining an intestinal mucosal barrier, antagonizing the colonization of pathogenic microorganisms, and contributing to metabolism and immune homeostasis [6].
The gut microbiota has been linked to various diseases, such as inflammatory bowel disease [7, 8], obesity [9], diabetes [10, 11], rheumatoid arthritis [12], atopic manifestations [13], liver cirrhosis [14], cardiovascular diseases [15], mental diseases [16, 17], and colorectal cancer [18, 19]. The gut microbiota exert an influence on both local and systemic metabolism and immunity [20, 21], and alterations of gut microbiota have been associated with extra-intestinal cancers including hepatocellular carcinoma, to which they may contribute by triggering chronic inflammation and altering microenvironment and metabolism [22, 23].
Increasing evidence suggests that microbe-host interactions have the potential to influence or serve as a biomarker of breast cancer pathogenesis [24, 25]. A comparison of 11 breast cancer patients and 7 healthy controls revealed differences in the gut microbiota, with Clostridia, Enterobacterium, Lactobacilli, Bacteroides, and Escherichia coli enriched in patients [24]. A comparison of 48 postmenopausal breast cancer case patients and 48 healthy controls [25] revealed an altered, less diverse gut microbiota in patients: Clostridiales, Clostridiaceae, Faecalibacterium, and Ruminococcaceae were enriched in patients, while Dorea and Lachnospiraceae were relatively less abundant in patients. Among controls, microbiota diversity correlated with total estrogen levels, suggesting that gut microbiota may be implicated in breast cancer by responding to or affecting estrogen metabolism.
Those previous studies have provided useful insights into the potential response of gut microbiota in breast cancer, but they have not been able to comprehensively catalog the taxonomies of the microbiota because they have relied on only biochemical analysis or 16S rRNA gene sequencing. In addition, previous studies did not explore the functional capacity of the gut microbiota in patients with breast cancer, which could provide more mechanistic insights into the role of the gut microbiota in this disease. As a result, how the gut microbiota and their biochemical and metabolic products change in breast cancer is unclear.
To address these questions, we used shotgun metagenomic analysis to compare the gut microbial community and its functional capabilities between breast cancer patients and healthy controls.
Methods
Subjects
The study was approved by the Ethics Committee of the Affiliated Tumor Hospital of Guangxi Medical University (Nanning, China). Fecal samples were collected from 18 premenopausal breast cancer patients, 25 premenopausal healthy controls, 44 postmenopausal breast cancer patients, and 46 postmenopausal healthy controls (Table 1). All patients with breast cancer were diagnosed by pathological examination at the Affiliated Tumor Hospital, and healthy controls were recruited from the Medical Examination Center of the First Affiliated Hospital of Guangxi Medical University. Controls were free of breast cancer at medical examination. None of the study subjects had diarrhea, diabetes, ulcerative colitis, Crohn’s disease, or other infectious diseases. No subjects took antibiotics, steroid hormones, Chinese herbal medicine (including oral, intramuscular, or intravenous injection), or probiotics such as yogurt during the 3 months before fecal sample collection. Breast cancer patients did not receive chemotherapy, radiation, or surgery prior to fecal sample collection.
Fecal samples were freshly collected from individuals and transported to the laboratory on ice. Samples were stored at − 80 °C until extraction. Bacterial DNA was extracted from fecal samples using the QIAampDNA Stool MiniKit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Metagenomic DNA sequencing and annotation
All samples were sequenced on the Illumina HiSeq × 10 platform. A paired-end library was constructed with 350-bp inserts for each sample. Low-quality reads and reads mapping to human DNA were removed from the raw data. For taxonomic assignments, the high-quality reads from each sample were aligned against the integrated reference catalog of the human gut microbiome (IGC) by bowtie2 using the criterion of “identity > 90%,” genes from the existing reference gene catalog IGC inherited their original taxonomic annotation, and the relative abundance of a taxon was calculated from the relative abundance of its genes [26, 27]. During KO profiling, genes from IGC inherited their original KO annotation, and KO abundance was calculated by summing the relative abundance of genes annotated to the same KO [26, 27].
Quantification of virulence factors and pathogen-host interaction genes
Genes in the catalog were aligned against proteins in the Virulence Factors of Pathogenic Bacteria database [28] using BLAST (version 2.2.24) set to default parameters, except that -p blastx -e 1e-5 -F F -a 4 -m 8. We selected the matches with the highest-scoring annotated hit containing identity > 40% and high-scoring segment pair scoring > 60 bits. The relative abundance of a virulence factor was calculated by summing the relative abundance of genes annotated to a feature. Genes in the gene catalog were aligned against the proteins in the Pathogen-Host Interactions database [29] using BLAST (version 2.2.24) set to default parameters except that -p blastx -e 1e-5 -F F -a 4 -m 8. We selected the matches with the highest-scoring annotated hits containing an identity > 40% and high-scoring segment pair scoring > 60 bits. The relative abundance of an interaction gene was calculated by summing the abundance of genes annotated to a feature.
Gut microbiota diversity
Based on the species profile, we calculated the within-sample (alpha) diversity to estimate gut microbiota richness and evenness based on the Shannon index and Chao1 index [30]. High alpha diversity indicates high diversity of gut microbiota within a sample. Between-sample differences in microbial composition (beta diversity) were assessed in terms of the Jensen-Shannon divergence (JSD) [31]. The JSD was calculated by the following steps: (1) We first calculated JSD between each two samples within one group. (2) The mean of all JSD values between a sample and others within one group was computed (the mean value represented the similarity of the sample to others). We compared the mean JSD values to find if the beta diversity is different or not among the groups.
Enterotyping
Samples were clustered based on relative genus abundances using JSD distance and the “partitioning around medoids” (PAM) clustering algorithm. The Calinski-Harabasz (CH) index was used to calculate the optimal number of clusters [32]. Principal component analysis was used to visualize the taxonomic drivers of clusters.
Statistical analysis
Demographics were compared among groups using Student’s t test or the chi-square test in SPSS 16.0 software (IBM, Chicago, IL, USA). R software (version 3.3.2) was used to perform other analyses. The Wilcoxon rank sum test was used to identify significant differences in abundance of genes, genera, virulence factors, interaction genes, and KOs. Differentially enriched pathways and modules were identified according to their reporter score from the Z scores of significant KOs. A module with a reporter score of Z > 1.6 was defined as differentially enriched [33, 34]. P values were adjusted based on the false discovery rate (FDR) using the method of Benjamini and Hochberg [35]. Permutational multivariate analysis of variance (PERMANOVA) using the “adonis” function in the R Vegan package was performed to assess effects of phenotype on gene/taxa profiles. The R package “ade4,” which involves instrumental principal component analysis [36], was used to visualize the taxonomic drivers of clusters during enterotyping. The “pheatmap” package (version 1.0.8) was used to generate heat maps, and the clustering method used in “pheatmap” function was “correlation.” Spearman’s rank correlation was used to find correlations of metagenomic features and clinical indices.
A species-based classifier was trained using the random forest package in R. A tenfold cross-validation was performed on a random forest model using the relative species abundance profile. The minimum error was calculated using fivefold cross-validation with the “rfcv” function, and the minimum error plus the s.d. at that point was used as the cutoff. The optimal number of species was selected by cross-validation with one SE rule. The case probability was calculated using this set of species and a receiver operating characteristic (ROC) curve within the pROC package in R. The model was tested on the testing set, and the prediction error was determined [37]. Differentially enriched genes were identified using the Wilcoxon rank test, and adjusted P values were estimated using the R package “q value” (version 2.2.2). All differentially enriched genes (q value < 0.05) were annotated to the butanoate metabolism pathways (using their original KO annotation which was inherited from the integrated reference catalog of the human gut microbiome database).
Results
Taxonomic characterization of gut microbiota in breast cancer patients and healthy controls
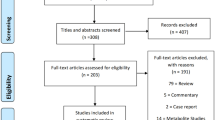
A total of 133 stool samples were sequenced from premenopausal breast cancer patients (n = 18), premenopausal healthy controls (n = 25), postmenopausal breast cancer patients (n = 44), and postmenopausal healthy controls (n = 46). The premenopausal breast cancer patients and controls were similar for age, BMI, and ethnicity (P > 0.05, Table 1); the postmenopausal breast cancer patients and controls were similar for age, BMI, age at menopause, and ethnicity (P > 0.05, Table 1). A total of 965 million 150-bp paired-end reads were generated, with an average (s.d.) of 7.25 ± 1.13 million reads for each sample. After quality control, we obtained 902 million high-quality reads free of adaptor and human DNA contaminants, with an average (s.d.) of 6.78 ± 1.08 million reads per sample (Additional file 1: Table S1).
To determine whether the sequencing adequately captured the gene diversity of the gut microbiota, rarefaction analysis was performed. The curves in all samples were near saturation, suggesting that the sequencing depth was sufficient to capture most gene diversity (Additional file 2: Figure S1).
A similar number of species was detected in premenopausal breast cancer patients and premenopausal healthy controls (P = 0.767, Wilcoxon rank sum test, Fig. 1a). Based on the species profile, we calculated the within-sample (alpha) diversity to estimate gut microbiota richness and evenness based on the Shannon index and Chao1 index. The mean Chao1 index was similar between premenopausal breast cancer patients and premenopausal controls (P = 0.777, Wilcoxon rank sum test, Fig. 1b). The mean Shannon index was higher for premenopausal patients than premenopausal controls (P = 0.027, Wilcoxon rank sum test, Fig. 1c).

Diversity of gut microbiota in breast cancer patients and healthy controls. a Total number of species in the four groups. b, c Alpha diversity of the four cohorts at species level, measured in terms of the Chao1 index and Shannon index. d Beta diversity of the four cohorts at species level. Each dot refers to a sample; if a sample has a high average JSD value, it indicates that the gut microbiota community structure of this sample is very different. Furthermore, if most samples of a group have high average JSD values, it indicates that the between-sample variability of the group is high. NS non-significant. *P < 0.05, **P < 0.01, ***P < 0.001
In contrast, the between-sample variability (beta diversity) of the gut microbiota community structure tended to be lower in premenopausal patients than in premenopausal controls (P = 0.056, Fig. 1d).
The number of species was significantly higher in postmenopausal breast cancer patients than in postmenopausal controls (P = 0.003, Wilcoxon rank sum test, Fig. 1a). Consistently, the mean Chao1 index was higher in postmenopausal patients than in postmenopausal controls (P = 0.007, Wilcoxon rank sum test, Fig. 1b). However, mean Shannon index was similar between postmenopausal breast cancer patients and postmenopausal controls (P = 0.502, Wilcoxon rank sum test, Fig. 1c). Beta diversity was higher for postmenopausal patients than postmenopausal controls (P < 0.001, Fig. 1d).
Previous studies have suggested that the human gut microbiome can be assigned to several robust enterotypes [38, 39]. To group the breast cancer patients and control samples into enterotype clusters, we applied the PAM method using JSD for the relative abundance of genera.
The optimal number of enterotypes was 2 as indicated by the CH index (Additional file 2: Figure S2a). Principal component analysis was used to cluster the samples of the four groups into two enterotypes (Additional file 2: Figure S2b). Enterotype 1 had a relatively high level of Bacteroides, enterotype 2, a relatively high level of Prevotella (Additional file 2: Figure S2c). These two enterotypes have been observed in European and Chinese populations [38, 39]. However, we found no significant relationship between enterotype and breast cancer disease status, either when we compared premenopausal patients with premenopausal controls (P = 0.141) or when we compared postmenopausal patients with postmenopausal controls (P = 0.445; Fisher’s exact test in both cases; Additional file 1: Table S2; Additional file 2: Figure S2d).
To further explore features of the gut microbial community in breast cancer patients, we compared the relative abundances of species between patients and controls. The taxonomic assignment for the metagenomic data was carried out using bowtie. The relative abundance of gut microbiota was calculated by summing the abundance of genes. Relative abundance of the gut microbiota in the four groups at the species level is shown in Fig. 2.

Relative abundance of the gut microbiota in the four groups at species level
There was no significant difference in gut microbiota species between premenopausal breast cancer patients and premenopausal healthy controls (q value > 0.05, Wilcoxon rank sum test; Additional file 1: Table S3). In contrast, 45 species differed significantly between postmenopausal patients and postmenopausal controls: 38 species were enriched in patients, including Escherichia coli, Klebsiella sp_1_1_55, and Prevotella amnii, while 7 species were reduced in patients, including Porphyromonas uenonis, Eubacterium eligens, and Lactobacillus vaginalis (q value < 0.05; Table 2; Additional file 1: Table S4, Fig. 3). PERMANOVA analysis showed that breast cancer status, menopause status, and age were significant factors for explaining the variation in the examined gut microbial samples (P < 0.05; Additional file 1: Table S5).

Relative abundance of 45 species differing significantly between postmenopausal breast cancer patients and postmenopausal healthy controls
Identification of postmenopausal breast cancer patients based on gut microbiota
To illustrate the potential diagnostic value of gut microbiota for breast cancer in postmenopausal women, we used a random forest classifier in an attempt to detect breast cancer samples from among a mixture of samples from postmenopausal patients and healthy controls. Tenfold cross-validation was repeated for five times with a training set consisting of 44 postmenopausal patients and 46 postmenopausal controls; 14 optimal species markers were selected, including Escherichia coli, Shigella sp_D9, Eubacterium eligens, Proteus mirabilis, and Fusobacterium varium (Additional file 2: Figure S6, Additional file 1: Table S6). ROC curves for the training set showed a remarkable performance in the training set when discriminating between postmenopausal breast cancer patients and postmenopausal healthy controls by specificity and sensitivity; the area under receiver operating curve (AUC) was 85.52; and 95% confidence interval (CI) is 77.57–93.47% (Fig. 4b). Next, we tested the same markers for their ability to detect breast cancer among 43 samples not used during training, comprising 18 premenopausal breast cancer patients and 25 premenopausal healthy controls. The AUC was 72% (95% CI 56.01–88.44%; Fig. 4d).

Classification to differentiate samples from postmenopausal breast cancer patients or postmenopausal healthy controls. a Probability of postmenopausal breast cancer in the training set. b ROC curves for the training set. The AUC was 87.25% (95% CI 79.82–94.68%). Classification of the test set consisted of 18 premenopausal breast cancer patients (red) and 25 premenopausal healthy controls (blue). c Classification of each sample. We used two cutoffs to assign the samples into three groups: 50% cases were classified into “Case” (probability of case > 50%), and 68% controls were classified into “Not case” (probability of case < 35%). Four cases and five controls stayed in “Uncertain.” d ROC for the test set. The AUC is 72% and the 95% CI is 56.01–88.44%
Quantification of virulence factors and pathogen-host interaction genes in the gut microbiota of postmenopausal breast cancer patients and postmenopausal healthy controls
To analyze proteins encoded by genes in gut microbiota, genes in the catalog were aligned against proteins in the Pathogen-Host Interactions (PHI) database and in the Virulence Factors of Pathogenic Bacteria database. The relative abundances of genes encoding virulence factors or pathogen-host interaction were calculated by summing the abundances of genes annotated to a feature.
PHI genes were relatively more abundant in postmenopausal breast cancer patients than in postmenopausal controls (P = 0.021, Fig. 5a). The top 15 representation of Pathogen-Host Interactions genes implicated in several human diseases, such as urinary tract infections, other infections, and tuberculosis (q value < 0.05, Wilcoxon rank sum test, Fig. 5b; Additional file 1: Table S7).

Relative abundance of genes encoding virulence factors and pathogen-host interactions in postmenopausal breast cancer patients and postmenopausal controls. a Relative abundance of pathogen-host interaction genes in the two groups. b The top 15 representation of Pathogen-Host Interactions genes in the two groups and their implication in human diseases. c Relative abundance of virulence factor genes in the two groups. d Relative abundance of the top 15 virulence factor genes in the two groups
Virulence factors were relatively more abundant in postmenopausal patients than in controls (P = 0.016, Fig. 5c); these factors included LOS glycosyltransferase, peritrichous flagella, and type I fimbriae (q value < 0.05, Wilcoxon rank sum test, Fig. 5d; Additional file 1: Table S8).
Association between gut microbiota and clinical indices
Next, we asked whether the 45 microbial species differing between postmenopausal breast cancer patients and postmenopausal controls correlated with well-established clinical indices of breast cancer based on Spearman correlation analysis (Fig. 6). Two gut microbiota species, Acinetobacter radioresistens (Spearman rho = 0.413, P = 0.015, q value > 0.05) and Vibrio cholerae (Spearman rho = 0.349, P = 0.043, q value > 0.05), positively but weakly correlated with C4 levels, whereas Yersinia enterocolitica (Spearman rho = − 0.345, P = 0.046, q value > 0.05) negatively but weakly correlated with C4 levels. Two species, Acinetobacter radioresistens (Spearman rho = 0.442, P = 0.009, q value > 0.05) and Enterococcus gallinarum (Spearman rho = 0.386, P = 0.024, q value > 0.05), positively but weakly correlated with levels of high-sensitivity C-reactive protein (hsCRP). Anaerococcus vaginalis (Spearman rho = 0.48, P = 0.002, q value > 0.05) and Porphyromonas uenonis (Spearman rho = 0.42, P = 0.009, q value > 0.05) positively but weakly correlated with CD19 levels, while Enterococcus gallinarum (Spearman rho = − 0.351, P = 0.031, q value > 0.05) negatively but weakly correlated with CD19 levels. Shewanella putrefaciens (Spearman rho = 0.379, P = 0.025, q value > 0.05) and Erwinia amylovora (Spearman rho = 0.351, P = 0.039, q value > 0.05) positively but weakly correlated with estradiol levels. Actinomyces sp. HPA0247 (Spearman rho = − 0.384, P = 0.017, q value > 0.05) negatively but weakly correlated with CD3+CD8+ T cell numbers. All these gut flora species were enriched in postmenopausal breast cancer patients relative to controls. Conversely, Eubacterium eligens was enriched in controls relative to patients, yet it negatively but weakly correlated with CD3+CD4+ T cell numbers (Spearman rho = − 0.349, P = 0.032, q value > 0.05) and IgA levels (Spearman rho = − 0.532, P = 0.001, q value > 0.05).

Correlation between gut microbiota species and clinical indices of breast cancer. Spearman’s rank correlation coefficient is indicated using a color gradient: red indicates positive correlation; blue, negative correlation. *P < 0.05
Metabolic functions of the gut microbiota in breast cancer patients and healthy controls
We explored functional features of the gut microbiota across the four groups in our study by annotating the gene catalog based on the KEGG modules. Modules differing between breast cancer patients and healthy controls with a reporter score > 1.6 were identified.
Among premenopausal women, 44 KEGG modules were significantly different between patients and controls (q value < 0.05, Wilcoxon rank sum test, Fig. 7a; Additional file 1: Table S9). Modules enriched in patients included the PTS system, secretion system, vitamin B12 transport system, amino acid transport system, and manganese/iron transport system. Modules enriched in controls included aminoacyl-tRNA biosynthesis, coenzyme A biosynthesis, nucleotide synthesis, and dicarboxylate-hydroxybutyrate cycle.

Functions of genes expressed in gut microbiota in pre- and postmenopausal breast cancer patients and healthy controls. a Gene functions in gut microbiota in premenopausal patients and controls; 26 KEGG modules were enriched in patients (red), and 18 were enriched in controls (blue). b Gene functions in gut microbiota in postmenopausal patients and controls; 43 KEGG modules were enriched in patients (blue)
Among postmenopausal women, 43 KEGG modules were enriched in patients, including lipopolysaccharide biosynthesis, iron complex transport system, vitamin B12 transport system, PTS system, secretion system, amino acid transport system, and beta-oxidation (q value < 0.05, Wilcoxon rank sum test, Fig. 7b; Additional file 1: Table S10).
The genes annotated to butanoate metabolism pathways differentially enriched in gut microbiome of postmenopausal patients and controls. Fourteen butyrate-synthesis genes were found: 10 genes were enriched in controls and 4 genes were enriched in postmenopausal patients (q value < 0.05, Wilcoxon rank sum test; Additional file 1: Table S11).
Discussion
Here, we performed a comprehensive metagenomic comparison of gut microbiota in breast cancer patients and healthy controls. Microbiota were analyzed in terms of taxonomic profile, genetic functional capacity, and associations with clinical indices of breast cancer. Our results identify various compositional and functional features of the gut microbiota metagenome that differ between postmenopausal patients and healthy controls, suggesting that they may be associated with postmenopausal breast cancer.
Significant taxonomic differences in gut microbiota were not detected between premenopausal breast cancer patients and controls. In contrast, several bacterial species were found to be enriched in postmenopausal patients relative to controls: Escherichia coli, Citrobacter koseri, Acinetobacter radioresistens, Enterococcus gallinarum, Shewanella putrefaciens, Erwinia amylovora, Actinomyces sp. HPA0247, Salmonella enterica, and Fusobacterium nucleatum. These results are consistent with previous research on several of these species, which has suggested associations with breast cancer [24]. In addition, we found a weak positive correlation of Shewanella putrefaciens and Erwinia amylovora with estradiol (P < 0.05), although there is no statistical significance after correction for multiple testing (q value > 0.05), which may be related to the small sample, but the association can still be considered exploratory. These results are consistent with the idea that the gut microbiota can influence or be affected by estrogen metabolism and thereby provide an independent biomarker of breast cancer. Recent literature has demonstrated that the gut microbiota is the modulation of systemic estrogens [40,41,42]. Elevated levels of circulating estrogens are associated with an increased risk of breast cancer [43,44,45,46,47].
We found that Eubacterium eligens and Roseburia inulinivorans were less abundant in postmenopausal breast cancer patients than in postmenopausal controls. Roseburia inulinivorans produces butyrate [48, 49]. In order to explore a potential association between butyrate-producing bacteria and breast cancer, we had added a differentially enriched gene analysis to show the potential association, all differentially enriched genes were annotated to the butanoate metabolism pathways, and finally, 14 butyrate-synthesis genes were found: 10 genes were enriched in controls and 4 genes were enriched in postmenopausal patients. Notably, these butyrate-synthesis genes were reduced in postmenopausal patients, which may be related to the decrease in butyrate-producing bacteria.
Butyrate acts as an anti-inflammatory agent, mainly by inhibiting the activation of nuclear factor κB (NF-κB) in intestinal epithelial cells [50]. Butyrate can also act on immune cells via specific G-protein-coupled receptors expressed on immune cells [51]. Reductions in colonic butyrate can promote inflammation. These findings provide further evidence for the idea that alterations in the gut microbial community are associated with breast cancer. For example, a decrease in Roseburia inulinivorans may render postmenopausal women more prone to inflammation and therefore at higher risk of breast cancer if the decrease in Roseburia had occurred at the time breast cancer was initiated.
Dysbiosis was detected in the gut microbiomes of postmenopausal breast cancer patients, but it was not detected in premenopausal patients. Therefore, the dysbiosis observed in the gut microbiomes of breast cancer patients may depend on age and menopause status.
Breast cancer-associated alterations in the gut microbial community likely translate into alterations in gut microbial functions. Among premenopausal women, breast cancer was associated in our study with enrichment in genes involved in the PTS system, secretion system, vitamin B12 transport system, and manganese/iron transport system. Among postmenopausal women, breast cancer was associated with enrichment in genes involved in lipopolysaccharide (LPS) biosynthesis, iron complex transport system, vitamin B12 transport system, PTS system, and secretion system. Iron enrichment affects the gut microbiome, increases pathogen abundance, and induces intestinal inflammation [52]. The PTS and secretion systems are associated with diabetes, liver cirrhosis, and rheumatoid arthritis [38, 53], while vitamin B12 status correlates positively with breast cancer risk in women [54]. Lipopolysaccharide is a potent trigger of macrophage-mediated systemic inflammation [55], which has been suggested to play an important role in promoting the transformation of inflammation into tumorigenesis [56,57,58]. Enrichment of the iron transport system and lipopolysaccharide biosynthesis in gut microbiota may cause systemic low-grade inflammation, thereby increasing the risk of breast cancer if the dysbiosis observed in the patient cohort was present in the same cohort prior to contracting breast cancer.
Conclusion
In conclusion, we have found alterations of gut microbial community and functions in postmenopausal breast cancer patients. The gut microbiota may regulate or respond to host immunity and metabolic balance. In this way, our study suggests an association between gut microbiota and development of postmenopausal breast cancer. However, our data do not allow us to determine whether the altered gut metagenome is the consequence of the disease process or is somehow involved in its pathogenesis.
References
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–67.
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–9.
Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–70.
Schloss PD, Handelsman J. Status of the microbial census. Microbiol Mol Biol Rev. 2004;68:686–91.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8.
O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–93.
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–5.
Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–93.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4.
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6:e25792.
Vaahtovuo J, Munukka E, Korkeamäki M, Luukkainen R, Toivanen P. Fecal microbiota in early rheumatoid arthritis. J Rheumatol. 2008;35:1500–5.
Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F, et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut. 2007;56:661–7.
Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–72.
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63.
Song Y, Liu C, Finegold SM. Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol. 2004;70:6459–65.
Parracho HM, Bingham MO, Gibson GR, McCartney AL. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J Med Microbiol. 2005;54:987–91.
Scanlan PD, Shanahan F, Clune Y, Collins JK, O'Sullivan GC, O'Riordan M, et al. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol. 2008;10:789–98.
Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393.
Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–41.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–41.
S Y, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101.
Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–16.
Minelli EB, Beghini AM, Vesentini S, Marchiori L, Nardo G, Cerutti R, et al. Intestinal microflora as an alternative metabolic source of estrogens in women with uterine leiomyoma and breast cancer. Ann N Y Acad Sci. 1990;595:473–9.
Goedert JJ, Jones G, Hua X, Xu X, Yu Q, Flores R, et al. Investigation of the association between the fecal microbiota and breast cancer in postmenopausal women: a population-based case-control pilot study. J Natl Cancer Inst. 2015;107:1-5.
Xie H, Guo R, Zhong H, Feng Q, Lan Z, Qin B, et al. Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome. Cell Syst. 2016;3:572–84.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41.
Chen LH, Zheng DD, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 2016;44:D694–7.
Urban M, Cuzick A, Rutherford K, Irvine A, Pedro H, Pant R, et al. PHI-base: a new interface and further additions for the multi-species pathogen-host interactions database. Nucleic Acids Res. 2017;45:D604–10.
Youssef NH, Elshahed MS. Species richness in soil bacterial communities: a proposed approach to overcome sample size bias. J Microbiol Methods. 2008;75:86–91.
Goedert JJ, Hua X, Yu G, Shi J. Diversity and composition of the adult fecal microbiome associated with history of cesarean birth or appendectomy: analysis of the American Gut Project. EBioMedicine. 2014;1:167–72.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80.
Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528.
Patil KR, Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc Natl Acad Sci U S A. 2005;102:2685–9.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Dray S, Dufour AB. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20.
Li J,Zhao F, Wang Y,Chen J,Tao J,Tian G,et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60.
Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. elife. 2013;2:e01202.
Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10:324–35.
Rose DP. Diet, hormones, and cancer. Annu Rev Public Health. 1993;14:1–17.
Flores R, Shi J, Fuhrman B, Xu X, Veenstra TD, Gail MH, et al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012;10:253.
Key TJ, Appleby PN, Reeves GK, Roddam AW, Helzlsouer KJ, Alberg AJ, et al. Circulating sex hormones and breast cancer risk factors in postmenopausal women: reanalysis of 13 studies. Br J Cancer. 2011;105:709–22.
Fuhrman BJ, Schairer C, Gail MH, Boyd-Morin J, Xu X, Sue LY, et al. Estrogen metabolism and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2012;104:326–39.
Eliassen AH, Spiegelman D, Xu X, Keefer LK, Veenstra TD, Barbieri RL, et al. Urinary estrogens and estrogen metabolites and subsequent risk of breast cancer among premenopausal women. Cancer Res. 2012;72:696–706.
Dallal CM, Tice JA, Buist DS, Bauer DC, Lacey JV, Cauley JA, et al. Estrogen metabolism and breast cancer risk among postmenopausal women: a case-cohort study within B~FIT. Carcinogenesis. 2014;35:346–55.
Key T, Appleby P, Barnes I, Reeves G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94:606–16.
Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int J Syst Evol Microbiol. 2002;52:1615–20.
Scott KP, Martin JC, Chassard C, Clerget M, Potrykus J, Campbell G, et al. Substrate-driven gene expression in Roseburia inulinivorans: importance of inducible enzymes in the utilization of inulin and starch. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4672–9.
Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology. 2000;118:724–34.
Meijer K, de Vos P, Priebe MG. Butyrate and other shortchain fatty acids as modulators of immunity: what relevance for health? Curr Opin Clin Nutr Metab Care. 2010;13:715–21.
Jaeggi T, Kortman GA, Moretti D, Chassard C, Holding P, Dostal A, et al. Iron fortification adversely affects the gut microbiome, increases pathogen abundance and induces intestinal inflammation in Kenyan infants. Gut. 2015;64:731–42.
Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the humangut microbiome in liver cirrhosis. Nature. 2014;513:59–64.
Matejcic M, de Batlle J, Ricci C, Biessy C, Perrier F, Huybrechts I, et al. Biomarkers of folate and vitamin B12 and breast cancer risk: report from the EPIC cohort. Int J Cancer. 2017;140:1246–59.
Seeley JJ, Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol. 2017;101:107–19.
Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766.
Tang XY, Zhu YQ, Wei B. Expression and functional research of TLR4 in human colon carcinoma. Am J Med Sci. 2010;339:319–26.
Cario E, Rosenberg IM, Brandwein SL, Beck PL, Reinecker HC, Podolsky DK. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing toll-like receptors. J Immunol. 2000;164:966–72.
Funding
This study was supported by the Guangxi Natural Science Foundation (2012GXNSFBA053117), Guangxi Natural Science Fund for Innovation Research Team (2013GXNSFFA019002), Guangxi Collaborative Innovation Center for Genomic and Personalized Medicine (201319), and the National Natural Science Foundation of China (81060169).
Author information
Authors and Affiliations
Contributions
ZNM and QYW designed the experiments. JZ, HSL, XJZ, SZH, JRT, and RQT obtained the samples. WYL carried out the metagenomic analyses. JZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Affiliated Tumor Hospital of Guangxi Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Generated data of the four groups. Table S2. Distribution of the samples of the four groups in the two enterotypes. Table S3. Relative abundance of the different species between premenopausal breast cancer patients and premenopausal healthy controls. Table S4. Relative abundance of the different species between postmenopausal breast cancer patients and postmenopausal healthy controls. Table S5. PERMANOVA analysis was performed to assess effects of different phenotypes on gene profile. Table S6. The optimal species markers in the classification of postmenopausal breast cancer patients and postmenopausal healthy controls. Table S7. The abundance of Pathogen-Host Interactions (PHI) gene coding for diseases in postmenopausal breast cancer patients and postmenopausal healthy controls. Table S8. The virulence factor in samples of postmenopausal breast cancer patients and postmenopausal healthy controls. Table S9. Relative abundance of the different KEGG modules between premenopausal breast cancer patients and premenopausal healthy controls. Table S10. Relative abundance of the different KEGG modules between postmenopausal breast cancer patients and postmenopausal healthy controls. Table S11. Differentially enriched genes which annotated to butanoate metabolism pathways between postmenopausal breast cancer patients and postmenopausal healthy controls. Table S12. Relative abundance of the species of all the samples. Table S13. The species counts of all the samples. (XLS 2530 kb)
Additional file 2:
Figure S1. Rarefaction for gut microbial gene in premenopausal breast cancer patients (n = 18), premenopausal healthy controls (n = 25), postmenopausal breast cancer patients (n = 44), and postmenopausal healthy controls (n = 46). Group 1 indicates premenopausal healthy controls, group 2 indicates premenopausal breast cancer patients, group 3 indicates postmenopausal healthy controls, and group 4 indicates postmenopausal breast cancer patients. Figure S2. The enterotypes of gut microbiota in breast cancer patients and healthy controls. (a) The optimal number of enterotypes was two of the four groups as indicated by Calinski-Harabasz (CH) index. The maximum CH index at two clusters (enterotypes) indicated the optimal enterotype number. (b) The gut microbiota of the four cohorts are clustered into two enterotypes at the genus level, dominated by either Bacteroides (enterotype 1) or Prevotella (enterotype 2). (c) Relative abundances of the top genera in the two enterotypes. (d) Distribution of the samples of the four groups in the two enterotypes. Figure S3. Relative abundance of the gut microbiota in the four groups at the phylum level. Figure S4. Relative abundance of the gut microbiota in the four groups at the genus level. Figure S5. Abundance distribution of the gut microbiota differed significantly between postmenopausal breast cancer patients and postmenopausal healthy controls at the genus level. Figure S6. Distribution of five trials of tenfold cross-validation error in random forest classification of postmenopausal breast cancer patients. The model was trained using the relative species abundances in patients and controls. The black line marks the average of the five trials (gray lines). The red line indicates the number of optimal species markers. Figure S7. Scatter plots for correlations between gut microbiota species and clinical indices. (DOCX 810 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhu, J., Liao, M., Yao, Z. et al. Breast cancer in postmenopausal women is associated with an altered gut metagenome. Microbiome 6, 136 (2018). https://doi.org/10.1186/s40168-018-0515-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-018-0515-3