
Abstract
Background
Although a number of recent studies have uncovered remarkable diversity of microbes associated with plants, understanding and managing dynamics of plant microbiomes remain major scientific challenges. In this respect, network analytical methods have provided a basis for exploring “hub” microbial species, which potentially organize community-scale processes of plant–microbe interactions.
Methods
By compiling Illumina sequencing data of root-associated fungi in eight forest ecosystems across the Japanese Archipelago, we explored hubs within “metacommunity-scale” networks of plant–fungus associations. In total, the metadata included 8080 fungal operational taxonomic units (OTUs) detected from 227 local populations of 150 plant species/taxa.
Results
Few fungal OTUs were common across all the eight forests. However, in each of the metacommunity-scale networks representing northern four localities or southern four localities, diverse mycorrhizal, endophytic, and pathogenic fungi were classified as “metacommunity hubs,” which were detected from diverse host plant taxa throughout a climatic region. Specifically, Mortierella (Mortierellales), Cladophialophora (Chaetothyriales), Ilyonectria (Hypocreales), Pezicula (Helotiales), and Cadophora (incertae sedis) had broad geographic and host ranges across the northern (cool-temperate) region, while Saitozyma/Cryptococcus (Tremellales/Trichosporonales) and Mortierella as well as some arbuscular mycorrhizal fungi were placed at the central positions of the metacommunity-scale network representing warm-temperate and subtropical forests in southern Japan.
Conclusions
The network theoretical framework presented in this study will help us explore prospective fungi and bacteria, which have high potentials for agricultural application to diverse plant species within each climatic region. As some of those fungal taxa with broad geographic and host ranges have been known to promote the survival and growth of host plants, further studies elucidating their functional roles are awaited.
Similar content being viewed by others

Background
Below-ground fungi in the endosphere and rhizosphere are key drivers of terrestrial ecosystem processes [1,2,3,4]. Mycorrhizal fungi, for example, are important partners of most land plant species, enhancing nutritional conditions and pathogen resistance of host plants [5,6,7]. In reward for the essential physiological services, they receive ca. 20% of net photosynthetic products from plants [8, 9]. Recent studies have also indicated that diverse taxonomic groups of endophytic fungi (e.g., endophytic fungi in the ascomycete orders Helotiales and Chaetothyriales) commonly interact with plant roots [10] and that those fungi play pivotal roles by providing soil nitrogen/phosphorous to their hosts [11,12,13,14,15], converting organic nitrogen into inorganic forms in the rhizosphere [16], and increasing plants’ resistance to environmental stresses [17,18,19]. Because of their fundamental roles, below-ground fungi have been considered as prospective sources of ecosystem-level functioning in forest management, agriculture, and ecosystem restoration [18,19,20,21]. However, due to the exceptional diversity of below-ground fungi [22,23,24] and the extraordinary complexity of below-ground plant–fungus interactions [25,26,27], we are still at an early stage of managing and manipulating plant-associated microbiomes [28,29,30].
In disentangling complex webs of below-ground plant–fungus associations, network analyses, which have been originally applied to human relations and the World-Wide Web [31, 32] and subsequently to biological systems [33, 34], provide crucial insights. By using network analytical tools, we can infer how plant species in a forest, grassland, or farmland are associated with diverse taxonomic and functional groups of fungi [25, 35,36,37]. Such information of network structure (topology) can be used to identify “hub” species, which are linked with many other species within networks depicting multispecies host–symbiont associations [38] (cf. [37, 39, 40]). Those hubs with broad host/symbiont ranges are expected to play key roles by mediating otherwise discrete ecological processes within a community [20, 25]. For example, although arbuscular mycorrhizal and ectomycorrhizal symbioses have been considered to involve distinct sets of plant and fungal lineages [41] (but see [42, 43]), hub endophytic fungi with broad host ranges may mediate indirect interactions between arbuscular mycorrhizal and ectomycorrhizal plant species through below-ground mycelial connections. As information of plant-associated fungal communities is now easily available with high-throughput DNA sequencing technologies [1, 22, 23], finding hub microbial species out of hundreds or thousands of species within a network has become an important step towards the understanding of ecosystem-scale phenomena.
Nonetheless, given that fungi can disperse long distances with spores, conidia, propagules, and animal vectors [44,45,46,47,48], information of local-scale networks alone does not provide thorough insights into below-ground plant–fungus interactions in the wild. In other words, no forests, grasslands, and farmlands are free from perturbations caused by fungi immigrating from other localities [49,50,51,52,53]. Therefore, to consider how local ecosystem processes are interlinked by dispersal of fungi, we need to take into account “metacommunity-scale” networks of plant–fungus associations [38]. Within a dataset representing multiple local communities (e.g., [26]), fungal species that occur in multiple localities may interlink local networks of plant–fungus associations. Among them, some species that not only have broad geographic ranges but also are associated with diverse host plant species would be placed at the core positions of a metacommunity-scale network [38]. Such “metacommunity hub” fungi would be major drivers of the synchronization and restructuring of local ecosystem processes (sensu [54]), and hence, their functional roles need to be investigated on a priority basis [38]. Moreover, in the screening of mycorrhizal and endophytic fungi that can be used in agriculture and ecosystem restoration programs [18, 21, 55], analytical pipelines for identifying metacommunity hubs will help us explore species that are potentially applied (inoculated) to diverse plant species over broad geographic ranges of farmlands, forests, or grasslands. Nonetheless, despite the potential importance of metacommunity hubs in both basic and applied microbiology, few studies have examined metacommunity-scale networks of plant–symbiont associations.
By compiling Illumina sequencing datasets of root-associated fungi [56], we herein inferred the structure of a metacommunity-scale network of below-ground plant–fungus associations and thereby explored metacommunity hubs. Our metadata consisted of plant–fungus association data in eight forest localities across the entire range of the Japanese Archipelago, including 150 plant species/taxa and 8080 fungal operational taxonomic units (OTUs) in temperate and subtropical regions. Based on the information of local- and metacommunity-scale networks, each of the fungal OTUs was evaluated in light of its topological position. We then examined whether fungal OTUs placed at the core of local-level plant–fungus networks could play key topological roles within the metacommunity-scale network. Overall, this study uncover how diverse taxonomic groups of mycorrhizal and endophytic fungi can form metacommunity-scale networks of below-ground plant–fungus associations, providing a basis for analyzing complex spatial processes of species-rich host–microbe systems.
Methods
Terminology
While a single type of plant–fungus interactions is targeted in each of most mycological studies (e.g., arbuscular mycorrhizal symbiosis or ectomycorrhizal symbiosis), we herein analyze the metadata including multiple categories of below-ground plant–fungus associations [56]. Because arbuscular mycorrhizal, ectomycorrhizal, and endophytic fungi, for example, vary in their microscopic structure within plant tissue [41], it is impossible to develop a general criterion of mutualistic/antagonistic interactions for all those fungal functional groups. Therefore, we used the term “associations” instead of “interactions” throughout the manuscript when we discuss patterns detected based on the Illumina sequencing metadata of root-associated fungi. Consequently, our network data could represent not only mutualistic or antagonistic interactions but also neutral or commensalistic interactions [25, 57, 58]. Our aim in this study is to gain an overview of the metacommunity-scale plant–fungus associations, while the nature of respective plant–fungus associations should be evaluated in future inoculation experiments.
Data
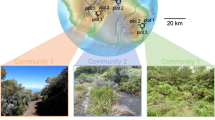
We compiled the Illumina (MiSeq) sequencing data collected in a previous study [56], in which community-scale statistical properties of below-ground plant–fungus associations were compared among eight forest localities (four cool-temperate, one warm-temperate, and three subtropical forests) across the entire range of the Japanese Archipelago (45.042–24.407 °N; Fig. 1). In each forest, 2 cm segment of terminal roots were sampled from 3 cm below the soil surface at 1 m horizontal intervals [56]. Those root samples were collected irrespective of their morphology and mycorrhizal type; hence, the samples as a whole represented below-ground relative abundance of plant species in each forest community. Host plant species were identified based on the sequences of the genes encoding the large subunit of ribulose-1,5-bisphosphate carboxylase (rbcL) and the internal transcribed spacer 1 (ITS1) of the ribosomal RNA region, although there were plant root samples that could not be identified to species with the rbcL and ITS1 regions [56]. The sequencing data are available through DDBJ Sequence Read Archives (accession DRA006339).

Study sites examined in this study. Across the entire range of the Japanese Archipelago, root samples were collected in four cool-temperate forests (sites 1–4), one warm-temperate forest (site 5), and three subtropical forests (sites 6–8)
The Illumina sequencing reads of the fungal ITS1 region were processed using the program Claident [59, 60] as detailed in the data-source study [56]: the UNIX scripts used are available as Additional file 1. After filtering and denoising processes, operational taxonomic units (OTUs) representing less than 10 sequencing reads were discarded. The primers used were designed to target not only Ascomycota and Basidiomycota but also diverse non-Dikarya (e.g., Glomeromycota) taxa [61]. In most studies analyzing community structure of Ascomycota and Basidiomycota fungi, OTUs of the ITS region are defined with a cut-off sequence similarity of 97% [23, 62, 63] (see also [64]). Meanwhile, Glomeromycota fungi generally have much higher intraspecific ITS-sequence variation than other taxonomic groups of fungi [65]. Consequently, we used 97% and 94% cut-off sequence similarities for defining non-Glomeromycota and Glomeromycota fungal OTUs, respectively [56]. The OTUs were then subjected to reference database search with the query-centric auto-k-nearest-neighbor algorithm [59, 60] and subsequent taxonomic assignment with the lowest common ancestor algorithm [66]. For the molecular identification process, the nt database ver. 2015-11-11 was downloaded from the NCBI FTP server (https://ftp.ncbi.nlm.nih.gov/blast/db/) and sequences lacking genus-level information were removed. Based on the inferred taxonomy, the functional group of each fungal OTU was inferred using the program FUNGuild 1.0 [67].
After a series of bioinformatics and rarefaction procedures, 1000 fungal ITS reads were obtained from each of the 240 samples collected in each forest locality (i.e., 1000 reads × 240 samples × 8 sites). In the process, plant–fungus associations whose read counts represented less than 0.1% of the total read count of each sample were removed to minimize the effects of PCR/sequencing errors [68]. A sample (row) × fungal OTU (column) data matrix, in which a cell entry depicted the number of sequencing reads of an OTU in a sample, was obtained for each local forest (“sample-level” matrix) (Additional file 2: Data S2). Each local sample-level matrix was then converted into a “species-level” matrix, in which a cell entry represented the number of root samples from which associations of a plant species/taxa (row) and a fungal OTU (column) was observed: the binary (presence/absence) information was used in the conversion from sample-level matrices to species-level matrices. In the species-level matrix of each forest, 17–55 plant species/taxa and 1149–1797 fungal OTUs were detected from the local species-level matrices (Additional file 3: Data S3). In total, the matrices included 150 plant species/taxa and 8080 fungal OTUs (Additional file 4: Data S4).
Local networks
Among the eight forest localities, variation in the order-level taxonomic compositions was examined with the permutational analysis of variance (PERMANOVA; [69]) and the permutational analysis for the multivariate homogeneity of dispersions (PERMDISP; [70]) with the “adonis” and “betadisper” functions of the vegan 2.4–3 package [71] of R 3.4.1 [72], respectively. The β-diversity values used in the PERMANOVA and PERMDISP analyses were calculated with the “Bray-Curtis” metric based on the sample-level matrices (Additional file 2: Data S2). Note that the “Raup-Crick” β-diversity metric [73], which controls α-diversity in community data but requires computationally intensive randomization, was not applicable to our large metadata. Geographic variation in the compositions of fungal functional groups was also evaluated by PERMANOVA and PERMDISP analyses. The R scripts for the PERMANOVA and PERMDISP analyses are available as Additional file 5.
For each of the eight local forests, the network structure of below-ground plant–fungus associations was visualized based on the species-level matrix (Additional file 3: Data S3) using the program GePhi 0.9.1 [74] with the “ForceAtlas2” layout algorithm [75]. Within the networks, the order-level taxonomy of fungal OTUs was highlighted. Although the dataset of each local forest had the information of plant–fungus association frequency (i.e., the number of root samples from which respective plant–fungus associations were observed) (Additional file 3: Data S3), all the links were shown equally in the network visualization because varying line (link) thickness could result in considerable overlaps of links, reducing the visibility of the network figure.
To evaluate host ranges of each fungal OTU in each local forest, we first calculated the d’ metric of interaction specificity [76]. However, estimates of the d’ metric varied considerably among fungal OTUs observed from small numbers of root samples (Additional file 6: Figure S1) presumably due to overestimation or underestimation of host preferences for those rare OTUs. Therefore, we scored each fungal OTU based on their topological positions within each local network by calculating network centrality indices (degree, closeness, betweenness, and eigenvector centralities metrics of network centrality; [32]). Among the centrality metrics, betweenness centrality, which measures the extent to which a given nodes (species) is located within the shortest paths connecting pairs of other nodes in a network [77], is often used to explore organisms with broad host or partner ranges [38]. Thus, in each local network, fungal OTUs were ranked based on their betweenness centrality scores (local betweenness). Note that there was clear correlation between degree and betweenness centrality scores in each of the eight forests studied (see below; Pearson’s correlation r, 0.853–0.950; P < 0.0001 for all the eight sites). While binary (presence/absence) link information is used in analyses with the original betweenness metric [77], quantitative (frequency) link information (i.e., the number of root samples from which respective plant–fungus associations were observed) can be taken into account by using a newly developed “weighted” betweenness metric [78]. However, as far as we know, few biological interpretations have been made on the use of weighted betweenness in analyses of plant–microbe associations. Therefore, we used the original betweenness metric for binary data in this study. Note that binary and quantitative betweenness scores (Additional file 4: Data S4) were highly correlated with each other in each local network (Pearson’s correlation r, 0.481–0.826; P < 0.0001 for all the eight sites).
Metacommunity-scale network
By compiling the species-level matrices of the eight local forests, the topology of the metacommunity-scale network of plant–fungus associations was inferred. In general, species interaction (association) networks of local communities can be interconnected by species that appear in two or more local networks, thereby merged into a metacommunity-scale network [38]. In our data across the eight local forests, 2109 OTUs out of the 8080 fungal OTUs appeared in two or more localities. Therefore, we could infer the topology of a metacommunity-scale network, in which the eight local networks were combined by the 2109 fungal OTUs. In the metacommunity-scale network, plant species/taxa observed in different localities were treated as different network nodes because our purpose in this study was to explore fungi that potentially play key roles in synchronizing local ecosystem processes [38]. In total, 227 plant nodes representing local populations of 150 plant species/taxa were included in the metacommunity-scale network.
We then screened for fungal OTUs with broad geographic and host ranges based on the betweenness centrality scores of respective fungal OTUs within the metacommunity network (metacommunity betweenness, Bmeta). In general, species with the highest metacommunity betweenness scores not only occur in local communities over broad biotic/abiotic environmental conditions but also are associated with broad ranges of host/partner species [38]. Possible relationship between local- and metacommunity-scale topological roles was then examined by plotting local and metacommunity betweenness scores (Blocal and Bmeta) of each fungal OTUs on a two-dimensional surface. To make the betweenness scores vary from 0 to 1, betweenness centrality of a fungal OTU i was standardized in the local- and metacommunity-scale networks, respectively, as follows:
where Blocal, i and Bmeta, iwere raw estimates of local- and metacommunity-scale betweenness of a fungal OTU i, and min() and max() indicated minimum and maximum values, respectively. For local betweenness of each OTU, a mean value across local networks was subsequently calculated (\( \overline{B}{\hbox{'}}_{\mathrm{local},i} \)): the local communities from which a target OTU was absent was omitted in the calculation of mean local betweenness. On the two-dimensional surface, the OTUs were then classified into four categories: metacommunity hubs having high betweenness in both local- and metacommunity-scale networks (\( \overline{B}{\hbox{'}}_{\mathrm{local},i} \) ≥ 0.5; B'meta, i ≥ 0.5), metacommunity connectors that had broad geographic ranges but displayed low local betweenness (\( \overline{B}{\hbox{'}}_{\mathrm{local},i} \) < 0.5; B'meta, i ≥ 0.5), local hubs that had high betweenness in local networks but not in the metacommunity-scale network (\( \overline{B}{\hbox{'}}_{\mathrm{local},i} \) ≥ 0.5; B'meta, i < 0.5), and peripherals with low betweenness at both local and metacommunity levels (\( \overline{B}{\hbox{'}}_{\mathrm{local},i} \) < 0.5; B'meta, i < 0.5) [38]. Approximately, 1–2% of fungal OTUs can show betweenness scores higher than 0.5 in each local or metacommunity network, while the threshold value may be changed depending on the purpose of each study [38].
In addition to metacommunity hubs within the metacommunity-scale network representing all the eight localities, those within the metacommunity-scale network representing northern (sites 1–4) or southern (sites 5–8) four localities were also explored. This additional analysis allowed us to screen for fungal OTUs that potentially adapted to broad ranges of biotic and abiotic environments within northern (cool-temperate) or southern (warm-temperate or subtropical) part of Japan.
Results
Local networks
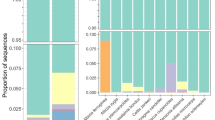
Among the eight forest localities, order-level taxonomic compositions of fungi varied significantly (PERMANOVA; Fmodel = 35.7, R2 = 0.116, P < 0.001), while the differentiation of community structure was attributed, at least partly, to geographic variation in among-sample dispersion (PERMDISP; F = 13.2, P < 0.001) (Fig. 2a). Compositions of fungal functional groups were also differentiated among the eight localities (PERMANOVA; Fmodel = 34.9, R2 = 0.113, P < 0.001), while within-site dispersion significantly varied geographically (PERMDISP; F = 9.2, P < 0.001) (Fig. 2b). The proportion of ectomycorrhizal fungal orders (e.g., Russulales, Thelephorales, and Sebacinales) was higher in temperate forests than in subtropical forests, while that of arbuscular mycorrhizal fungi increased in subtropical localities (Fig. 2). The proportion of the ascomycete order Helotiales, which has been known to include not only ectomycorrhizal but also endophytic, saprotrophic, and ericoid mycorrhizal fungi [79], was higher in northern localities. In contrast, Diaporthales, which has been considered as predominantly plant pathogenic taxon [80] (but see [81]), was common in subtropical forests but not in others.

Compositions of fungal taxa and functional groups in each forest. a Order-level taxonomic composition of fungal OTUs in each locality. The number of fungal OTUs detected is shown in a parenthesis for each forest. b Functional-group composition. The fungal functional groups were inferred by the program FUNGuild [67]
In each of the eight local networks depicting plant–fungus associations, some fungal OTUs were located at the central positions of the network, while others are distributed at peripheral positions (Additional file 7: Figure S2). Specifically, fungal OTUs belonging to the ascomycete orders Chaetothyriales (e.g., Cladophialophora and Exophiala) and Helotiales (e.g., Rhizodermea, Pezicula, Rhizoscyphus, and Leptodontidium) as well as some Mortierella OTUs had high betweenness centrality scores in each of the cool-temperate forests (Fig. 3a, b). In contrast, arbuscular mycorrhizal fungi (Glomeromycota) were common among OTUs with the highest betweenness scores in subtropical forests (Fig. 3a–c). Some fungi in the ascomycete order Hypocreales (e.g., Trichoderma, Ilyonectria, Simplicillium, and Calonectria) also had high betweenness scores in some temperate and subtropical forests (Fig. 3b).

Fungal OTUs with highest local betweenness. a Order-level taxonomic composition of top 20 OTUs with highest local betweenness in each forest. (Additional file 4; Data S4) for betweenness scores of all fungal OTUs in respective local forests. b Genus-level taxonomic composition of top 20 OTUs with highest local betweenness. c Functional-group composition of top 20 OTUs with highest local betweenness
Metacommunity-scale network
In the metacommunity-scale network representing the connections among the eight local networks, not only arbuscular mycorrhizal but also saprotrophic/endophytic fungi were placed at the central topological positions (Fig. 4; Additional file 8: Figure S3). Among non-Glomeromycota OTUs, Mortierella (Mortierellales), Cryptococcus [Trichosporonales; the Blast top-hit fungus in the NCBI database was recently moved to Saitozyma (Tremellales); [82]], Malassezia (Malasseziales), Oidiodendron (incertae sedis), Trichoderma (Hypocreales), and a fungus distantly allied to Melanconiella (Diaporthales) displayed the highest metacommunity betweenness (Table 1). Among the OTUs with high metacommunity betweenness, only a Mortierella OTU was designated as a metacommunity hub (i.e., \( \overline{B}{\hbox{'}}_{\mathrm{local},i} \) ≥ 0.5; B'meta, i ≥ 0.5) and others had low betweenness scores at the local community level (\( \overline{B}{\hbox{'}}_{\mathrm{local},i} \) < 0.5; Fig. 5a).

Metacommunity-scale network including all the eight local forests. The size of circles roughly represents relative scores of betweenness centrality. The functional groups of fungi inferred with the program FUNGuild [67] were organized into six categories, i.e., arbuscular mycorrhizal (bue), ectomycorrhizal (red), ericoid mycorrhizal (skyblue), saprotrophic/endophytic (yellow), plant pathogenic (purple), and other/unknown fungi (gray) (Additional file 4: Data S4). For plant species/taxa (green), the geographic information of source populations is indicated in (Additional file 8: Figure S3)

Relationship between local- and metacommunity-level betweenness. a Full meatcommunity. On the horizontal axis, the mean values of betweenness centrality scores across all the eight local forests are shown for respective fungal OTUs. On the vertical axis, the betweenness scores within the metacommunity-scale network consisting of the eight localities (Fig. 4) are shown for respective OTUs. b Metacommunity of cool-temperate forests. For the sub-dataset consisting of the four cool-temperate forests (Additional file 9: Figure S4), mean local betweenness and metacommunity betweenness are shown on the horizontal and vertical axes, respectively. c Metacommunity of warm-temperate and subtropical forests. For the sub-dataset consisting of the warm-temperate forest and the three subtropical forests (Additional file 10: Figure S5), mean local betweenness and metacommunity betweenness are shown on the horizontal and vertical axes, respectively
In the metacommunity-scale network representing the four cool-temperate forests (sites 1–4), many saprotrophic/endophytic fungal OTUs were associated with diverse plant species/taxa, located at the central topological positions within the network topology (Additional file 9: Figure S4; Fig. 5b). The list of these fungi with high metacommunity betweenness involved OTUs in the genera Mortierella, Cladophialophora (Chaetothyriales), Pezicula (Helotiales), and Oidiodendron as well as OTUs allied to Ilyonectria protearum (Nectriales) and Cadophora orchidicola (Helotiales) (Table 1). Most of these fungal OTUs also had high metacommunity betweenness designated as metacommunity hubs (Fig. 5b).
In the metacommunity-scale network consisting of the warm-temperate and subtropical forests (sites 5–8), arbuscular mycorrhizal and saprotrophic/endophytic fungi had the highest betweenness scores (Additional file 10: Figure S5; Fig. 5c). The list of non-Glomeromycota OTUs with the highest metacommunity betweenness included Saitozyma (Cryptococcus), Mortierella, Trichoderma, and Tomentella as well as OTUs allied to Cladophialophora, Scleropezicula (Helotiales), Melanconiella (Diaporthales), and Rhexodenticula (incertae sedis) (Table 1). Among the taxa, Saitozyma and Mortierella included OTUs classified as metacommunity hubs (Fig. 5c; Table 1). In an additional analysis of a metacommunity-scale network including only the three subtropical forests (sites 6–8), similar sets of fungal taxa were highlighted (Additional file 11: Table S1). Results also showed that similar sets of fungal taxa were highlighted in the analyses with binary and weighted betweenness metrics (Additional file 12: Table S2). The detailed information of the network index scores examined in this study is provided in Additional file 4; Data S4 .
Discussion
Based on the metadata of root-associated fungi across the Japanese Archipelago, we herein inferred the structure of a network representing metacommunity-scale associations of 150 plant species/taxa and 8080 fungal OTUs. Our analysis targeted diverse functional groups of fungi such as arbuscular mycorrhizal, ectomycorrhizal, ericoid-mycorrhizal, saprotrophic/endophytic, and pathogenic fungi, which have been analyzed separately in most previous studies on plant–fungus networks. The comprehensive analysis of below-ground plant–fungus associations allowed us to explore metacommunity hub fungi, which not only occurred over broad geographic ranges but also had broad host ranges in respective local communities. Consequently, this study highlights several taxonomic groups of fungi potentially playing key roles in synchronizing metacommunity-scale processes of temperate and/or subtropical forests.
In the metacommunity-scale network representing all the eight local forests (Fig. 4), fungi in several saprotrophic or endophytic taxa showed higher betweenness centrality scores than other fungi (Table 1). Mortierella is generally considered as a saprotrophic lineage [83], but it also includes fungi contributing to the growth and pathogen resistance of plants [84,85,86]. A phosphate solubilizing strain of Mortierella, for example, increases shoot and root growth of host plants under salt stress, especially when co-inoculated with an arbuscular mycorrhizal fungus [84]. In addition, polyunsaturated fatty acids produced by some Mortierella species are known to increase resistance of plants against phytopathogens [85, 86]. Fungi in the genus Trichoderma are commonly detected and isolated from the rhizosphere [83, 87]. Many of them inhibit the growth of other fungi, often used in the biological control of phytopathogens [88,89,90]. Some of them are also reported to suppress root-knot nematodes [91] or to promote root growth [92]. The analysis also highlighted basidiomycete yeasts in the genus Saitozyma or Cryptococcus (teleomorph = Filobasidiella), which are often isolated from soil [23, 93] and above-/below-ground parts of plants [94,95,96,97].
Along with possibly saprotrophic or endophytic taxa, ericoid mycorrhizal and phytopathogenic taxa of fungi displayed relatively high betweenness scores within the metacommunity-scale network representing all the eight local forests (Table 1). Specifically, Oidiodendron (teleomorph = Myxotrichum) is a taxon represented by possibly ericoid mycorrhizal species (O. maius and O. griseum) [98, 99], although fungi in the genus have been found also from roots of non-ericaceous plants and soil [100]. On the other hand, fungi in the family Nectriaceae are known to cause black foot disease [101], often having serious damage on economically important woody plants [102, 103]. Although we collected seemingly benign roots in the eight forests studied, some samples may be damaged by those pathogens. Alternatively, some fungi in the family Nectriaceae may be associated with plant hosts non-symptomatically, having adverse effects context-dependently.
Although the fungi mentioned above are candidates of metacommunity hubs, which are characterized by broad geographic ranges and host plant ranges, none except but a Mortierella OTU had high betweenness scores at both local and metacommunity levels (Fig. 5a). This result suggests that even if some fungi have broad geographic ranges across the Japanese Archipelago, few played important topological roles in each of the local networks representing plant–fungus associations. In other words, fungi that can adapt to biotic and abiotic environments in forest ecosystems throughout cool-temperate, warm-temperate, and subtropical regions are rare.
Therefore, we also explored fungi with broad geographic and host ranges within each metacommunity representing northern (cool-temperate) or southern (warm-temperate and subtropical) part of Japan. In the metacommunity consisting of the four cool-temperate forests (Additional file 9: Figure S4), fungal OTUs in the genera Mortierella, Cladophialophora, and Pezicula as well as those allied to Ilyonectria and Cadophora had the highest betweenness at both local and metacommunity levels, classified as metacommunity hubs (Fig. 5b; Table 1). Among them, Cladophialophora is of particular interest because it has been known as a lineage of “dark septate endophytes” [104,105,106] (sensu [15, 16, 107]). A species within the genus, C. chaetospira (= Heteroconium chaetospira), to which highest-betweenness OTUs in our data were closely allied, has been known not only to provide nitrogen to host plants but also to suppress pathogens [13, 17, 108]. Likewise, the Helotiales genus Pezicula (anamorph = Cryptosporiopsis) includes endophytic fungi [109,110,111], some of which produce secondary metabolites suppressing other microbes in the rhizosphere [112, 113]. Our finding that some of Cladophialophora and Pezicula fungi could be associated with various taxonomic groups of plants over broad geographic ranges highlights potentially important physiological and ecological roles of those endophytes at the community and metacommunity levels.
In the southern metacommunity network consisting of warm-temperate and subtropical forests (Additional file 10: Figure S5), some arbuscular mycorrhizal OTUs and Saitozyma (Cryptococcus) and Mortierella OTUs had high betweenness scores at both local and metacommunity levels, designated as metacommunity hubs (Fig. 5c; Table 1). Given the abovementioned prevalence of fungal OTUs allied to Cladophialophora chaetospira in the cool-temperate metacommunity, the contrasting list of metacommunity hubs in the southern (warm-temperate–subtropical) metacommunity implies that different taxonomic and functional groups of fungi play major metacommunity-scale roles in different climatic regions. This working hypothesis is partially supported by previous studies indicating endemism and vicariance in the biogeography of fungi and bacteria [114, 115], promoting conceptual advances beyond the classic belief that every microbe is everywhere but the environment selects microbial species/taxa colonizing respective local communities [116].
The roles of possible metacommunity hubs highlighted in this study are of particular interest from the aspect of theoretical ecology. Hub species connected to many other species in an ecosystem often integrate “energy channels” [117] within species interaction networks, having great impacts on biodiversity and productivity of the ecosystems [38]. The concept of “keystone” or “foundation” species [118, 119] can be extended to the metacommunity level, thereby promoting studies exploring species that restructure and synchronize ecological (and evolutionary) dynamics over broad geographic ranges [38]. Given that below-ground plant–fungus symbioses are key components of the terrestrial biosphere [1, 2], identifying fungal species that potentially have great impacts on the metacommunity-scale processes of such below-ground interactions will provide crucial insights into the conservation and restoration of forests and grasslands. We here showed that the list of metacommunity hubs could involve various lineages of endophytic fungi, whose ecosystem-scale functions have been underappreciated compared to those of mycorrhizal fungi. As those endophytic fungi are potentially used as inoculants when we reintroduce plant seedlings in ecosystem restoration programs [21, 55], exploring fungi with highest potentials in each climatic/biogeographic region will be a promising direction of research in conservation biology.
The finding that compositions of metacommunity hubs could vary depending on climatic regions also gives key implications for the application of endophytes in agriculture. Although a number of studies have tried to use endophytic fungi and/or bacteria as microbial inoculants in agriculture [18, 19, 120], such microbes introduced to agroecosystems are often outcompeted and replaced by indigenous (resident) microbes [121, 122]. Moreover, even if an endophytic species or strain increases plant growth in pot experiments under controlled environmental conditions, its effects in the field often vary considerably depending on biotic and abiotic contexts of local agroecosystems [18] (see also [123]). Therefore, in the screening of endophytes that can be used in broad ranges of biotic and abiotic environmental conditions, the metacommunity-scale network analysis outlined in this study will help us find promising candidates out of thousands or tens of thousands microbial species in the wild. Consequently, to find promising microbes whose inocula can persist in agroecosystems for long periods of time, exploration of metacommunity hubs needs to be performed in respective climatic or biogeographic regions.
For more advanced applications in conservation biology and agriculture, continual improvements of methods for analyzing metacommunity-scale networks are necessary [10]. First, while the fungal OTUs in our network analysis was defined based on the cut-off sequence similarities used in other studies targeting “species-level” diversity of fungi [63, 65], physiological functions can vary greatly within fungal species or species groups [15, 124]. Given that bioinformatic tools that potentially help us detect single-nucleotide-level variation are becoming available [125], the resolution of network analyses may be greatly improved in the near future. Second, although some computer programs allow us to infer functions of respective microbial OTUs within network data [67, 126], the database information of microbial functions remains scarce. To increase the coverage and accuracy of automatic annotations of microbial functions, studies describing the physiology, ecology, and genomes of microbes should be accelerated. With improved reference databases, more insights into the metacommunity-scale organization of plant–fungus associations will be obtained by reanalyzing the network data by compiling enhanced information of fungal functional groups. Third, as the diversity and compositions of plant–fungus associations included in a network can depend on how we process raw samples, special care is required in the selection of methods for washing and preparing root (or soil) samples. By sterilizing root samples with NaClO [127], for example, we may be able to exclude fungi or bacteria that are merely adhering to root surfaces. Meanwhile, some of those fungi and bacteria on root surfaces may play pivotal physiological roles in the growth and survival of plants [128]. Accordingly, it would be productive to compare network topologies of plant–microbe associations among different source materials by partitioning endosphere, rhizoplane, and rhizosphere microbial samples with a series of sample cleaning processes using ultrasonic devices [129]. Fourth, although this study targeted fungi associated with roots, our methods can be easily extended to network analyses involving other groups of microbes. By simultaneously analyzing the prokaryote 16S rRNA region [129,130,131] with the fungal ITS region, we can examine how bacteria, archaea, and fungi are involved in below-ground webs of symbioses. Fifth, not only plant–microbe associations but also microbe–microbe interactions can be estimated with network analytical frameworks. Various statistical pipelines have been proposed to infer how microbes interact with each other in facilitative or competitive ways within host macroorganisms [40, 132, 133]. Overall, those directions of analytical extensions will enhance our understanding of plant microbiome dynamics in nature.
Conclusions
By compiling datasets of below-ground plant–fungus associations in temperate and subtropical forest ecosystems, we explored metacommunity-hub fungi, which were characterized by broad geographic and host ranges. Such metacommunity-scale analyses are expected to provide bird’s-eye views of complex plant–microbe associations, highlighting plant-growth-promoting microbes that can be applied to diverse plant taxa in various environments. Given that endophytic fungi promoting the growth and pathogen resistance of host plants can be isolated from forest soil (e.g., Cladophialophora chaetospira [105]), the list of metacommunity-hub endophytic fungi featured in this study itself may include prospective species to be used in agriculture. By extending the targets of such network analyses to diverse types of plant-associated microbes (e.g., phyllosphere fungi and bacteria [81, 130, 134]) in various climatic/biogeographic regions, a solid basis for managing plant microbiomes will be developed.
Abbreviations
- DDBJ:
-
DNA Data Bank of Japan
- ITS:
-
Internal transcribed spacer
- NCBI:
-
National center for biotechnology information
- OTU:
-
Operational taxonomic unit
- PERMANOVA:
-
Permutational analysis of variance
- PERMDISP:
-
Permutational analysis for the multivariate homogeneity of dispersions
- rRNA:
-
Ribosomal ribonucleic acid
References
Peay KG, Kennedy PG, Talbot JM. Dimensions of biodiversity in the Earth mycobiome. Nat Rev Microbiol. 2016;14:434–47.
van der Heijden MG, Bardgett RD, van Straalen NM. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett. 2008;11:296–310.
van der Heijden MG, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T, et al. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature. 1998;396:69–72.
Wardle DA, Bardgett RD, Klironomos JN, Setälä H, van der Putten WH, Wall DH. Ecological linkages between aboveground and belowground biota. Science. 2004;304:1629–33.
Nara K. Ectomycorrhizal networks and seedling establishment during early primary succession. New Phytol. 2006;169:169–78.
Finlay R, Read D. The structure and function of the vegetative mycelium of ectomycorrhizal plants. II. The uptake and distribution of phosphorus by mycelial strands interconnecting host plants. New Phytol. 1986;103:157–65.
Klironomos JN, McCune J, Hart M, Neville J. The influence of arbuscular mycorrhizae on the relationship between plant diversity and productivity. Ecol Lett. 2000;3:137–41.
Högberg MN, Högberg P. Extramatrical ectomycorrhizal mycelium contributes one-third of microbial biomass and produces, together with associated roots, half the dissolved organic carbon in a forest soil. New Phytol. 2002;154:791–5.
Högberg P, Nordgren A, Buchmann N, Taylor AF, Ekblad A, Högberg MN, et al. Large-scale forest girdling shows that current photosynthesis drives soil respiration. Nature. 2001;411:789–92.
Toju H, Peay KG, Yamamichi M, Narisawa K, Hiruma K, Naito K, et al. Core microbiomes for sustainable agroecosystems. Nat Plants. 2018;4:247–57.
Almario J, Jeena G, Wunder J, Langen G, Zuccaro A, Coupland G, et al. Root-associated fungal microbiota of nonmycorrhizal Arabis alpina and its contribution to plant phosphorus nutrition. Proc Natl Acad Sci U S A. 2017;114:E9403–12.
Hiruma K, Gerlach N, Sacristán S, Nakano RT, Hacquard S, Kracher B, et al. Root endophyte Colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent. Cell. 2016;165:464–74.
Hashiba T, Narisawa K. The development and endophytic nature of the fungus Heteroconium chaetospira. FEMS Microbiol Lett. 2005;252:191–6.
Addy H, Piercey M, Currah R. Microfungal endophytes in roots. Can J Bot. 2005;83:1–13.
Jumpponen A. Dark septate endophytes-are they mycorrhizal? Mycorrhiza. 2001;11:207–11.
Newsham KK. A meta-analysis of plant responses to dark septate root endophytes. New Phytol. 2011;190:783–93.
Narisawa K, Tokumasu S, Hashiba T. Suppression of clubroot formation in Chinese cabbage by the root endophytic fungus, Heteroconium chaetospira. Plant Pathol. 1998;47:206–10.
Calvo P, Nelson L, Kloepper JW. Agricultural uses of plant biostimulants. Plant Soil. 2014;383:3–41.
Porras-Alfaro A, Bayman P. Hidden fungi, emergent properties: endophytes and microbiomes. Annu Rev Phytopathol. 2011;49:291–315.
Mandyam K, Jumpponen A. Seeking the elusive function of the root-colonising dark septate endophytic fungi. Stud Mycol. 2005;53:173–89.
Fuchs B, Haselwandter K. Red list plants: colonization by arbuscular mycorrhizal fungi and dark septate endophytes. Mycorrhiza. 2004;14:277–81.
Tedersoo L, Bahram M, Põlme S, Kõljalg U, Yorou NS, Wijesundera R, et al. Global diversity and geography of soil fungi. Science. 2014;346:1256688.
Buée M, Reich M, Murat C, Morin E, Nilsson R, Uroz S, et al. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol. 2009;184:449–56.
Öpik M, Vanatoa A, Vanatoa E, Moora M, Davison J, Kalwij J, et al. The online database MaarjAM reveals global and ecosystemic distribution patterns in arbuscular mycorrhizal fungi (Glomeromycota). New Phytol. 2010;188:223–41.
Toju H, Guimarães PR Jr, Olesen JM, Thompson JN. Assembly of complex plant–fungus networks. Nat Commun. 2014;5:5273.
Toju H, Guimarães PR Jr, Olesen JM, Thompson JN. Below-ground plant–fungus network topology is not congruent with above-ground plant–animal network topology. Sci Adv. 2015;1:e1500291.
Kadowaki K, Yamamoto S, Sato H, Tanabe AS, Hidaka A, Toju H. Plant−soil feedbacks between arbuscular- and ecto-mycorrhizal communities. bioRxiv. 2017; https://doi.org/10.1101/228387
Schlaeppi K, Bulgarelli D. The plant microbiome at work. Mol Plant-Microbe Int. 2015;28:212–7.
Berendsen RL, Pieterse CM, Bakker PA. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012;17:478–86.
Busby PE, Soman C, Wagner MR, Friesen ML, Kremer J, Bennett A, et al. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol. 2017;15:e2001793.
Albert R, Jeong H, Barabási AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–82.
Newman MEJ. Networks: an introduction. Oxford: Oxford University Press; 2010.
Bascompte J, Jordano P, Melián CJ, Olesen JM. The nested assembly of plant-animal mutualistic networks. Proc Natl Acad Sci U S A. 2003;100:9383–7.
Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–4.
Toju H, Tanabe A, Ishii H. Ericaceous plant–fungus network in a harsh alpine–subalpine environment. Mol Ecol. 2016;25:3242–57.
Bennett AE, Daniell TJ, Öpik M, Davison J, Moora M, Zobel M, et al. Arbuscular mycorrhizal fungal networks vary throughout the growing season and between successional stages. PLoS One. 2013;8:e83241.
Bahram M, Harend H, Tedersoo L. Network perspectives of ectomycorrhizal associations. Fungal Ecol. 2014;7:70–7.
Toju H, Yamamichi M, Guimarães PR Jr, Olesen J, Mougi A, Yoshida T, et al. Species-rich networks and eco-evolutionary synthesis at the metacommunity level. Nat Ecol Evol. 2017;1:24.
Agler MT, Ruhe J, Kroll S, Morhenn C, Kim S-T, Weigel D, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14:e1002352.
Toju H, Yamamoto S, Tanabe AS, Hayakawa T, Ishii HS. Network modules and hubs in plant-root fungal biome. J R Soc Interface. 2016;13:20151097.
Smith SE, Read DJ. Mycorrhizal symbiosis. New York: Academic press; 2008.
Dickie IA, Koide RT, Fayish AC. Vesicular-arbuscular mycorrhizal infection of Quercus rubra seedlings. New Phytol. 2001;151:257–64.
Plattner I, Hall I. Parasitism of non-host plants by the mycorrhizal fungus Tuber melanosporum. Mycol Res. 1995;99:1367–70.
Hallenberg N, Kuffer N. Long-distance spore dispersal in wood-inhabiting basidiomycetes. Nord J Bot. 2001;21:431–6.
Nagarajan S, Singh D. Long-distance dispersion of rust pathogens. Annu Rev Phytopathol. 1990;28:139–53.
Brown JK, Hovmøller MS. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science. 2002;297:537–41.
Hovmøller M, Justesen A, Brown J. Clonality and long-distance migration of Puccinia striiformis f. sp. tritici in North-West Europe. Plant Pathol. 2002;51:24–32.
Herrera J, Poudel R, Khidir HH. Molecular characterization of coprophilous fungal communities reveals sequences related to root-associated fungal endophytes. Microb Ecol. 2011;61:239–44.
Jousimo J, Tack AJ, Ovaskainen O, Mononen T, Susi H, Tollenaere C, et al. Ecological and evolutionary effects of fragmentation on infectious disease dynamics. Science. 2014;344:1289–93.
Parker MA. Mutualism in metapopulations of legumes and rhizobia. Am Nat. 1999;153:S48–60.
Thrall PH, Burdon JJ. Evolution of virulence in a plant host-pathogen metapopulation. Science. 2003;299:1735–7.
Lekberg Y, Koide RT, Rohr JR, ALDRICH-WOLFE L, Morton JB. Role of niche restrictions and dispersal in the composition of arbuscular mycorrhizal fungal communities. J Ecol. 2007;95:95–105.
Peay KG, Schubert MG, Nguyen NH, Bruns TD. Measuring ectomycorrhizal fungal dispersal: macroecological patterns driven by microscopic propagules. Mol Ecol. 2012;21:4122–36.
Gouhier TC, Guichard F, Gonzalez A. Synchrony and stability of food webs in metacommunities. Am Nat. 2010;175:E16–34.
Miller R, Jastrow J. The application of VA mycorrhizae to ecosystem restoration and reclamation. New York: Springer; 1992.
Toju H, Sato H, Yamamoto S, Tanabe AS. Structural diversity across arbuscular mycorrhizal, ectomycorrhizal, and endophytic plant–fungus networks. bioRxiv. 2018; https://doi.org/10.1101/269563
Caruso T, Rillig MC, Garlaschelli D. On the application of network theory to arbuscular mycorrhizal fungi–plant interactions: the importance of basic assumptions. New Phytol. 2012;194:891–4.
Thompson JN. The geographic mosaic of coevolution. Chicago: University of Chicago Press; 2005.
Tanabe AS. Claident v0.2.2016.07.05, a software distributed by author at http://www.fifthdimension.jp/. 2016.
Tanabe AS, Toju H. Two new computational methods for universal DNA barcoding: a benchmark using barcode sequences of bacteria, archaea, animals, fungi, and land plants. PLoS One. 2013;8:e76910.
Toju H, Tanabe AS, Yamamoto S, Sato H. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS One. 2012;7:e40863.
Smith DP, Peay KG. Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS One. 2014;9:e90234.
O'Brien HE, Parrent JL, Jackson JA, Moncalvo J-M, Vilgalys R. Fungal community analysis by large-scale sequencing of environmental samples. Appl Environ Microbiol. 2005;71:5544–50.
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AF, Bahram M, et al. Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol. 2013;22:5271–7.
Thiéry O, Vasar M, Jairus T, Davison J, Roux C, Kivistik PA, et al. Sequence variation in nuclear ribosomal small subunit, internal transcribed spacer and large subunit regions of Rhizophagus irregularis and Gigaspora margarita is high and isolate-dependent. Mol Ecol. 2016;25:2816–32.
Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–86.
Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016;20:241–8.
Peay KG, Russo SE, McGuire KL, Lim Z, Chan JP, Tan S, et al. Lack of host specificity leads to independent assortment of dipterocarps and ectomycorrhizal fungi across a soil fertility gradient. Ecol Lett. 2015;18:807–16.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62:245–53.
Oksanen J, Blanachet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, et al. Vegan: community ecology package. R package version 2.0–3 available at http://CRAN.R-project.org/package=vegan, vol. 10; 2012. p. 2008.
R-Core-Team. R 3.4.1: A language and environment for statistical computing available at http://www.R-project.org/. Vienna: R Foundation for Statistical Computing; 2015.
Chase JM, Kraft NJ, Smith KG, Vellend M, Inouye BD. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere. 2011;2:1–11.
Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks, Proceedings of the 3rd International AAAI Conference on Weblogs and Social media, vol. 8; 2009. p. 361–2.
Jacomy M, Heymann S, Venturini T, Bastian M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi Software. PLoS One. 2011;96:e98679.
Blüthgen N, Menzel F, Blüthgen N. Measuring specialization in species interaction networks. BMC Ecol. 2006;6:9.
Freeman LC. A set of measures of centrality based on betweenness. Sociometry. 1977;40:35–41.
Brandes U. A faster algorithm for betweenness centrality. J Math Sociol. 2001;25:163–77.
Tedersoo L, Pärtel K, Jairus T, Gates G, Põldmaa K, Tamm H. Ascomycetes associated with ectomycorrhizas: molecular diversity and ecology with particular reference to the Helotiales. Environ Microbiol. 2009;11:3166–78.
Rossman AY, Farr DF, Castlebury LA. A review of the phylogeny and biology of the Diaporthales. Mycoscience. 2007;48:135–44.
Sieber TN. Endophytic fungi in forest trees: are they mutualists? Fungal Biol Rev. 2007;21:75–89.
Liu X-Z, Wang Q-M, Göker M, Groenewald M, Kachalkin A, Lumbsch HT, et al. Towards an integrated phylogenetic classification of the Tremellomycetes. Stud Mycol. 2015;81:85–147.
Watanabe T. Pictorial atlas of soil and seed fungi: morphologies of cultured fungi and key to species. Boca Raton: CRC press; 2010.
Zhang H, Wu X, Li G, Qin P. Interactions between arbuscular mycorrhizal fungi and phosphate-solubilizing fungus (Mortierella sp.) and their effects on Kostelelzkya virginica growth and enzyme activities of rhizosphere and bulk soils at different salinities. Biol Fertil Soils. 2011;47:543.
Eroshin V, Dedyukhina E. Effect of lipids from Mortierella hygrophila on plant resistance to phytopathogens. World J Microbiol Biotechnol. 2002;18:165–7.
Melo IS, Santos SN, Rosa LH, Parma MM, Silva LJ, Queiroz SC, et al. Isolation and biological activities of an endophytic Mortierella alpina strain from the Antarctic moss Schistidium antarctici. Extremophiles. 2014;18:15–23.
Papavizas G. Trichoderma and Gliocladium: biology, ecology, and potential for biocontrol. Annu Rev Phytopathol. 1985;23:23–54.
Datnoff L, Nemec S, Pernezny K. Biological control of Fusarium crown and root rot of tomato in Florida using Trichoderma harzianum and Glomus intraradices. Biol Control. 1995;5:427–31.
Bailey B, Bae H, Strem M, Crozier J, Thomas S, Samuels G, et al. Antibiosis, mycoparasitism, and colonization success for endophytic Trichoderma isolates with biological control potential in Theobroma cacao. Biol Control. 2008;46:24–35.
Bae H, Roberts DP, Lim H-S, Strem MD, Park S-C, Ryu C-M, et al. Endophytic Trichoderma isolates from tropical environments delay disease onset and induce resistance against Phytophthora capsici in hot pepper using multiple mechanisms. Mol Plant-Microbe Int. 2011;24:336–51.
AL-Shammari TA, Bahkali AH, Elgorban AM, El-Kahky MT, Al-Sum BA. The use of Trichoderma longibrachiatum and Mortierella alpina against root-knot nematode, Meloidogyne javanica on tomato. J Pure Appl Microbiol. 2013;7:199–207.
Ming Q, Su C, Zheng C, Jia M, Zhang Q, Zhang H, et al. Elicitors from the endophytic fungus Trichoderma atroviride promote Salvia miltiorrhiza hairy root growth and tanshinone biosynthesis. J Exp Bot. 2013;64:5687–94.
Mašínová T, Bahnmann BD, Větrovský T, Tomšovský M, Merunková K, Baldrian P. Drivers of yeast community composition in the litter and soil of a temperate forest. FEMS Microbiol Ecol. 2016;93:fiw223.
Nutaratat P, Srisuk N, Arunrattiyakorn P, Limtong S. Plant growth-promoting traits of epiphytic and endophytic yeasts isolated from rice and sugar cane leaves in Thailand. Fungal Biol. 2014;118:683–94.
Renker C, Blanke V, Börstler B, Heinrichs J, Buscot F. Diversity of Cryptococcus and Dioszegia yeasts (Basidiomycota) inhabiting arbuscular mycorrhizal roots or spores. FEMS Yeast Res. 2004;4:597–603.
Gai CS, Lacava PT, Maccheroni W, Glienke C, Araújo WL, Miller TA, et al. Diversity of endophytic yeasts from sweet orange and their localization by scanning electron microscopy. J Basic Microbiol. 2009;49:441–51.
Cloete KJ, Valentine AJ, Stander MA, Blomerus LM, Botha A. Evidence of symbiosis between the soil yeast Cryptococcus laurentii and a sclerophyllous medicinal shrub, Agathosma betulina (Berg.) Pillans. Microb Ecol. 2009;57:624–32.
Couture M, Fortin J, Dalpe Y. Oidiodendron griseum Robak: an endophyte of ericoid mycorrhiza in Vaccinium spp. New Phytol. 1983;95:375–80.
Vohník M, Albrechtová J, Vosátka M. The inoculation with Oidiodendron maius and Phialocephala fortinii alters phosphorus and nitrogen uptake, foliar C: N ratio and root biomass distribution in Rhododendron cv. Azurro Symbiosis. 2005;40:87–96.
Rice AV, Currah RS. Oidiodendron: a survey of the named species and related anamorphs of Myxotrichum. Stud Mycol. 2005;53:83–120.
Lombard L, Van Der Merwe NA, Groenewald JZ, Crous PW. Lineages in Nectriaceae: re-evaluating the generic status of Ilyonectria and allied genera. Phytopathol Mediterr. 2014;53:515–32.
Whitelaw-Weckert M, Rahman L, Appleby L, Hall A, Clark A, Waite H, et al. Co-infection by Botryosphaeriaceae and Ilyonectria spp. fungi during propagation causes decline of young grafted grapevines. Plant Pathol. 2013;62:1226–37.
Vitale A, Aiello D, Guarnaccia V, Perrone G, Stea G, Polizzi G. First report of root rot caused by Ilyonectria (= Neonectria) macrodidyma on avocado (Persea americana) in Italy. J Phytopathol. 2012;160:156–9.
Usuki F, Narisawa K. Formation of structures resembling ericoid mycorrhizas by the root endophytic fungus Heteroconium chaetospira within roots of Rhododendron obtusum var. kaempferi. Mycorrhiza. 2005;15:61–4.
Narisawa K, Hambleton S, Currah RS. Heteroconium chaetospira, a dark septate root endophyte allied to the Herpotrichiellaceae (Chaetothyriales) obtained from some forest soil samples in Canada using bait plants. Mycoscience. 2007;48:274–81.
Vohník M, Albrechtová J. The co-occurrence and morphological continuum between ericoid mycorrhiza and dark septate endophytes in roots of six European Rhododendron species. Folia Geobotanica. 2011;46:373–86.
Jumpponen A, Trappe JM. Dark septate endophytes: a review of facultative biotrophic root-colonizing fungi. New Phytol. 1998;140:295–310.
Usuki F, Narisawa K. A mutualistic symbiosis between a dark septate endophytic fungus, Heteroconium chaetospira, and a nonmycorrhizal plant, Chinese cabbage. Mycologia. 2007;99:175–84.
Wang W, Tsuneda A, Gibas CF, Currah RS. Cryptosporiopsis species isolated from the roots of aspen in Central Alberta: identification, morphology, and interactions with the host, in vitro. Botany. 2007;85:1214–26.
Kowalski T, Bartnik C. Cryptosporiopsis radicicola sp. nov. from roots of Quercus robur. Mycol Res. 1995;99:663–6.
Sigler L, Allan T, Lim SR, Berch S, Berbee M. Two new Cryptosporiopsis species from roots of ericaceous hosts in western North America. Stud Mycol. 2005;53:53–62.
Schulz B, Sucker J, Aust H, Krohn K, Ludewig K, Jones P, et al. Biologically active secondary metabolites of endophytic Pezicula species. Mycol Res. 1995;99:1007–15.
Strobel GA, Miller RV, Martinez-Miller C, Condron MM, Teplow DB, Hess W. Cryptocandin, a potent antimycotic from the endophytic fungus Cryptosporiopsis cf. quercina. Microbiology. 1999;145:1919–26.
Talbot JM, Bruns TD, Taylor JW, Smith DP, Branco S, Glassman SI, et al. Endemism and functional convergence across the North American soil mycobiome. Proc Natl Acad Sci U S A. 2014;111:6341–6.
Martiny JBH, Bohannan BJ, Brown JH, Colwell RK, Fuhrman JA, Green JL, et al. Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–12.
De Wit R, Bouvier T. ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ Microbiol. 2006;8:755–8.
Rooney N, McCann K, Gellner G, Moore JC. Structural asymmetry and the stability of diverse food webs. Nature. 2006;442:265–9.
Ellison AM, Bank MS, Clinton BD, Colburn EA, Elliott K, Ford CR, et al. Loss of foundation species: consequences for the structure and dynamics of forested ecosystems. Front Ecol Environ. 2005;3:479–86.
Paine RT. Food web complexity and species diversity. Am Nat. 1966;100:65–75.
Waller F, Achatz B, Baltruschat H, Fodor J, Becker K, Fischer M, et al. The endophytic fungus Piriformospora indica reprograms barley to salt-stress tolerance, disease resistance, and higher yield. Proc Natl Acad Sci U S A. 2005;102:13386–91.
Castro-Sowinski S, Herschkovitz Y, Okon Y, Jurkevitch E. Effects of inoculation with plant growth-promoting rhizobacteria on resident rhizosphere microorganisms. FEMS Microbiol Lett. 2007;276:1–11.
Streeter JG. Failure of inoculant rhizobia to overcome the dominance of indigenous strains for nodule formation. Can J Bot. 1994;40:513–22.
Hartman K, van der Heijden MG, Roussely-Provent V, Walser J-C, Schlaeppi K. Deciphering composition and function of the root microbiome of a legume plant. Microbiome. 2017;5:2.
Jumpponen A, Trappe JM. Performance of Pinus contorta inoculated with two strains of root endophytic fungus, Phialocephala fortinii: effects of synthesis system and glucose concentration. Can J Bot. 1998;76:1205–13.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21.
Vohník M, Mrnka L, Lukešová T, Bruzone MC, Kohout P, Fehrer J. The cultivable endophytic community of Norway spruce ectomycorrhizas from microhabitats lacking ericaceous hosts is dominated by ericoid mycorrhizal Meliniomyces variabilis. Fungal Ecol. 2013;6:281–92.
Richardson AE, Barea J-M, McNeill AM, Prigent-Combaret C. Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil. 2009;321:305–39.
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90.
Bai Y, Müller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature. 2015;528:364–9.
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci U S A. 2015;112:E911–20.
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–50.
Kurtz ZD, Mueller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLOS Comp Biol. 2015;11:e1004226.
Arnold AE, Mejía LC, Kyllo D, Rojas EI, Maynard Z, Robbins N, et al. Fungal endophytes limit pathogen damage in a tropical tree. Proc Natl Acad Sci U S A. 2003;100:15649–54.
Acknowledgements
We thank Teshio Experimental Forest (Hokkaido University), Tomakomai Experimental Forest (Hokkaido University), Sugadaira Research Station (Tsukuba University), Yona Field (Ryukyu University), Tropical Biosphere Research Center (Ryukyu University), and Forestry Agency of Japan for the permission of fieldwork. We are also grateful to three reviewers for their constructive comments that improved the manuscript.
Funding
This work was financially supported by JSPS KAKENHI Grant (26711026), JST PRESTO (JPMJPR16Q6), and the Funding Program for Next Generation World-Leading Researchers of Cabinet Office, the Government of Japan (GS014) to HT.
Availability of data and materials
The Illumina sequencing data were deposited to DNA Data Bank of Japan (DDBJ Sequence Read Archive DRA006339). The raw data of fungal community structure and the fungal community matrices analyzed are available with the source study [56] and Additional files 2, 3, and 4, respectively. The UNIX scripts for the bioinformatic pipeline and the R scripts for the PERMANOVA and PERMDISP analyses are available as Additional files 1 and 4, respectively.
Author information
Authors and Affiliations
Contributions
HT designed the work. HT, AST, and HS conducted fieldwork. HT performed the molecular experiments. HT wrote the manuscript with AST and HS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Data S1. UNIX scripts for the bioinformatic pipeline. (ZIP 6 kb)
Additional file 2:
Data S2. Sample-level matrices of the eight forests examined. (XLSX 8388 kb)
Additional file 3:
Data S3. Species-level matrices of plant–fungus associations. (XLSX 9256 kb)
Additional file 4:
Data S4. Information of 8080 fungal OTUs analyzed. (XLSX 4319 kb)
Additional file 5:
Data S5. R scripts for the PERMANOVA and PERMDISP analyses. (ZIP 65 kb)
Additional file 6:
Figure S1. Number of sequencing reads, interaction specificity, and local betweenness. (DOCX 428 kb)
Additional file 7:
Figure S2. Structure of plant–fungus networks in each local forest. (DOCX 4270 kb)
Additional file 8:
Figure S3. Locality information within the full metacommunity-scale network. (DOCX 4949 kb)
Additional file 9:
Figure S4. Metacommunity-scale network of cool-temperate forests. (DOCX 3939 kb)
Additional file 10:
Figure S5. Metacommunity-scale network of warm-temperate and subtropical forests. (DOCX 4792 kb)
Additional file 11:
Table S1. Top 10 list of non-Glomeromycota OTUs with the highest betweenness within the subtropical metacommunity network. (DOCX 114 kb)
Additional file 12:
Table S2. Top 10 list of non-Glomeromycota OTUs with the highest weighted betweenness within the metacommunity networks. (DOCX 125 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Toju, H., Tanabe, A.S. & Sato, H. Network hubs in root-associated fungal metacommunities. Microbiome 6, 116 (2018). https://doi.org/10.1186/s40168-018-0497-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-018-0497-1