
Abstract
Background
Perturbations to the composition and function of bronchial bacterial communities appear to contribute to the pathophysiology of asthma. Unraveling the nature and mechanisms of these complex associations will require large longitudinal studies, for which bronchoscopy is poorly suited. Studies of samples obtained by sputum induction and nasopharyngeal brushing or lavage have also reported asthma-associated microbiota characteristics. It remains unknown, however, whether the microbiota detected in these less-invasive sample types reflect the composition of bronchial microbiota in asthma.
Results
Bacterial microbiota in paired protected bronchial brushings (BB; n = 45), induced sputum (IS; n = 45), oral wash (OW; n = 45), and nasal brushings (NB; n = 27) from adults with mild atopic asthma (AA), atopy without asthma (ANA), and healthy controls (HC) were profiled using 16S rRNA gene sequencing. Though microbiota composition varied with sample type (p < 0.001), compositional similarity was greatest for BB-IS, particularly in AAs and ANAs. The abundance of genera detected in BB correlated with those detected in IS and OW (r median [IQR] 0.869 [0.748–0.942] and 0.822 [0.687–0.909] respectively), but not with those in NB (r = 0.004 [− 0.003–0.011]). The number of taxa shared between IS-BB and NB-BB was greater in AAs than in HCs (p < 0.05) and included taxa previously associated with asthma.
Of the genera abundant in NB, only Moraxella correlated positively with abundance in BB; specific members of this genus were shared between the two compartments only in AAs. Relative abundance of Moraxella in NB of AAs correlated negatively with that of Corynebacterium but positively with markers of eosinophilic inflammation in the blood and BAL fluid. The genus, Corynebacterium, trended to dominate all NB samples of HCs but only half of AAs (p = 0.07), in whom abundance of this genus was negatively associated with markers of eosinophilic inflammation.
Conclusions
Induced sputum is superior to nasal brush or oral wash for assessing bronchial microbiota composition in asthmatic adults. Although compositionally similar to the bronchial microbiota, the microbiota in induced sputum are distinct, reflecting enrichment of oral bacteria. Specific bacterial genera are shared between the nasal and the bronchial mucosa which are associated with markers of systemic and bronchial inflammation.
Similar content being viewed by others

Background
A growing body of evidence implicates perturbations to the composition and function of bronchial bacterial communities in the pathophysiology of asthma across a broad breadth of clinical presentations [1,2,3,4,5,6,7]; relative enrichment of members of the Proteobacteria, such as Haemophilus and Neisseria in the lower airways, is a consistent feature of inflammatory airway disease [1, 3,4,5, 7]. We confirmed this enrichment in our recent study of mild atopic asthmatic subjects, in conjunction with bronchial expansion of the oral commensals, Fusobacterium and Porphyromonas [3], whose relative abundance was associated with sputum eosinophilia. Like most studies of the bronchial microbiota in asthma, our study relied on the invasive procedure of bronchoscopy. We used this procedure to obtain protected bronchial brushings, whereas others have used it to obtain unprotected brushings or broncho-alveolar lavage (BAL) for microbial analysis [1, 4]. Taken together, these independent observations have provided tantalizing insights into the relationships between the bronchial microbiota and asthma but suggest great complexity, with a highly heterogeneous microbial community differing across subjects with distinct phenotypes of asthma and possibly within subjects over time. Unraveling these complex relationships will likely require longitudinal studies of large numbers of subjects, but the costs, discomforts, and expertise required to perform bronchoscopy significantly limit subject recruitment. The studies done so far have thus mostly enrolled only adults and have been cross-sectional in design and small in size.
Correcting these deficiencies will require less invasive sampling approaches for studying the airway microbiome in asthma. Several studies have reported asthma-associated differences in bacterial community composition of sputum samples from asthmatic subjects [8,9,10,11], characterized by expansion of Haemophilus, Moraxella, and Streptococcus. Enrichments in these genera have also been described in studies of hypopharyngeal aspirates from children with wheezy respiratory illness [12], of hypopharyngeal and nasopharyngeal samples from infants who went on to develop asthma [13, 14], and in nasal samples of asthmatic children [15] including those who experienced more severe rhinovirus infections and related exacerbations of asthma [14, 16].
The associations of asthma and of asthma exacerbations with perturbations of the bacterial communities in the nose, nasopharynx, or induced sputum raise questions as to the relationships between the microbiota of the upper and lower airways. In healthy adults, dispersal of microorganisms from the oral (but not the nasal) cavity is considered the dominant source of bronchial microbiota [17,18,19,20], through naturally occurring microaspiration rather than carry over during sample collection [18]. It remains unclear, however, whether the bacterial topography of healthy airways is also characteristic of asthmatic airways. It is also unclear how representative the bacterial communities described in less-invasive sample types are of the composition of bronchial microbiota in asthma.
To address these questions, we took advantage of protected bronchial brush (BB), induced sputum (IS), oral wash (OW), and nasal brush (NB) samples obtained in a subset of a larger cohort of adults in whom the relationships between the bacterial microbiota in BB samples and disease category (atopy with mild asthma—AAs; atopy without asthma—ANAs; and neither atopy nor asthma—HCs) were studied [3]. We anticipated that the bacterial microbiota detected in the less-invasive sample types would differ from those found in BB samples, and we sought to address two main questions in our analysis: first, to determine which of these samples most closely resembled protected bronchial brushings in community composition, and, second, to determine which, if any, of these samples also revealed known asthma-related taxa or taxa associated with clinical or inflammatory features of atopic asthma in our subjects.
Results
Study group characteristics
Atopic asthmatic (AA) subjects in this cohort had mild well-controlled disease, significantly higher serum total IgE, blood and sputum eosinophil cell counts, and were more likely to report a history of allergic rhinitis compared to atopic non-asthmatics (ANAs) and non-atopic healthy control (HC) subjects (Table 1 and Additional file 1 Table S1).
The nasal airways harbor less complex bacterial communities than the lower airways
To determine compositional similarity between less-invasive airway sample types and BBs, we first focused on alpha diversity indices for the subset of samples (n = 27 pairs) from our larger study cohort [3], for which NB samples were collected and all four samples were profiled (NB, IS, OW, and BB; Additional file 1: Table S2). Comparison of alpha diversity indices across these paired specimen types showed that the richness and diversity of the microbiota of BB was significantly higher than NB, but significantly lower than IS and OW (Fig. 1a–d). Expanding the comparison of alpha diversity indices between BB, IS, and OW to the larger subset of samples (i.e., including subjects from whom NB samples were not obtained; n = 45; Additional file 1: Table S2), confirmed that bacterial communities detected in IS samples were more diverse than both BB and OW (Additional file 1: Figure S1a–d). These observations suggest that the microbiota found in IS is likely sourced in variable degrees from the upper and lower airways. In contrast, the nasal airways comprise of less complex communities than those detected in the bronchi of adult subjects, findings that are in agreement with those from other studies [5, 17]. We additionally found the microbiota of IS to be more diverse than that of oral wash, indicating that this sample type reflects a more complex community. Importantly, this relationship in alpha diversity across sample types remained conserved in samples from AA and HC subjects analyzed independently (Additional file 1: Figure S2a–b).

Alpha diversity in the microbiota of different specimen types demonstrating that the upper airway harbors significantly sparser bacterial communities than the lower airways or the oral cavity. a Bacterial richness as indicated by the total number of taxa detected in each sample type. b Shannon index of bacterial diversity in each sample type. c Phylogenetic Faith’s index of bacterial diversity in each sample type. d Pielou’s index of community evenness in each sample type. Statistical significance was determined using Wilcoxon matched-pairs signed rank test
As reported previously [3], phylogenetic diversity in the subset of BBs examined in this study (n = 27 and n = 45) tended to be higher in AAs compared to HC subjects (Additional file 1: Figure S2a–b). No such trend was observed in comparison of the other sample types from the same individuals, nor was a difference in other alpha-diversity indices found from comparison of AAs and HCs (data not shown). These findings suggest that the phylogenetic expansion reported in our earlier study of mild atopic asthma is confined to protected BB samples; the signature is not detected in other sample types from our very mild group of asthmatic adults.
Bacterial microbiota in BB, IS, OW, and NB across all subjects is compositionally distinct
We next assessed the overall phylogenetic community composition by principal coordinate analysis and found that microbiota composition co-varied with sample type (Fig. 2a; unweighted UniFrac; LME p < 0.001). Bronchial microbiota was compositionally distinct from all other sample types (Additional file 1: Figure S3a–c; unweighted UniFrac; LME p < 0.001), although intra-subject paired distance between BB and other sample types was significantly shorter for OW than NB (Fig. 2b and Additional file 1: Figure S4a), suggesting less compositional similarity between the microbiota of the bronchi and the nasal cavity. Bacterial microbiota membership (Fig. 2c) and to a lesser extent community structure (Additional file 1: Figure S4a–b) in BB were more similar to those detected in IS samples than between BB and NB or OW samples.

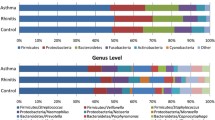
Compositionally lower airway microbiota is more similar to the oral than nasal cavity. a Principal coordinate analysis (unweighted UniFrac) shows compositional dissimilarity between paired samples (linear mixed-effects model, p < 0.0001). b Mean intra-subject paired distance to BB shows that the NB microbiota are most distinct from BB (Wilcoxon matched-pairs signed rank test; Whiskers extend to 95% confidence interval). c Shorter mean intra-subject paired distance between IS and BB compared to OW and BB, suggests that the IS microbiota are more representative of BB than OW is of BB (Wilcoxon matched-pairs signed rank test; Whiskers extend to 95% confidence interval). d Shorter mean intra-subject paired distance is observed between BB and IS compared to OW in AAs and ANAs but not HCs (Paired t test). e Shorter mean intra-subject paired distance between IS and BB in AAs compared to HCs (Welch’s corrected t test; Whiskers extend to 95% confidence interval) suggests that IS microbiota are more representative of BB in asthmatic subjects. f Summary of the relative abundance of taxa identified in paired samples (n = 27) shows NB as being the most compositionally distinct. Bacterial taxonomic classification is shown at a family (genus) level
Using paired samples from 45 participants, we demonstrate that although the membership of the microbiota in IS resembled OW more closely than BB (unweighted UniFrac; Fig. 2c), this was not the case for the community structure (weighted UniFrac; Additional file 1: Figure S4b). Overall, bacterial communities detected in IS were distinct from OW samples (Additional file 1: Figure S3d; unweighted UniFrac LME p = 0.019), indicating that IS microbiota are not simply mirroring OW bacterial communities. To determine whether this observation was consistent across the three subject groups, we examined the mean paired distance between BB-IS and BB-OW across AAs, ANAs, and HCs. Mean paired distance to BB was shorter for paired IS than OW in AAs (unweighted UniFrac; Fig. 2d and weighted UniFrac; Additional file 1: Figure S4c), indicating that the resemblance in bacterial membership of IS to BB was greater in AA than in HC subjects (Fig. 2e). Community membership in IS samples of ANAs was also more similar to BB than those detected in OW (unweighted UniFrac; Fig. 2d) though this relationship did not extend to the overall community structure (weighted UniFrac; Additional file 1: Figure S4c). However, in the HC subjects, mean paired distance between BB-OW and BB-IS was indistinguishable (Fig. 2d and Additional file 1: Figure S4c). Unfortunately, likely due to the small number of HCs in whom NB samples were collected, confounded further by greater dissimilarity in community composition between NB and BB, we found no difference in the BB-NB paired distance between AAs and HC subjects (Additional file 1: Figure S4d–e).
Taxonomic architecture in IS samples more closely resembles that of the bronchial microbiota in asthmatic than in healthy adults
Taxonomic comparison across samples (Fig. 2f) indicated that the overall relative abundance of bacterial genera detected in BB samples correlated more strongly with those detected in IS and OW (Pearson r median [IQR] = 0.869 [0.748–0.942] and r = 0.822 [0.687–0.909] respectively Additional file 1: Table S3) than with those present in the NB samples (r = 0.004 [− 0.003–0.011]; Additional file 1: Table S3 and Figure S5a). Consistent with our earlier observation of a shorter mean paired distance between BB-IS compared to BB-OW, taxonomic composition in BB correlated more strongly with those in IS than OW (Additional file 1: Figure S5b) in the AAs but not ANA or HC subjects (Additional file 1: Figure S5c–e). Together, these findings suggest that asthma and to a lesser extent atopy may be associated with heightened predisposition of the airways to bacterial colonization resulting in communities which are less like those colonizing the oral cavity.
The bronchial microbiota was predominantly colonized by Prevotella, Actinomyces, and to a lesser extent Porphyromonas and Streptococcus (Fig. 3), a pattern similar to that observed for IS samples. Specifically, the relative abundance of Prevotella, Actinomyces, Corynebacterium, and Staphylococcus was indistinguishable between paired BB and IS samples (Fig. 3) but differed between these two samples and the OW. Furthermore, the overall relative abundance of Prevotella and Corynebacterium correlated between BB and IS paired samples (Additional file 1: Table S4) as did those of Porphyromonas, Streptococcus, and Leptotrichia. Although the relative abundance of all highly prevalent genera examined (present at ≥ 3% in either of the samples) was distinct between BB and OW samples, a significant correlation was observed in the relative abundance of Leptotrichia, Porphyromonas, Corynebacterium, and Streptococcus (Additional file 1: Table S4). It should be noted, though, that the relative abundance of all highly prevalent genera examined (present at ≥ 3% in either of the samples) also correlated between IS and OW (Additional file 1: Table S4), attesting to the unsurprising similarity in community composition between these sample types. In contrast, NB samples were predominantly colonized by Corynebacterium followed by Staphylococcus, Streptococcus, Alloiococcus, and Moraxella (Fig. 3). Notably, the relative abundance of only Moraxella correlated strongly between BB and NB samples without also showing a correlation between BB-IS and BB-OW (Additional file 1: Table S4). No significant difference in the relative abundance of the above genera were observed in any of the four samples between AAs and HC subjects, although that of a Staphylococcus trended to be higher in NBs of AAs (Additional file 1: Figure S6a), This enrichment was confirmed at operational taxonomic unit (OTU) level in the NB of AA subjects (Additional file 1: Figure S6b).

Distribution of the relative abundance for most prevalent genera (present at ≥ 3% in any one of the samples) in the four paired (n = 27) specimen types, illustrates the dissimilarity in bacterial composition of NB compared to other samples. Significance was determined using Wilcoxon matched-pairs signed rank test (p > 0.05 not shown)
Next, we evaluated the number of specific OTUs per participant in a larger number of samples (BB, IS, and OW n = 45 pairs), which were exclusive or shared across sample types in our three subject groups (Fig. 4a). We found that a large and similar number of OTUs was shared between all three sample types in all subject groups (Kruskal-Wallis p = 0.48), but that the median number of OTUs shared between IS-BB but not between OW-BB samples was higher in AAs and ANAs compared to HC subjects (Fig. 4b). Interestingly, within the AAs, the number of IS-BB-shared OTUs was higher than those shared between OW-BB paired samples (Fig. 4b). Additionally, phylogenetic diversity for taxa shared between IS-BB samples trended to be higher in AAs and ANAs compared to HCs (Fig. 4c), which was not observed for taxa shared between OW-BB samples (Mann Whitney; p > 0.2). The OTUs predominantly shared (in at least 20% of subjects) between BB-IS samples in AAs included members of Prevotella and a number of other genera previously reported to be associated with asthma, specifically Haemophilus, Fusobacterium, and Neisseria [3], which were not shared at the same frequency between BB-IS samples in the HC subjects (Fig. 4d). Overall, these findings suggest that though significant differences are apparent between all non-invasive sample types and protected bronchial brush bacterial communities, IS microbiota more closely resemble that of BB than do those in OW and NB samples in atopic asthmatic and atopic non-asthmatic adults. This is not to say that taxa detected in IS represented all those detected in the BB, since a number of taxa detected in each sample included sample-specific members of Fusobacterium, Neisseria, Aggregatibacter, Streptococcus, and Prevotella (Additional file 1: Figure S5f). Additionally, among taxa identified in BB but not IS were members of genera whose enrichment has previously been associated with asthma [1, 3, 7, 9], such as Pseudomonas, Sphingomonas, and Moraxella; this was true as well for Lactobacillus whose depletion has been similarly associated with asthma [3]. Conversely, among taxa detected in IS that were not detected in BB were specific members of Haemophilus and Porphyromonas (Additional file 1: Figure S5f), whose enrichment has been reported in asthmatic airways in previous studies.

A greater number of operational taxonomic units (OTUs) was exclusively shared between IS-BB in AAs compared to HC subjects and included taxa previously associated with asthma. a Number of OTUs associated with specific sample type in AAs, ANAs, and HC subjects. Values are shown as a median (IQR). b A greater number of OTUs was shared exclusively (in at least 20% of subjects) between IS and BB in AAs and ANAs compared to HCs, which was greater than those shared in BB-OW (Welch’s corrected t test and &Wilcoxon matched-pairs signed rank test; p > 0.2 not shown). c Faith’s Phylogenetic diversity (PD) of taxa shared between paired IS-BB samples trended to be higher in AAs and ANAs compared to HCs (Mann Whitney test; p > 0.2 not shown). d Frequency distribution of specific genera (present in at least 20% of participants for each group) for OTUs shared exclusively between IS-BB was distinct in AAs and HCs and included known asthma-associated taxa (Chi-square test; p > 0.05 not shown)
Bronchial microbiota comprise taxa shared with both the oral and to a lesser extent the nasal cavity and include a suite of asthma-associated bacterial genera
Considering that the bronchial microbiota in healthy subjects has been reported to simply reflect dispersal from the oral cavity [17,18,19,20,21,22], we next thought to evaluate the contribution of the oral and nasal bacterial communities to the bronchial microbiota in our subjects. Although bronchial microbiota was found to be compositionally distinct from both the OW and NB, we hypothesized that a proportion of taxa detected in BB samples would also be detected in paired OW and NB samples from each individual. Indeed, we identified a number of specific OTUs that were shared with BB samples, significantly greater for OW samples (median/IQR 290/230–348; Fig. 5a) than for NB samples (median/IQR 26/13–48). Taxa also found in OW represented a large proportion (Fig. 5b; median/IQR 61/57–67%) of the total taxa detected in BB samples, a dramatically larger proportion than for the taxa shared between BB-NB samples (median/IQR 6/3–11%). Similarly, the phylogenetic diversity of the taxa shared between OW-BB (Additional file 1: Figure S7a; median/IQR 22/17–24) accounted for a larger proportion of the overall phylogenetic diversity of BB samples (Additional file 1: Figure S7b; median/IQR 66/63–76%), compared to the much smaller contribution to the diversity of taxa shared with NB (median/IQR 15/9–20%). Asthmatic subjects shared a greater number of OTUs between NB and BB samples than did HCs (Fig. 5c), but this was not the case for the number of OW-BB-shared OTUs, although the phylogenetic diversity of the NB-BB-shared taxa was similar for AAs and HCs (Mann-Whitney, p > 0.20). These results indicate that the oral cavity may serve as a source of bacteria for the lower airway microbiota in both healthy and asthmatic subjects, accounting for over 60% of the bronchial microbiota complexity. A novel observation of this study is that the nasal airway may also share microbiota members with the bronchial microbiota in adults and that in AA and ANA subjects more taxa are shared between the two compartments. Additionally, we show that OW and NB share distinct bacterial genera with the bronchial microbiota, since a comparison of the taxa shared between each of these niches and the bronchial microbiota was found to have no significant phylogenetic correlation (Mantel test; r = − 0.031, p = 0.69).

A proportion of all the taxa detected in the BB is shared with paired NB and OW samples, with each compartment contributing distinct taxa to the bronchial community. a The number of taxa shared with BB is higher for OW compared to NB (Wilcoxon matched-pairs signed rank test). b Richness of taxa shared represents a larger proportion of the overall richness detected in BB sample for paired OW compared to NB (Wilcoxon matched-pairs signed rank). c Richness of the shared taxa between paired BB and NB was greater in AAs compared to HCs (&Mann-Whitney test). d Distribution of specific taxa shared with paired BB is distinct between NB and OW samples in AAs. (Genera identified in at least 20% of subjects in the same sample type; Statistical significance between OW and NB genera was determined using Fisher’s exact test where ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05)
Of the known asthma-associated bacteria, a number were identified among those shared between BB and either or both nasal and oral compartments in AAs. As expected, the taxa shared between BB-OW samples included oral commensal genera such as Fusobacterium, Porphyromonas, and Neisseria (Fig. 5d), whereas Streptococcus, Corynebacterium, Staphylococcus, and to a lesser extent Haemophilus were among taxa detected with equal frequency as shared between BB-NB and BB-OW (Fig. 5d). It is important to note, however, that HCs also exhibited the presence of these genera among taxa shared between BB and either or both NB and OW samples (Additional file 1: Figure S7c). Taxa exclusively shared between bronchial and nasal microbiota included members of Pseudomonas and Moraxella in AAs (Fig. 5d). Importantly, Moraxella was not detected among the taxa shared between NB and BB of healthy subjects (Additional file 1: Figure S7c). Collectively, these findings indicate that the nasal taxa shared with bronchial airways in asthma may be few in number but great in importance, as they include bacterial genera previously associated with asthma in adults [1, 7] and asthma exacerbation in children [14,15,16].
Nasal microbiota of asthmatics is associated with clinical and immunological features of allergic inflammation
Recent studies of adult airway microbiota in respiratory disease have described specific patterns of bacterial colonization with distinct dominant genera in both the upper [23] and lower [24] airway compartments, differentially associated with divergent clinical outcomes. Accordingly, we assessed the distribution of bacterial genera dominating each of the paired samples and found that Prevotella was most frequently found to dominate BB and IS (Additional file 1: Figure S6c) and that there was no significant difference in its prevalence across these sample types (21/27 and 23/27, respectively; Fisher’s exact p = 0.73; Additional file 1: Figure S6c). In contrast, this genus dominated OWs with lower frequency (12/27) and did not dominate any of the NB samples (0/27; Fisher’s exact p = 0.024 and p < 0.0001, respectively; Additional file 1: Figure S6c). The microbiota of NBs also differed dramatically in the frequency with which Corynebacterium dominated these samples (19/27 Additional file 1: Figure S6c), further highlighting niche specificity in bacterial microbiota of the nasal airways. Our analysis of the frequency of Prevotella dominance in the four sample types from AAs and HC subjects showed them not to differ (Additional file 1: Figure S6d), and the consistency between BB and IS samples was observed in both groups. There was, however, a difference in the frequency of dominance by Corynebacterium in NB samples, which was found in all HCs but in only half of the AA subjects (Fig. 6a; Fisher’s exact p = 0.07). Possibly because of the small sample size and the mildness of the asthma of our subjects, we did not detect differences in the frequency distribution of other dominant genera between AAs and HC subjects, although we note that Moraxella-dominated communities in NB samples were detected only in asthmatic subjects (3/18 AAs, Additional file 1: Figure S6c).

Nasal microbiota in asthmatic patients are associated with markers of systemic and bronchial inflammation. a Frequency distribution of Corynebacterium dominant communities in NB samples was lower in AAs (n = 18) compared to HC (n = 6) subjects (Fisher’s exact test). b The relative abundance of Corynebacterium in NB samples in AAs correlated negatively with a number of inflammatory markers of atopic asthma (Spearman correlation). c The relative abundance of Moraxella in NB samples in AAs correlated positively with a number of inflammatory markers of atopic asthma (Spearman correlation)
In light of these observations, we enquired whether the relative abundance of the prevalent bacterial genera in the AAs was associated with a number of clinical and immunological features characteristic of the disease. Indeed, the relative abundance of Actinomyces in both BB and OW samples was inversely associated with serum IgE levels, as well as BAL neutrophil counts and G-CSF levels, respectively (Table 2). No significant associations were found between the inflammatory and clinical parameters examined and the relative abundance of the prevalent genera in IS samples, likely due to the complexity of the microbiota detected in this specimen type. Surprisingly, the most associations with features of atopic asthma was observed with bacterial genera detected in the nasal airways (Table 2) and included those previously implicated in asthma in pediatric studies [14]. For instance, the relative abundance of Alloiococcus positively correlated with PC20 (i.e., with lower bronchial reactivity) and that of Corynebacterium negatively correlated with a number of systemic and bronchial inflammatory markers including BAL eosinophil counts, IL-6, IL-7, and IL-21 levels (Table 2 and Fig. 6b). In contrast, the relative abundance of Moraxella and Streptococcus correlated positively with systemic and lower airway eosinophilia (Table 2 and Fig. 6c) and bronchial pro-inflammatory cytokines including TNF and IL-7, respectively. We did not detect a correlation between the relative abundance of these two genera (Spearman, p > 0.20), but both were negatively associated with the relative abundance of Corynebacterium (Spearman rho = − 0.51 and − 0.53, respectively, p = 0.030).
Additionally, asthmatic subjects whose nasal microbiota was dominated by Corynebacterium exhibited significantly lower levels of pro-inflammatory BAL cytokines, specifically IL-6, IL-7, and IL-21 (Additional file 1: Table S5) compared to subjects exhibiting nasal microbiota dominated by other bacterial genera. Nasal microbiota of the Corynebacterium-dominated subgroup of asthmatic subjects were enriched for specific taxa belonging to Corynebacterium, Streptococcus, and Lactobacillus genera and were highly depleted for members of Moraxella, Neisseriaceae, Streptococcus, Staphylococcus, and Haemophilus compared to asthma subjects whose nasal microbiota was dominated by non-Corynebacterium genera (Additional file 1: Table S6). Whilst the bronchial microbiota in the same subjects was enriched for specific but unique members of Microbacteriaceae, Lactobacillus, and Leptotrichia and depleted for a number of taxa including Streptococcus, Treponema, Fusobacterium, and Neisseria (Additional file 1: Table S7). Thus, it appears that bronchial microbiota in asthmatics whose nasal microbiota is dominated by non-Corynebacterium bacterial genera are enriched for asthma-associated taxa, which may explain the elevated levels of pro-inflammatory cytokines in these subjects. However, apart from the observation that nasal Corynebacterium dominance of asthmatic subjects was more frequently observed in males (80%, Fisher’s exact p = 0.06; Additional file 1: Table S5), we were limited by a small sample size in this study to identify any other patient characteristics associated with this microbiological signature. Nevertheless, our observations suggest that predominant colonization of the nasal airways with members of Corynebacterium may serve as a biomarker for dampened inflammation associated with atopic asthma and are in agreement with prior observations of infant nasopharyngeal colonization with these genera being associated with lower risk for acute respiratory illness [14, 25], a risk factor for asthma development later in life [26,27,28]. Collectively, these findings suggest that the nasal microbiota in adult asthmatics, although distinct from bronchial bacterial communities, may prove useful biomarkers of bronchial and systemic inflammation associated with atopic asthma.
Discussion
In this study, we aimed to address the compositional similarity between the bacterial microbiota detected in samples obtained by minimally invasive sampling approaches (nasal brushing, oral wash, sputum induction) to those detected in protected bronchial brushings, focusing on adults with mild, corticosteroid-naïve atopic asthma and adults without atopy or asthma. Our findings confirm niche specificity of microbiota in adult airways but indicate that the architecture of the bacterial communities in induced sputum, particularly in asthmatic subjects, resembles that in protected bronchial brushings more closely than do those in oral or nasal samples. Of the sampling methods examined, our observations collectively support the use of IS for inferring lower airway microbial community composition in asthmatic individuals, recognizing that IS microbiota may provide an incomplete picture of the bronchial microbiota, and inaccurately bias the microbiota composition to reflect oral taxa enrichment, especially in very mild airway disease, as in our cohort. However, our analyses by multiple approaches consistently found evidence in asthmatic subjects of greater similarity in IS and BB bacterial microbiota compared to non-asthmatic subjects. Further, while the bacterial communities found in the nasal mucosa were distinct from those in the bronchial mucosa, their composition was associated with markers of systemic and bronchial inflammation clinically relevant to asthma. Another novel observation we report in this study is that nasal airways are a potential source of distinct bacterial taxa for lower airways in adults, and that this inoculum is richer in atopic asthmatic subjects.
Although our findings provide evidence in support of using less-invasive sampling methods to study the airway microbiota in larger numbers of asthmatic patients, important limitations of these findings must be acknowledged. The number of subjects from our cohort who had all paired sample types available for analysis was relatively small, especially a paucity of NB samples from atopic subjects without asthma, an important control group to compare against subjects with atopic asthma. Thus, the observation of a richer shared microbiota composition between NB and BB samples in asthmatic than in healthy subjects could reflect associations with atopic status and immune responses. Also, a relatively constricted scope of asthma phenotype was captured in our cohort who all had mild, corticosteroid-naïve atopic asthma. The similarities and dissimilarities we quantified between upper airway and bronchial samples in bacterial microbiota composition may or may not hold true in other asthma phenotypes or with worsened disease activity. Our analysis also focused only on adult subjects, and it is possible that findings may differ in infants and young children due to differences in anatomical distance between upper and lower airway compartments. A major strength of this study is the standardized collection and analysis of paired upper and lower airway sample types, all of which were collected either on the same day (NB, OW and BB at bronchoscopy visit) or within 2 weeks (IS) prior to the bronchoscopy.
Our findings confirm that different compartments of the airways harbor compositionally distinct microbiota in patients with asthma, as they do in healthy adults [17]. They further reveal the bacterial communities colonizing the nasal cavity to be significantly scarcer than those colonizing the lower airways. That we should find the microbial composition of induced sputum to differ overall from bronchial brush is not surprising, given that an earlier study reported distinct differences in the microbial communities detected in BAL and brush obtained from the same subjects [1]. We expected comparison of the induced sputum and protected bronchial brushes collected in our study to reveal dissimilarities, considering the unavoidable contamination of bronchial secretions by saliva during the process of sputum collection, as well as the difference in spatial topography of the airways sampled by either method. Indeed, we found the microbiota of induced sputum to be compositionally most similar to oral wash but still to resemble the bronchial brush microbiota more closely than did oral wash or nasal brush samples. The taxonomic architecture of induced sputum samples resembled that of the bronchial brush samples, particularly in atopic subjects, and the taxa shared between the two sample types included those previously reported to be associated with asthma, including members of Haemophilus, Fusobacterium, and Neisseria [1, 3,4,5, 7]. These observations suggest that the bacterial communities in induced sputum, although distinct from those of the bronchial mucosa, capture useful aspects of the latter in asthma. This finding is encouraging for future mechanism-oriented studies aimed at elucidating the contribution of the airway microbiome to asthma heterogeneity, enabling expansion of studies to include larger cohorts of patients and the ability to study patients repeatedly over time.
Nasal colonization with S. aureus has previously been reported as a risk factor for asthma diagnosis in children and young adults [29], while Staphylococcus-derived products have been observed to directly induce pro-inflammatory Th2 inflammation in human nasal epithelium [30] and exacerbate allergic airway inflammation in experimental model of asthma in mice [31]. Our observation of enrichment of Staphylococcus in the nasal microbiota of atopic asthmatics in our cohort further supports a possible role for this pathobiont in asthma. Further, despite our finding that nasal and bronchial microbiota are compositionally distinct in asthmatic as well as healthy [17] adults, we identified a suite of bacterial taxa shared between these two compartments that accounted for over 15% of the bronchial phylogenetic diversity in our atopic asthma subjects. Although this contribution of the nasal compartment is dwarfed by the contribution of taxa from the oral cavity, both compartments contribute distinct and important asthma-associated taxa to the lower airways in adult asthma, as has been shown in children with chronic lung disease [32]. In particular, we show that the relative abundance of Moraxella correlates positively between paired nasal and bronchial brush samples, and members of this genus are among the taxa shared between the two compartments in some of our asthmatic but in none of the healthy subjects. Intriguingly, the relative abundance of nasal Moraxella was positively associated with systemic and airway eosinophil counts, suggesting associations with allergic inflammation, as well as with asthma-associated [33, 34] pro-inflammatory chemokine CXCL11 and TNF levels in bronchial lavage fluid. Increased expression of nasal epithelial TNF has also been reported in children and adolescents with asthma whose nasal microbiome was enriched for M. catarrhalis [35].
This association of nasal Moraxella with inflammatory cells and cytokines in our asthmatic subjects contrasted with our findings related to detection of Corynebacterium in nasal brushings. Corynebacterium was detected as a dominant genus in the nasal samples from all healthy subjects, but in only half of our asthmatic subjects, in whom the abundance of this genus was negatively associated with the presence of Moraxella and Streptococcus in nasal brushes and of eosinophils in the blood and of asthma-associated [30, 33] inflammatory cytokines in BAL fluid. Previous findings from a number of pediatric studies have shown differences in nasal microbiota to be related to differences in asthma-related outcomes [12,13,14,15,16, 25], suggesting that the microbiota of the nasal airways may be a biomarker of asthma risk in children. Of particular interest in this context is the finding of an inverse relationship in the abundance of Corynebacterum and both Moraxella [25, 36] and Streptococcus [36,37,38] in the nasal microbiota of young children and inverse association with respiratory health [14, 25], which appear to be sustained in adult airways of asthmatic subjects. The mechanisms driving these antagonistic interactions may involve commensal bacteria directly inhibiting outgrowth of a pathobiont, as was shown for commensal Corynebacterium’s inhibition of S. pneumoniae growth in- vitro [38], but much more work is needed to explore these relationships and their subsequent associations with host immune function. At the least, our findings suggest that additional studies of the nasal microbiota associations to asthma-related inflammation, phenotype, clinical course, or response to therapy are warranted.
Conclusion
We conclude that in adults, induced sputum is superior to nasal brush or oral wash for studying bronchial microbiota in asthma. Although compositionally similar, the microbiota in induced sputum are distinct from bronchial microbiota in reflecting enrichment of oral bacterial taxa. Although the nasal microbiota was highly distinct from that of the oral or bronchial compartments, the abundance of specific bacterial genera in nasal brushes from asthmatic subjects (Corynebacerium and Moraxella) was associated with abundance or depletion in bronchial brushes of genera previously reported as associated with asthma, and further to be associated with markers of systemic and bronchial inflammation. Future studies designed to elucidate bacterial mechanisms contributing to the pathophysiology of asthmatic airway inflammation may therefore benefit from parallel sampling of lower and upper airway compartments.
Methods
Study population and sample collection
The study cohort consisted of a subset of subjects enrolled in the NHLBI AsthmaNet microbiome study NCT01537133 [3] and included 22 atopic asthmatics (AA), 12 atopic non-asthmatics (ANA) and 11 non-atopic healthy control subjects (HC) whose characteristics are summarized in Table 1 and Additional file 1: Table S1. Atopy was defined by serologic evidence (> 0.35 kU/l) of sensitivity to ≥ 1 of 12 aeroallergens (specific IgE by ImmunoCap; Thermo-Scientific). Asthma was confirmed by measurement of airway responsiveness (methacholine PC20 ≤ 8 mg/mL or FEV1 improvement ≥ 12% post-albuterol). Allergic inflammation in asthmatic subjects was assessed by measurement of circulating and sputum inflammatory cells and BAL levels of inflammatory cytokines [33]. At enrollment, asthmatics had been clinically stable for 3 months, without the use of a controller medication in the preceding 6 months. Exclusion criteria included a history of smoking, respiratory infection within 6 weeks or antibiotic use within 3 months of enrollment. Each subject signed informed consent approved by their center’s IRB; an NHLBI-appointed Data Safety Monitoring Board (DSMB) oversaw the study conduct.
Samples processed for microbiota analysis were collected across nine clinical centers and included 45 paired protected bronchial brushings (BB), oral wash (OW) and induced sputum (IS) samples. In subset of 27 of these 45 subjects, intranasal brushings (NB) were also collected (Additional file 1: Table S2). The distribution of samples did not vary significantly across clinical centers (Chi-square; p > 0.10). IS samples were collected as previously described [39] within a 2-week window (median 8 [IQR 8–10] days) from the date of bronchoscopy and those with > 80% squamous epithelial cell counts were excluded from the study. All samples intended for microbial analysis were stored in RNAprotect Saliva Reagent (QIAgen) at − 80 °C until processing.
BAL cytokine multiplex testing
Bronchoalveolar lavage (BAL) supernatants collected during bronchoscopy visit were concentrated by applying 4 mL of sample to Amicon Ultra-4 Centrifugal Filter Unit tubes (Millipore) following centrifugation at 4000g for 40 min at 4 °C. Concentrated BAL fluid was assayed using a MILLIPLEX MAP human high sensitivity T cell panel 21-plex immunology multiplex assay (Millipore) with antibody-coated beads for detection of Fractalkine, TNF, MIP-3α, MIP-1β, MIP-1α, ITAC, IL-23, IL-21, IL-17A, IL-13, IL-12 (p70), IL-10, IL-8, IL-7, IL-6, IL-5, IL-4, IL-2, IL-1β, IFNγ, and GM-CSF. Standards and experimental samples were tested in duplicate. Results were acquired on a Labscan 200 analyzer (Luminex) using Bio-Plex manager software 6.1 (Bio-Rad). A 5-point logistic regression curve was used to calculate the concentration from the fluorescence intensity of the bead measurements. Samples that were below the level of detection were assigned one-half the lowest detectable value for that analyte. Out of the 21 targeted cytokines, only 11 had detectable levels in at least 4 (i.e., 20%) subjects; these were included in the comparisons with the microbiological data.
Nucleic acid extraction and 16S rRNA-based sequencing
Nucleic acids were extracted as previously described [7] using a modified bead-beating protocol and the AllPrep kit (QIAgen). The variable region 4 (V4) of the 16S rRNA gene was amplified using 515F/806R primer combination as previously described [40, 41]. Amplicons were purified using SPRI beads (Beckman Coulter) or in the presence of multiple bands, gel-extracted with a QIAgen Gel Extraction kit (QIAgen), analyzed on Bioanalyzer (Aligent), and quantified using the Qubit HS dsDNA kit (Invitrogen). Only samples with amplicons ≥ 10 ng were sequenced. Blank control DNA extracts were also amplified, bead purified, and sequenced. Samples with sufficient amplicon were pooled at 50 ng per sample, with a blank control per 30–35 samples. The barcoded, pooled library was quantified using the Qubit HS dsDNA kit (Invitrogen), denatured and 5pM was loaded onto the Illumina MiSeq cartridge (V3) in combination with a 15% (v/v) of denatured 12.5pM PhiX for sequencing.
Sequence data processing and quality control
Paired-end sequences were combined using FLASh version 1.2.7 [42]. Sequence analysis was performed using the Quantitative Insights into Microbial Ecology (QIIME) pipeline [43]. Raw sequences were de-multiplexed by barcode and quality filtered by removing low-quality sequences. Sequences with three or more consecutive bases with a Q score < 30 were truncated and discarded if the length was less than 75% of the original 250 bp read length. Sequences were aligned using PyNAST [44], and operational taxonomic units (OTUs) were picked at 97% sequence identity using UCLUST [45] against the Greengenes database [46]. Reads that failed to hit the reference sequence collection were retained and clustered de novo. PyNAST-aligned sequences were chimera checked using ChimeraSlayer and putative chimeras as well as OTUs identified in negative controls (Additional file 1: Table S8) were removed from the working OTU table. A phylogenetic tree was built using FastTree [47] and used to compute Faith’s Phylogenetic Diversity and UniFrac distances on OTU table multiply rarefied [48] to 26,185 sequences per sample.
Statistical analysis
All statistical analyses were performed as indicated in QIIME, R environment or using PRISM software. Statistical tests used were selected based on the outcome of Shapiro-Wilk’s normality test coupled with a graphical approach. Wilcoxon matched-pairs rank test was used for comparison between correlated sample types. Welch’s corrected t test, or Mann-Whitney test and Chi-square or Fisher’s exact test where appropriate were used to determine significant differences in comparisons between independent study groups. Spearman correlation was used to test for an association between the relative abundance of bacterial genera in paired sample types and immunological and clinical parameters in AA subjects. Ordination was visualized using principal coordinate analysis (PCoA) on unweighted UniFrac distance matrix and plotted using Emperor [49]. Significant differences in beta-diversity between paired samples were calculated on unweighted UniFrac PC1 coordinates (as a response variable) using Linear mixed effects (LME) model ([50] using lmerTest package in R). Three-model approach (Poisson, negative binomial, and zero-inflated negative binomial mixed-effect models) corrected [51] for multiple testing (q < 0.10) as previously described [52] was used to determine specific OTUs in NBs or BB which differed in relative abundance between groups. Mantel test [53] based on unweighted UniFrac distance was used to compare phylogenetic contribution of nasal and oral compartments to BB microbiota based on OTUs shared between paired samples.
Abbreviations
- AA:
-
Atopic adults with mild asthma
- ANA:
-
Atopic non-asthmatic adults
- BAL:
-
Broncho-alveolar lavage
- BB:
-
Protected bronchial brushings
- HC:
-
Healthy controls
- IS:
-
Induced sputum
- LEM:
-
Linear mixed effects model
- NB:
-
Nasal brushings
- OTU:
-
Operational taxonomic unit
- OW:
-
Oral wash
References
Denner DR, Sangwan N, Becker JB, Hogarth DK, Oldham J, Castillo J, Sperling AI, Solway J, Naureckas ET, Gilbert JA, et al. Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. J Allergy Clin Immunol. 2015;137(5):1398–1405.e3.
Durack J, Boushey HA, Lynch SV. Airway microbiota and the implications of dysbiosis in asthma. Curr Allergy Asthma Rep. 2016;16(8):52.
Durack J, Lynch SV, Nariya S, Bhakta NR, Beigelman A, Castro M, Dyer AM, Israel E, Kraft M, Martin RJ, et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol. 2017;140(1):63–75.
Goleva E, Jackson LP, Kirk Harris J, Robertson CE, Sutherland ER, Hall CF, Good JTJ, Gelfand EW, Martin RJ, Leung DYM. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188(10):1193–201.
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578.
Huang YJ, Nariya S, Harris JM, Lynch SV, Choy DF, Arron JR, Boushey H. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol. 2015;136(4):874–84.
Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127(2):372–381.e1–3.
Green BJ, Wiriyachaiporn S, Grainge C, Rogers GB, Kehagia V, Lau L, Carroll MP, Bruce KD, Howarth PH. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS One. 2014;9(6):e100645.
Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. 2013;131(2):346–352.e341–343.
Taylor SL, Leong LEX, Choo JM, Wesselingh S, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Jenkins C, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2017;S0091-6749(17):30743–1.
Zhang Q, Cox M, Liang Z, Brinkmann F, Cardenas PA, Duff R, Bhavsar P, Cookson W, Moffatt M, Chung KF. Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS One. 2016;11(4):e0152724.
Bisgaard H, Hermansen MN, Bonnelykke K, Stokholm J, Baty F, Skytt NL, Aniscenko J, Kebadze T, Johnston SL. Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ. 2010;341:c4978.
Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bonnelykke K, Brasholt M, Heltberg A, Vissing NH, Thorsen SV, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. 2007;357(15):1487–95.
Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, Holt BJ, Hales BJ, Walker ML, Hollams E. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17(5):704–15.
Depner M, Ege MJ, Cox MJ, Dwyer S, Walker AW, Birzele LT, Genuneit J, Horak E, Braun-Fahrländer C, Danielewicz H, et al. Bacterial microbiota of the upper respiratory tract and childhood asthma. J Allergy Clin Immunol. 2017;139(3):826–834.e813.
Kloepfer KM, Lee WM, Pappas TE, Kang TJ, Vrtis RF, Evans MD, Gangnon RE, Bochkov YA, Jackson DJ, Lemanske RF, et al. Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. J Allergy Clin Immunol. 2014;133(5):1301–7. 1307.e1301–1303
Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6(2):e00037.
Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, Curtis JL. Bacterial topography of the healthy human lower respiratory tract. MBio. 2017;8(1):e02287-16.
Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, Schmidt TM. Application of a neutral community model to assess structuring of the human lung microbiome. MBio. 2015;6(1):e02284–14.
Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–63.
Segal LN, Alekseyenko AV, Clemente JC, Kulkarni R, Wu B, Chen H, Berger KI, Goldring RM, Rom WN, Blaser MJ, et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome. 2013;1(1):1–12.
Segal LN, Clemente JC, Tsay J-CJ, Koralov SB, Keller BC, Wu BG, Li Y, Shen N, Ghedin E, Morris A, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. 2016;1:16031.
Cope EK, Goldberg AN, Pletcher SD, Lynch SV. Compositionally and functionally distinct sinus microbiota in chronic rhinosinusitis patients have immunological and clinically divergent consequences. Microbiome. 2017;5(1):53.
Shenoy MK, Iwai S, Lin DL, Worodria W, Ayakaka I, Byanyima P, Kaswabuli S, Fong S, Stone S, Chang E, et al. Immune response and mortality risk relate to distinct lung microbiomes in patients with HIV and pneumonia. Am J Respir Crit Care Med. 2017;195(1):104–14.
Bosch AA, de Steenhuijsen Piters WA, van Houten MA, Chu M, Biesbroek G, Kool J, Pernet P, de Groot PCM, Eijkemans MJC, Keijser BJF, et al. Maturation of the infant respiratory microbiota, environmental drivers and health consequences: a prospective cohort study. Am J Respir Crit Care Med. 2017;196(12):1582–90.
Beigelman A, Bacharier LB. Early-life respiratory infections and asthma development: role in disease pathogenesis and potential targets for disease prevention. Curr Opin Allergy Clin Immunol. 2016;16(2):172–8.
Busse WW, Lemanske RF, Gern JE. The role of viral respiratory infections in asthma and asthma exacerbations. Lancet. 2010;376(9743):826–34.
Rantala AK, Jaakkola MS, Mäkikyrö EMS, Hugg TT, Jaakkola JJK. Early respiratory infections and the development of asthma in the first 27 years of life. Am J Epidemiol. 2015;182(7):615–23.
Davis MF, Peng RD, McCormack MC, Matsui EC. Staphylococcus aureus colonization is associated with wheeze and asthma among US children and young adults. J Allergy Clin Immunol. 2015;135(3):811–813.e815.
Patou J, Gevaert P, Van Zele T, Holtappels G, van Cauwenberge P, Bachert C. Staphylococcus aureus enterotoxin B, protein A, and lipoteichoic acid stimulations in nasal polyps. J Allerg Clin Immunol. 2008;121(1):110–5.
Huvenne W, Callebaut I, Plantinga M, Vanoirbeek JA, Krysko O, Bullens DM, Gevaert P, Van Cauwenberge P, Lambrecht BN, Ceuppens JL, et al. Staphylococcus aureus enterotoxin B facilitates allergic sensitization in experimental asthma. Clin Exp Allergy. 2010;40(7):1079–90.
Marsh RL, Kaestli M, Chang AB, Binks MJ, Pope CE, Hoffman LR, Smith-Vaughan HC. The microbiota in bronchoalveolar lavage from young children with chronic lung disease includes taxa present in both the oropharynx and nasopharynx. Microbiome. 2016;4(1):37.
Brasier AR, Victor S, Boetticher G, Ju H, Lee C, Bleecker ER, Castro M, Busse WW, Calhoun WJ. Molecular phenotyping of severe asthma using pattern recognition of bronchoalveolar lavage-derived cytokines. J Allergy Clin Immunol. 2008;121(1):30–37.e36.
Hartl D, Griese M, Nicolai T, Zissel G, Prell C, Konstantopoulos N, Gruber R, Reinhardt D, Schendel DJ, Krauss-Etschmann S. Pulmonary chemokines and their receptors differentiate children with asthma and chronic cough. J Allergy Clin Immunol. 2004;115(4):728–36.
Castro-Nallar E, Shen Y, Freishtat RJ, Perez-Losada M, Manimaran S, Liu G, Johnson WE, Crandall KA. Integrating microbial and host transcriptomics to characterize asthma-associated microbial communities. BMC Med Genet. 2015;8:50.
Pettigrew MM, Laufer AS, Gent JF, Kong Y, Fennie KP, Metlay JP. Upper respiratory tract microbial communities, acute otitis media pathogens, and antibiotic use in healthy and sick children. Appl Environ Microbiol. 2012;78(17):6262–70.
Laufer AS, Metlay JP, Gent JF, Fennie KP, Kong Y, Pettigrew MM. Microbial communities of the upper respiratory tract and otitis media in children. MBio. 2011;2(1):e00245–10.
Bomar L, Brugger SD, Yost BH, Davies SS, Lemon KP. Corynebacterium accolens releases antipneumococcal free fatty acids from human nostril and skin surface triacylglycerols. MBio. 2016;7(1):e01725–15.
Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol. 2014;133(2):388–394.e385.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621–4.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(1):4516–22.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–7.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72.
Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–50.
Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, Panzer AR, LaMere B, Rackaityte E, Lukacs NW. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. 2016;22:1187–91.
Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience. 2013;2(1):16.
Pinheiro JC. Linear mixed effects models for longitudinal data. Encyclopedia of biostatistics. Wiley; 2005.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc. 1995;57(1):289–300.
Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Nikita L, Galuppi M, Lamont RF, Chaemsaithong P, Miranda J, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. 2014;2(1):4.
Diniz-Filho JAF, Soares TN, Lima JS, Dobrovolski R, Landeiro VL, de Campos Telles MP, Rangel TF, Bini LM. Mantel test in population genetics. Genet Mol Biol. 2013;36(4):475–85.
Acknowledgements
We would like to thank Sheila Keating (The Core Immunology Laboratory, Blood Systems Research Institute, San Francisco, CA) for her help with quantifying cytokines in BAL fluids for participants in this study. We additionally would like to thank the following AsthmaNet coordinators and staff at each of the participating study sites: Robert Pedicini8; Kathy Zheng8; Duanny Alva11; Assel Biyasheva11; Jenny Hixon11; Lucius Robinson11; Mary Gill10; James T. Good10; Christena Kolakowski10; Allen Stevens10; E. Rand Sutherland10; Julia Bach13; Rich Cornwell13; Holly Eversoll13; Tiffany Huard13; Keith Meyer13; Barbara Miller13; Ann Sexton13; Michele Wolff13; Merritt Fajt14; Sherri Hill14; Lisa Lane14; Russell Traister14; Cathy Vitari14; Vanessa Curtis6; Brenda Patterson6; Cheryl Shelton6; Kelly Norsworthy7; Kelsey Wollen3; Eugene Bleeker16; Christopher Barrios16; Suzan Farris16; Jeffrey Krings16; Victor Ortega16; Cheryl Wilmoth16; Matthew Bowman7; Linda Engle7; Jennifer Lucier7; Aimee J. Merchlinski7; Kathryn Trasatt7; Angela Updegrave7; Rachel Weber7; and Ronald R. Zimmerman7.
Funding
This study was supported by National Heart, Lung, and Blood Institute (NHLBI)/AsthmaNet [HL098107] and National Institute of Allergy and Infectious Disease (NIAID)/Inner City Asthma Consortium (ICAC) [AI114271]. YJH was supported by NIAID 1R01AI129958 and NHLBI 1R03HL138310.
Availability of data and materials
All sequence data related to this study are available from the European Nucleotide Archive (ENA) under accession numbers PRJEB15534 and PRJEB22676.
Author information
Authors and Affiliations
Consortia
Contributions
All authors but SN and LSC participated in discussions of the parent trial and contributed to the design, conduct, and interpretation of the current study. All authors, but JD, SN, LSC, SVL, AB, and KMA, helped examine the subjects and collect clinical samples. Laboratory analysis was performed by JD, SN, and LSC. Data analysis was performed by JD. All authors have contributed to the manuscript drafting and have approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
JD has no conflict of interest. YJH has received grants from the NHLBI, NIAID, and the Michigan Institute of Health and Clinical Research; has received travel and lodging compensation from the NIH and National Academy of Science; and has received payment for lectures from the American Academy of Allergy, Asthma & Immunology; the Massachusetts Institute of Technology; the European Respiratory Society; the Microbiome R&D Business Forum; and the European Academy of Allergy, Asthma, and Clinical Immunology. SN has no conflict of interest. LSC has no conflict of interest. KMA has no conflict of interest. AB reports grants from NHLBI. MC reports grants from NIH, ALA; personal fees from Astra-Zeneca, Aviragen, Boehringer-Ingelheim, Boston Scientific, Elsevier, 4DPharma, Genentech, Mallinckrodt, Neutronic, Nuvaira, Teva, Theravance and VIDA; and grants from Boehringer-Ingelheim, Chiesi, Gilead, Novartis, Sanofi-Aventis, Vectura (all unrelated to the current work). AMD reports grants from NHLBI. EI reports personal fees from AstraZeneca, Novartis, Philips, Respironics, Regeneron Pharmaceuticals, Research in Real Life (RiRL), TEVA Specialty Pharmaceuticals, Bird Rock Bio, Nuvelution, Pharmaceuticals, Vitaeris, Inc., Sanofi, Merck, Entrinsic Health Solutions and GlaxoSmithKline; non-financial support from Boehringer Ingelheim, GlaxoSmithKline, Merck, Sunovion and TEVA Specialty Pharmaceuticals; and grants from Sanofi, Genentech and Boehringer Ingelheim. MK reports grants from NIH; personal fees from Teva Pharmaceuticals, Astra Zenec, FDA LABA Trials Joint DSMB and Elsevier; and grants from Chiesi and Sanofi. RJM reports grants from NIH; personal fees from AstraZeneca, PMD Healthcare and Respiratory Effectiveness Group; and grants from MedImmune and CHiesi FarmaceuticiSpA. DTM reports grants from NIH and non-financial support from GlaxoSmithKIlne, Merck and Boehringer Ingelheim. SRR reports grants from NIH. TSK reports grants from NIH and personal fees from Pearl Therapeutics, KaloBios, and Insmed Inc. SRW reports grants from NIH. LCD reports grants from NIH and personal fees from Sanofi and GSK. FH has no conflict of interest. SCL reports grants from NIH. NL reports personal fees from TEVA, Astrazeneca and GSK and grants from GSK, SANOFI and AstraZeneca. SPP reports grants from NIH. LJS reports grants from NIH and personal fees from Merck. MEW reports personal fees from AstraZeneca, BSCI, Novartis, Sanofi, Vectura, Regeneron, Meda, Mylan, Gilacure, Tunitas, Genentech, Theravance, Neurotronic, Sentien, Teva, Boehringer INgelheim, GlaxoSmithKline and grants from Sanofi and GlaxoSmithKlin. SVL reports grants from NIH/NIAID, NIH/NICHD, NIH/Office of the Director, NIH/NIDA, Broad Foundation, NIH/NIAID, Sloan Foundation, Pfizer Inc., Gilead Sciences and Janssen; personal fees from Janssen, Boston Consulting Group, Regeneron, MedImmune, Siolta Therapeutics; has a patent Reductive prodrug cancer chemothera (Stan449-PRV) issued, a patent Combination antibiotic and antibody therapy for the treatment of Pseudomonas aeruginosa infection (WO 2010091189 A1) with royalties paid to KaloBios Inc., a patent Therapeutic microbial consortium for induction of immune tolerance with royalties paid to Siolta Therapeutics, a patent Systems and methods for detecting antibiotic resistance (WO 2012027302 A3) issued, a patent Nitroreductase enzymes (US 7687474 B2) issued, a patent Sinusitis diagnostics and treatments (WO 2013155370 A1) issued, and a patent Methods and systems for phylogenetic analysis (US 20120264637 A1) issued; has Co-founded Siolta Therapeutics and is currently a board member and paid consultant for the company; and owns 25% stock. HAB has received grants including travel and lodging compensation from the NIH/NHBI and the NIH/NIAID and has received royalty payments from the McGraw-Hill Companies; he is a consultant for Siolta Therapeutics, Inc. of San Francisco, CA.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplemental information includes Figures S1-7 and Tables S1-8. (DOCX 10754 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Durack, J., Huang, Y.J., Nariya, S. et al. Bacterial biogeography of adult airways in atopic asthma. Microbiome 6, 104 (2018). https://doi.org/10.1186/s40168-018-0487-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-018-0487-3