
Abstract
Background
Clostridium difficile disease is the leading antibiotic-associated cause of diarrhea and nosocomial acquired infection in the western world. The per annum burden in the USA alone amounts to 250,000 cases with 14,000 ascribed deaths and medical costs in excess of a billion dollars. Novel models for the study of C. difficile infection are therefore pertinent.
Results
Germ free C57BL/6 mice gavaged with a healthy human fecal microbiota maintained a stable “humanized” microbiota over multiple generations when housed under specific pathogen-free (SPF) conditions. As with mice containing a conventional microbiota, treatment with a five-antibiotic cocktail followed by a single dose of clindamycin renders the animals susceptible to C. difficile infection (CDI). Interestingly, after recovery from the initial CDI infection, a single intraperitoneal injection of clindamycin is sufficient to induce CDI relapse. Relapse of CDI can be induced up to 35 days postinfection after recovery from the initial infection, and multiple episodes of relapse can be induced.
Conclusions
This model enables the study of recurrent C. difficile disease in a host containing a human-derived microbiota. Probiotic treatments using human-derived microbes, either prophylactic or curative, can be tested within the model. The identification and testing of human-derived microbial communities within a humanized microbiota mouse model may enable a higher rate of successful transfer of bacteria-based treatments from the lab to human patients due to the microbes involved initiating from, and being adapted to, the human GI tract.
Similar content being viewed by others
Background
Clostridium difficile infection (CDI) is the principle cause of antibiotic-associated diarrhea (AAD) and nosocomial infection in the western world. Severe cases lead to pseudomembranous colitis and toxic megacolon which can require drastic surgical intervention. Recurrence occurs in approximately 25 % of cases treated with metronidazole or vancomycin with 12 % percent of patients experiencing at least two recurrences and 6 % an excess of two [1]. The per annum burden in the USA amounts to 250,000 cases with 14,000 ascribed deaths and medical costs in excess of a billion dollars [2].
Disruption of the host microbiota, whose functions include the prevention and limitation of pathogen colonization and growth, is the most prominent risk factor in CDI [3]. Recently, the recapitulation of this barrier, via fecal microbial transplantation (FMT), has shown great promise as a treatment for recurrent CDI cases, recalcitrant to other treatments [4]. FMT, however, has not yet been approved by the FDA and there are safety concerns over the use of a complex mix of bacterial, viral, and fungal components in addition to prions and potentially unknown biologically active substances [5].
Numerous animal [6] and in vitro [7, 8] models have been used to study C. difficile biology, virulence factors, and associated disease. Until recently, the golden Syrian hamster was the most widely used model of CDI. However, hamsters rapidly succumb to disease, often becoming moribund within 48 h, and are not suitable for studies of recurrent disease. Therefore, researchers have shifted towards the use of mouse models of CDI, and a few different models for the study of C. difficile relapse/recurrence have been developed. These models rely upon repeat antibiotic treatment with or without re-infection [9, 10] or suppression of initial infection with antibiotics [11] to achieve a relapse-like state. To our knowledge, animal models to study CDI relapse/recurrence in the presence of a human-derived microbiota have not been described. Here, we characterize a humanized microbiota mouse (HMbmouse) model of CDI with the ability to induce relapse/recurrence of disease in otherwise healthy mice by the administration of a single dose of clindamycin. Investigation of the changes in microbiota that lead to susceptibility to CDI may facilitate a targeted approach to identifying therapeutic microbes. The benefit of this recurrence model is the potential to identify human-derived microbes or microbial communities with prophylactic or curative properties. Bacterial communities that have coevolved within their host are likely to out-compete those from an extraneous source within that niche [12]. The use of an HMbmouse model colonized with human-derived microorganisms, un-adapted to the murine environment, overcomes this inherent problem when adding back potential therapeutic human microbes [11].
Results and discussion
Humanized microbiota mice
To produce humanized microbiota mice (HMbmice), pooled human-derived feces was administered, via gavage, to germ free (GF) C57BL/6 mice. Microbiota was subsequently passed to progeny via maternal transfer and coprophagy. To monitor and compare the fecal microbial communities, we pyrosequence bar-coded and amplified bacterial V3–V5 variable region 16S rDNA in fecal samples from conventional microbiota mice (CMbmice, n = 6), HMb founder mice (GF gavaged with human feces, n = 6), and HMb progeny (n = 16). Following clustering of sequences (97 % similarity) and removal of singletons (operational taxonomic units (OTUs) that only appear once), 371 OTUs were identified across all samples (mean 3886 reads per sample). Fifty-four OTUs were shared between CMb/HMb mice with 137 unique to CMbmice and 180 to HMbmice. The reduced number of unique sequences found in CMbmice may be attributed to the smaller sample population (n = 6 vs n = 22), limiting opportunity for species of low abundance to be sampled. This is borne out by comparison of alpha indices which show higher observed OTUs in CMbmouse communities compared to HMbmouse communities (mean 131.3 ± 11.4 vs 103.3 ± 18.4, p = <0.001). This could be anticipated as certain species from the human microbiota may not survive the transfer process or may be ill suited to the murine gastrointestinal tract. The reduction, albeit slight, is concordant with data of xenomicrobiota transferred to the murine host [13]. There was no significant difference in the Chao1 estimator, a predictor of true richness, suggesting that deeper sequencing would reveal more comparable OTU numbers. CMbmouse communities possessed greater diversity (inverse Simpson index) and evenness (Pielou) of species (Fig. 1).

Comparison of α diversity between conventional and HMbmice reveals higher diversity within conventional microbiota. A Welch two-sample t test was used to determine significance between the groups: observed OTUs, p < 0.001; Chao1 estimator (richness), p = 0.143; inverse Simpson (biodiversity), p < 0.001; and Pielou’s evenness index, p <0.001 (evenness of relative abundance). Holm correction applied to p values to account for multiple inference. Singletons were not removed for α analysis as they are required for the Chao1 estimator
Temporal examination of the HMbmouse microbiota revealed a significant reduction in richness (though not biodiversity or evenness of relative abundance) in progeny mice compared to founder mice (gavaged fecal material). Twenty-two OTUs present in at least four of six founder mice were absent in all progeny mice (Table 1). This may indicate the inability of certain species to be transferred via the parent—offspring route or an inability of these species to remain competitive within the murine host. Following an initial drop between founder and progeny, alpha diversity trended upward over generations, though not significantly (Fig. 2).

HMbMice progeny have a lower α diversity than gavaged founder mice. Pairwise t tests with pooled SD and Holm correction for multiple tests were used to determine significant differences. Singletons were not removed for α analysis as they are required for correct calculation of the Chao1 estimator. Descendant HMbmouse samples were taken from mice aged 6–10 weeks from offspring of founder mice at 4, 9, and 11 months post initial gavage of fecal material
To test for invasion of the HMbmice communities following multiple generations under specific pathogen-free (SPF) conditions, 16S rDNA in fecal samples from HMbmice (2.5 years following initial colonization of founder mice), were sequenced on the Illumina MiSeq platform and compared to the original pooled human fecal sample via reciprocal BLAST search [14]. HMbMouse communities remained remarkably immutable with ~95 % of reads from the deep sequenced HMbmice matching sequences identified in the original fecal sample. Reads that could not be matched clustered into OTUs predominantly identified as Lachnospiraceae and may represent invasive species or those found below the level of detection in the original fecal material.
Visualization of the beta diversity of both CMb and HMb samples (sub-sampled to an even depth of 3224 reads per sample), using principal coordinate analysis (PCoA) of Bray-Curtis dissimilarities followed by a boot strapped Gap-Statistic analysis, identified three clusters: CMbmice, gavaged founder HMbmice, and progeny HMbmice (Fig. 3). An independent PCoA visualization of the HMbmice groups, following removal of CMbmice, maintains the separation of gavaged founder HMbmice. No distinction was evidenced between the multi-generation progeny (Additional file 1: Figure S1).

Clustering of conventional and humanized mouse microbiota based upon principal coordinates analysis (PCoA) of Bray-Curtis dissimilarities. A clear distinction can be visualized along axis 1, accounting for 74.6 % dissimilarity between conventional mouse microbiota (blue, n = 6) and humanized mouse microbiota (green, founder mice 3 weeks post gavage n = 6. Orange, offspring of gavaged mice with microbiota derived by maternal transfer, n = 16). A goodness of clustering measure, the “gap” statistic independently identified three clusters belonging to CMbmice, gavaged founder HMbmice, and their offspring
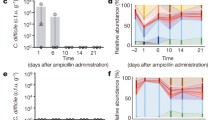
Similarities and differences between CMb/HMbmouse communities at the family level are easily visualized in relative abundance plots (Fig. 4). The largest difference is within the Bacteroidales, which although similar in abundance differs markedly in its constituents between CMb/HMbmouse communities. CMbmouse communities are dominated by the Porphyromondaceae which account for almost 70 % of all sequences compared to ~10 % in HMbmouse communities. Within HMbmice the Bacteroidaceae account for the dominant group, comprising ~50 % of all sequences.

Relative abundance of major bacterial orders in the gut microbiota from CMb and HMb mice. Data are average relative abundances of OTUs assigned at the family level of sequenced communities from CMbmice, n = 6, founder HMbmice, n = 6; HMboffspring at 4 months, n = 4; HMboffspring at 9 months, n = 6; HMboffspring at 11 months, n = 6
In summary, HMbmice are distinguishable from CMbmice by sequencing, are resistant to widespread invasion by murine species, and remain stable over multiple generations when housed under SPF conditions.
HMbMouse C. difficile infection model
Models of C. difficile infection, initially developed in the Syrian hamster [15], have been established in conventional and gnotobiotic mice as well as other animals encompassing a broad host range [6, 16]. To produce a robust infection model in stably colonized HMbmice, a number of antibiotic pretreatment regimes were tested (see “Methods” section). Only the use of a five-antibiotic cocktail administered ad libitum in drinking water followed by intraperitoneal (IP) injection of clindamycin resulted in consistent results. To examine whether the duration of antibiotic pretreatment affected C. difficile disease susceptibility or severity, HMbmice were administered the five-antibiotic cocktail for a period of 3, 4, or 5 days followed by clindamycin IP injection. Mice were challenged 24 h later with 5 × 105 spores of either the high toxin producing lab strain VPI10463 or a clinically relevant NAP1/B1/027 strain CD3017 [8] (see experimental outline, Fig. 5). Disease severity, measured here by maximum weight loss (48 h post challenge), was significantly different across antibiotic pretreatment groups when challenged with VPI10463 (Kruskal-Wallis test, χ 2 = 9:84, p = 0.007; post hoc Mann-Whitney tests with Holm correction: 3 vs 5 days, p = 0.015; Fig. 6). Furthermore, all animals in the 5-day antibiotic pretreatment (n = 6) reached a moribund state requiring euthanasia, whereas animals in the 3- (n = 6) and 4 (n = 6)-day groups recovered. Of the mice challenged with CD3017, 6 of 18 (1/6, 3/6, and 2/6 from the 3-, 4-, and 5-dayantibiotic treatment groups, respectively) reached a moribund state within 48 h. Weight loss was higher in the groups with longer antibiotic treatment; however, due to the reduced number of mice (and therefore statistical power), the difference was not significant (Fig. 7).

Experiment design. A five-antibiotic cocktail consisting of kanamycin (0.4 mg ml−1), gentamicin (0.035 mg ml−1), colistin (850 U ml−1), metronidazole (0.215 mg ml−1), and vancomycin (0.045 mg ml−1)) was administered ad libitum in drinking water for 3, 4, or 5 days. Antibiotic water was changed to sterile antibiotic free water and 24 h later mice were administered an intra-peritoneal injection of clindamycin (10 mg kg−1). Twenty-four hours post clindamycin IP, mice are challenged with C. difficile spores by oral gavage and monitored for signs of disease. Recurrent CDI is induced by administration of a further clindamycin injection (10 mg kg−1)

Mean relative mouse weights and spore counts after C. difficile VPI10463 challenge. Mice were administered a five-antibiotic cocktail in drinking water ad libitum for 3, 4, or 5 days followed by an IP injection of clindamycin (10 mg kg−1). Twenty-four hours after IP injection, mice were challenged with the high toxin producing strain VPI10463 (n = 6 for each group). Antibiotic and C. difficile control groups showed no signs of weight loss or disease (data not shown). Fecal samples were homogenized in 500 μl PBS and heated to 65 °C for 30 min to kill vegetative cells. Samples were serially diluted and plated in replicate on TCCFA plates to enumerate C. difficile spore numbers. No spores were detected in the negative control group or the no antibiotics control group even with 106 spores administered by gavage (not shown). Shaded area and error bars indicate standard deviation from mean. *6/6 mice became moribund. ND spores non-detectable

Mean relative mouse weights and spore counts after C. difficile CD3017 challenge. Mice were administered a five-antibiotic cocktail in drinking water ad libitum for 3, 4, or 5 days followed by an IP injection of clindamycin (10 mg kg−1). Twenty-four hours after IP injection mice were challenged with the clinical NAP1/B1/027 ribotype strain, CD3017 (n = 6 for each group). Fecal samples were homogenized in 500 μl PBS and heated to 65 °C for 30 min to kill vegetative cells. Samples were serially diluted and plated in replicate on TCCFA plates to enumerate C. difficile spore numbers. Shaded area and error bars indicate standard deviation from mean. *1/6, 3/6, and 2/6 mice became moribund in the 3-, 4-, and 5-day Abx groups, respectively. ND spores non-detectable
A Kruskal-Wallis test on spore count data identified a significant difference in VPI10463 spore numbers 24 h post challenge between antibiotic pretreatment groups (χ 2 = 12:91, p = 0.002). Post hoc Mann-Whitney tests with Holm correction established significant differences between groups 3 day (below level of detection) and 5 day (mean = 8.00 × 104 spores g−1 feces), p = 0.008, and groups 4 day (mean = 2.78 × 103 spores g−1 feces) and 5 day, p = 0.023. After 48 h, only mice in the 3- and 4-day antibiotic pretreatment groups survived with no significant difference in spore counts (Fig. 6). Mice challenged with CD3017 did not have a significant difference in spore production between antibiotic pretreatment groups (Fig. 7). However, spore levels were higher in mice that became moribund compared to those that recovered (mean = 4.61 × 106 vs mean = 1.41 × 106 spores g−1 feces), though this difference was not statistically significant due to a large standard deviation from the mean. Across all time points sampled, CD3017 consistently produced more spores than VPI10463 and continued producing detectable spore levels for a longer period of time. CD3017 spores were observable 14 days post challenge (mean across all CD3017 groups day 14 = 4.42 × 103 spores g−1 feces), whereas VPI10463 spores were undetectable by day 11. The VPI10463 strain is known to be a poor spore former both in vitro [17] and in vivo [18]. Conversely, increased spore production has been proposed as a mechanism for the spread of NAP1/B1/027 strains [19], though this is controversial with a wide range of sporulation efficiencies seen in vitro [20].
To examine the effect of tissue damage within the HMbmice, histopathology was undertaken on portions of the distal colon. Samples were scored for edema, cellular infiltration, and epithelial damage on a scale of 0–4 for each metric (0 being no abnormality). The sum of scores (maximum of 12) was taken as an indicator of overall severity. Control mice that received no antibiotic pretreatment (i.e., were not susceptible to CDI) but were administered 5 × 105 VPI10463 spores or that received antibiotic treatment without C. difficile challenge showed minimal pathology (n = 3, mean score 2.33 ± 0.58); mice that had recovered from CDI presented mild disease signs (n = 3, mean score 3.33 ± 0.58) while moribund mice had severe intestinal pathology (n = 6, mean score 10.5 ± 0.55) (Fig. 8).

Histopathology of disease. Histological sections of distal colon were scored blindly by an independent researcher. Three criteria, edema, cellular infiltration, and epithelial damage, were individually scored from 0–4 (a score of 0 indicates no pathology), and the three values summed to give a disease severity score
Recurrent model of CDI
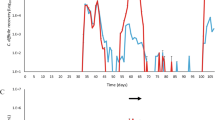
Recurrent CDI is a considerable complication of infection, occurring in as many as 25 % of patients. In a separate experiment, HMbmice were challenged with 5 × 104 CD3017 spores following 4 days of the five-antibiotic pretreatment and clindamycin IP injection. The reduced number of spores administered resulted in less weight loss across all mice during the initial infection (n = 13, mean % bodyweight lost 48 h post challenge = 8.16 % ± 5.9). Mice were allowed to recover from signs of disease (weight loss, detectable spores). After recovery from initial infection, a recurrence of CDI was triggered by a single IP injection of clindamycin. No additional spores were administered. Groups of mice were found to be susceptible to relapse when administered clindamycin 11, 19, or 35 days after the first C. difficile challenge (Fig. 9a–c). HMbMouse weight immediately prior to relapse was normalized to 100 %. Weight loss did not significantly differ between groups upon relapse, regardless of when triggered (11–35 days post initial challenge, combined weight loss 48 h after relapse: n = 13, 9.87 % ± 3.43). Following an initial clindamycin induced relapse (11 days postinfection), a further CDI episode could be provoked (41 days post the initial infection and 30 days after the first relapse) upon an additional clindamycin injection (n = 5, mean weight loss 48 h post second relapse 4.97 % ± 2.11) (Fig. 9c).

HMbMouse model of recurrent CDI. Mice were administered a five-antibiotic cocktail in drinking water ad libitum for 4 days followed by an IP injection of clindamycin (10 mg kg−1). Twenty-four hours after IP injection, mice were challenged with the clinical NAP1/B1/027 ribotype strain, CD3017 (a: n = 5, b: n = 4, c: n = 4). Relapse was induced by further IP injection of clindamycin (10 mg kg−1). Shaded area and error bars indicate standard deviation from mean. Star = clindamycin-induced relapse. ND spores non-detectable. C. difficile Spore numbers were enumerated by homogenizing fecal samples in 500 μl PBS and heating to 65 °C for 30 min to kill vegetative cells followed by serial dilution and plating in replicate on TCCFA plates
C. difficile spores in feces were below the level of detection at the time of relapse induction (as determined by plating). One possibility is that spores adhered to the coat of HMbmice (cages were changed with sterile replacements weekly), triggering relapse upon ingestion in post clindamycin-susceptible animals. Another hypothesis is that vegetative cells or spores remained associated, at low levels, within the murine gastrointestinal (GI) tract. Within 24 h of triggering CDI relapse, spores were detected at levels ≥107 g−1 feces, where they remained for ~5 days before waning to undetectable levels (Fig. 9). This rapid increase suggests that vegetative cells may already be present within the intestine, allowing for rapid growth and spore production when conditions permit. An association of C. difficile and the gut mucosa/epithelium has been observed previously in both animal [21] and in vitro experiments [22].
Effect of antibiotics and C. difficile infection on the HMbmouse microbiota
The use of antibiotics can significantly reduce or ablate sensitive species while simultaneously allowing resistant opportunists to bloom [23]. To examine this effect in HMbmice, fecal samples were sequenced by Illumina MiSeq (average reads per sample 35,505, sub-sampled to an even depth of 23,057) from three time points: base composition, prior to antibiotic insult; post-antibiotics, immediately following the five-antibiotic cocktail and clindamycin IP injection; recovered, following challenge with C. difficile and subsequent recovery from CDI, 14–17 days post cessation of antibiotics.
Comparison of microbial community structures for changes in abundance enables identification of bacteria that potentially facilitate susceptibility/increased severity or resistance/amelioration of C. difficile disease. Principal coordinate analysis of Bray-Curtis dissimilarity of communities prior to antibiotics (n = 38), following 3, 4, or 5 days of antibiotics (n = 36), and ensuing recovery from C. difficile infection (n = 31) was undertaken. Examination of communities from the antibiotic time courses revealed little obvious difference in community structure regardless of duration. Furthermore, communities that recovered from the antibiotic assault (14–17 days post-antibiotic cessation) and C. difficile challenge look remarkably similar to unperturbed, pre-antibiotic, communities (Fig. 10) despite their susceptibility to clindamycin-induced CDI relapse.

Clustering of HMbmice communities: pre-antibiotics, post-antibiotics, and following recovery from C. difficile infection based upon principal coordinates analysis (PCoA) of Bray-Curtis dissimilarities. HMbMouse communities that have undergone 3, 4, or 5 days of antibiotic treatment (3 day, 4 day, 5 day) separate clearly from pre-antibiotic or recovered groups, though do not form distinct clusters of their own as determined by the gap statistic. Communities that have recovered from antibiotic assault only (5 day recov B) and antibiotic assault plus C. difficile invasion (3 day recov, 4 day recov, 5 day recov) return to a pre-antibiotic-like state as evidenced by the close grouping of these communities
Pre- vs post-antibiotic treated microbiota
To examine the effect of antibiotics on the HMbmouse microbiome in more detail, data were subset into pre-antibiotic and immediately post-antibiotic cessation samples. Differentially abundant OTUs between the two groups were detected using negative binomial generalized linear models with DESeq2 in R. Originally designed to detect differentially abundant gene expression from RNA-Seq data, this package has been shown to work well with 16S sequencing count data [24]. p values were corrected for multiple inference using the Benjamini-Hochberg false discovery rate (FDR) procedure and a strict adjusted alpha cutoff value of 0.01 set as criteria for inclusion. Two hundred and eighteen OTUs were found to have significantly different abundances. One hundred and ninety-eight OTUs decreased while 20 OTUs increased post-antibiotic use (see Additional file 2: Table S1). Of those OTUs that decreased, 44.4 % belonged to the Lachnospiraceae, 16.7 % to the Ruminococcaceae, 9.1 % Bacteroidaceae, and the remaining 29.8 % were split across 12 family groups or unclassified. This wide-ranging reduction of taxa can be explained by the fact that the five-antibiotic cocktail possesses a broad spectrum of antimicrobial activity, targeting both Gram positive and negative species via multiple modes of action.
Proposed mechanisms of colonization resistance in respect to C. difficile include competition for sparse resources within a niche; microbially produced short-chain fatty acids, specifically butyrate; and the conversion of primary bile acids into secondary acids via 7α-dehydroxylating bacteria. Overall, 69.7 % of the OTUs significantly decreased by antibiotic treatment belonged to the Clostridia, potentially freeing a niche for C. difficile to exploit. Butyrate possesses anti-inflammatory properties, provides the preferred energy source in colonocytes, and can directly inhibit C. difficile growth in vitro [25]. Members of the Lachnospiraceae and Ruminococcaceae are known to be primary butyrate producers [26], so their significant reduction likely leads to reduced butyrate production, increasing susceptibility to CDI. Primary bile acids, such as taurocholic acid, are known to induce C. difficile germination while secondary bile acids can exert inhibitory effects on germination or kill vegetative cells [27]. The majority of known bile acid 7α-dehydroxylating bacteria belong to Clostridium cluster XIVa [28], of which six OTUs were significantly reduced following antibiotics. Removal of 7α-dehydroxylating bacteria can alter the proportions of primary and secondary bile acids, switching the environment from inhibitory to stimulatory in respect to C. difficile spore germination [29, 30].
Of the OTUs that increased, the largest increase (base mean = 159, log2 fold change = 8.48) belonged to an Enterococcaceae which are known to bloom in humans undergoing certain antibiotic treatments [31, 32] and have been shown to correlate positively with CDI [33]. These blooms may be incidental or may have a synergistic effect on disease severity.
Microbiota in moribund vs recovered mice
Indicator species analysis can be applied to identify OTUs that differ in their occurrence or relative abundances between groups. The fidelity (probability an OTU occurs within a group) and specificity (the proportion of groups that contain an OTU) of all OTUs were calculated and the statistical significance of their associations inferred by permutation followed by Benjamini-Hochberg correction for multiple inference [34]. An OTU is highly characteristic of a group if it is significantly more likely to occur in that group than in another or is considerably more abundant in that group. Mice challenged with C. difficile following antibiotic pretreatment can be split into two groups: “alive” (those that recover) and “dead” (those that become moribund). By carrying out indicator species analysis on these groups, it is possible to correlate OTUs to outcomes that, with further empirical testing, may be shown to reduce or increase the effect of CDI. Four OTUs were associated with mice that survived and 13 with moribund animals (Table 2, alpha cutoff for inclusion = 0.1). Of the four OTUs indicative of mice that survived, two stood out: OTU44, classified to the genus level as Clostridium cluster XIVa, a group associated with butyrate production [35]; and OTU91, a Blautia whose sequence matched Blautia hansenii by BLAST search and is annotated to possess bile-acid 7α-dehydroxylase activity. Recently, Blautia hansenii was predicted to have a negative, inhibitory, interaction with C. difficile in both humans and mouse models [36]. Furthermore, the addition of a bacterial species, Clostridium scindens, containing bile-acid 7α-dehydroxylase activity was shown to ameliorate CDI in a mouse model [36]. No obvious associations could be made for OTUs indicative to moribund animals, in part because eight of the 13 were unclassified at the family level. Those that were identified comprised Parabacteroides (2), Parasutterella, Clostridium sensu stricto, and an unclassified Ruminococcaceae. The isolation and testing of these strains for their protective, in the first instance, or synergistic deleterious effect, in the latter, may provide further insight into the treatment or progression of CDI.
Recovery of HMbmouse microbiota following antibiotic treatment and C. difficile infection
It has been proposed that just a single course of antibiotic use can lead to long-term shifts in the microbiota [37]. Visualization of family level abundances reveals similarity between the untreated and the recovered groups suggesting that HMbmice communities return to their stable state within 14–17 days of antibiotic cessation (Fig. 11). To examine the effect of antibiotic treatment followed by CDI in HMbmice communities, the data were subset into pre-antibiotic (n = 38) and recovered (n = 25) samples (taken 14–17 days post-antibiotic cessation and C. difficile challenge); samples that received antibiotics but were not challenged with C. difficile were excluded from analysis. Forty-two OTUs showed a significant difference, considerably less than the 218 in the pre vs post antibiotic treatment groups, including a decrease in 19 OTUs and an increase in 23 OTUs in the recovered communities compared to the pre-antibiotic-treated communities (Additional file 3: Table S2). Eighteen of the 19 OTUs with reduced abundance were also diminished immediately following antibiotic cessation indicating that they had not yet recovered, though in all cases were trending back towards their base abundance (i.e., the log2 fold change was closer to zero). Just one OTU, classifying as a Clostridium cluster XlVb, had reduced abundance in the recovered samples that was not seen to be reduced immediately following antibiotic cessation. Of the 23 OTUs with increased abundance, 14 were OTUs that have been significantly reduced by antibiotics and had rebounded during recovery to a higher than baseline state. Of the remaining OTUs, one, present in 18 of 25 recovered samples, was C. difficile as determined by BLAST search. The presence of C. difficile, albeit at low levels (0–24 reads in non-sub-sampled/normalized samples or 0–0.07 % of reads, median 0.003 %), is evidence that it is not always cleared following recovery from disease, providing a potential reservoir for relapse when conditions permit. The remaining eight OTUs were diversely composed of Lachnospiraceae, Bacteroidaceae, Streptococcaceae, Clostridiaceae, and Verrucomicrobiaceae. Examination of the alpha metrics associated with pre-antibiotic and recovered samples show a significant increase in estimated richness while evenness is reduced in recovered samples, though the difference in both cases is slight. There was no disparity in diversity between the two groups (Table 3). Taken together, it could be concluded that HMbmice communities would return to their base constitution given enough time following antibiotic insult.

Relative abundance of major bacterial orders broken down at family level in the gut microbiota from HMbmice. Data are average relative abundances of OTUs assigned at the family level of sequenced communities from: 0, pre-antibiotics (n = 38); 3, 3 day Abx (n = 8); 4, 4 day Abx (n = 11); 5, 5 day Abx (n = 17), R3, 3 day Abx group 14–17 days postinfection (n = 12); R4, 4 day Abx group 14–17 days postinfection (n = 9); R5, 4 day Abx group 14–17 days postinfection (n = 4); R5b, 5 day Abx group 14–17 days post cessation of antibiotics—no C. difficile challenge (n = 6)
Conclusion
Fecal microbial transplantation (FMT) as a treatment for C. difficile disease is not a contemporary idea, having been reported in scientific literature for over 50 years [38]. Recently, the use of FMT for the treatment of recurrent CDI has burgeoned with a reported efficacy of >90 % [4]. Despite this, CDI remains the most prevalent nosocomal disease in the developed world. There are tangible risks associated with administering an unknown microbiota to a patient. Increased use of FMT will bring with it a heightened likelihood of seeing complications such as infection, allergic response, or long-term unsuspected sequelae such as weight gain [39]. An alternative to using the mélange of bacteria, fungi, and virons present in FMT samples is to create a defined cocktail of well characterized, sequenced, bacteria that is shown to be efficacious in its treatment or prevention of CDI. This approach has been successfully applied in conventional mouse models using indigenous murine microbes [11] and to ameliorate disease using specific human derived microbiota [36].
The use of HMbmice as a model of both CDI and recurrent CDI in combination with sequencing technology and advanced bacterial isolation techniques provides a resource for the identification of beneficial microbes and the means to test them among other human-derived microbes.
Methods
Humanized microbiota mice
Germ-free C57BL/6 mice were administered, via oral gavage, a human-derived fecal microbiota from a pooled fecal slurry of 12 healthy adults as described in Robinson et al. [8] and classified as HMbmice. HMbmice derived by gavage were housed and bred in gnotobiotic isolators. Progeny and descendants of the founder HMbmice acquired their microbiota via maternal transfer and were maintained under SPF conditions. To limit the introduction of invasive species, gloves were changed between handling conventional microbiota mice (CMbmice) and all chow, water, and bedding autoclaved prior to use. All descendant HMbmouse samples were taken from mice aged 6–10 weeks from offspring of founder mice at time points 4, 9, and 11 months post initial gavage of fecal material. A separate sample was taken from mice 2.5 years, and multiple generations, out for comparison to the original starting fecal material. The animal use protocol was approved by the Animal Ethics Committee of Baylor College of Medicine (Protocol no. AN-6675).
Microbiome analysis
Mouse fecal samples were collected in sterile microfuge tubes and stored at −80 °C until needed. DNA was extracted by bead beating and modified extraction and cleanup with the Qiagen DNEasy Tissue Kit as described previously [8]. For temporal analysis of the HMbmice, the V3–V5 region of the 16S rRNA gene was amplified by PCR using the 357F/962R primers with unique barcodes designed by the Human Microbiome Project [40] and sequenced using a 454 GS Junior (Roche Diagnostics). Analysis of microbial communities following antibiotic administration and C. difficile challenge experiments was carried out by amplifying the V4 region of the 16S rRNA gene with primers F515/R806, using a dual indexing approach (4 forward primer; 96 reverse primer) and subsequent sequencing by Illumina MiSeq. The 96-indexed R806 primers used were previously described in Caparaso et al. [41] (806rbc0–806rbc96). The indexed F515 primers were essentially as described in Kozich et al. [42], except that we generated four barcodes that balanced the nucleotide composition at each position (atcgatgg, tcacgaca, ggtatctc, and cagtcgat) in place of those described in Kozich et al.
PCR reactions were carried out in triplicate. Each reaction contained 4 μl of diluted template, 1X Phusion High-Fidelity Buffer (New England Biolabs), 200 μM dNTPs (Promega or Invitrogen), 10 nM primers, 0.2 units of Phusion DNA Polymerase (New England Biolabs), and PCR grade water to a final volume of 20 μl. The amplification cycle consisted of an initial denaturation at 98 °C for 30 s, followed by 30 cycles of 10 s at 98 °C, 20 s at 51 °C, and 1 min at 72 °C. Replicates were pooled and cleaned using AMPure beads as previously described [8]. DNA sample concentrations were determined with Quant-iT (Life Technologies) and pooled at equimolar ratios. Prior to sequencing, the quality of pooled DNA was assessed by analysis on a Bioanalyzer High Sensitivity DNA Kit (Agilent).
Sequence data analysis
Sequence data were processed using platform-specific (454/MiSeq) pipelines in Mothur v.1.34.3 [43] based upon published SOPs [44, 45]. Alignment was achieved using the Silva 16S rRNA gene reference database. Chimeric sequences and any classifying as chloroplast, mitochondria, Archaea, or Eukaryota were removed after identification by the Mothur implementation of uchime or mothur-formatted ribosomal database project (version 9) classifier, respectively. Sequences were clustered by the average-neighbor method into operational taxonomic units (OTUs) of ≥97 % sequence identity. Analysis of OTU data were performed in R [46] using the phyloseq [47], DESeq2 [48], and indicspecies [49] packages. Sequence data are available via the Sequence Read Archive (SRA) accession numbers SRP061089 and SRP061088.
Alpha diversity
To examine differences in community alpha measurements, several metrics were compared. Richness: both a count of observed OTUs and an estimate of true species richness of a sample with Chao1. Evenness: a measure of how similarly taxa are represented in abundance. Pielou’s evenness index ranges from zero to one with low values indicating uneven communities in which one or a few taxa are highly represented and high values indicating communities with more even representation among taxa. Diversity: the quantification of both richness and evenness. The inverse Simpson index values are higher when either many taxa are present, or when they are more similar in abundance.
S obs = the number of species in the sample, F 1 = the number of singletons (single occurrence within a sample) and F 2 = the number of doubletons.
pi = proportion of Sobs made up of the ith species.
ni = the number of individuals in the ith OTU and N = the total number of individuals in the community.
Beta diversity
The Bray-Curtis dissimilarity matrix was used to describe differences in microbial community structure. Patterns of similarity among the different HMb/CMbmouse fecal communities were examined using principal coordinates analysis (PCoA). Robustness of results was tested by recomputing output using alternative dissimilarity measures (Jaccard, thetayc) and ordinations (non-metric multi-dimensional scaling (NMDS)). No significant changes were observed (data not shown).
Spore preparation and enumeration
Individual C. difficile colonies were picked into pre-reduced BHIS media and propagated anaerobically at 37 °C overnight. Spores were cultivated by spread plating overnight BHIS cultures onto BHIS medium and incubating anaerobically at 37 °C for 5 days. Spores were scraped from plates, resuspended in sterile water, and heated to 65 °C for 30 min to kill vegetative cells. Viable spores were enumerated by plating serial dilutions on BHIS plates supplemented with 0.1 % taurocholic acid. Spore preparations were diluted in sterile water to a final concentration of 5 × 106 spores ml−1 and maintained at 4 °C until used. To enumerate C. difficile spores from HMbmouse feces, fecal pellets were weighed, homogenized in 500 μl sterile water, and incubated at 65 °C for 30 min to kill vegetative cells. Following heat killing, spores were serially diluted and spot plated in duplicate onto selective TCCFA plates containing 0.1 % taurocholic acid.
Establishment of robust C. difficile infection model in HMbmice
To produce a robust infection model in HMbmice, a number of antibiotic pretreatment regimes, known to work well in CMbmice, were tested including the following: cefoperazone (0.5 mg ml−1) in sterile drinking water for 10 days [50] and clindamycin (0.25 mg ml−1) in drinking water for 7 days [51]. However, only the use of a five-antibiotic cocktail developed by Chen et al. [9] produced reproducible results in HMbmice. HMbMice were administered kanamycin (0.4 mg ml−1), gentamicin (0.035 mg ml−1), colistin (850 U ml−1), metronidazole (0.215 mg ml−1), and vancomycin (0.045 mg ml−1) ad libitum in drinking water for 3–5 days followed 24 h later by IP injection of clindamycin (10 mg kg−1). Twenty-four hours post IP injection, mice were challenged, via oral gavage, with 5 × 105 spores of either the high toxin producing strain VPI10463 or the clinically relevant NAP1/B1/027 ribotype strain CD3017. Symptoms were similar to those observed in humans: diarrhea, weight loss, and histologic damage.
CDI recurrence/relapse
HMbMice were administered the five-antibiotic cocktail for 4 days ad libitum in drinking water followed by IP clindamycin injection. Twenty-four hours later, mice were challenged with 5 × 104 CD3017 spores (tenfold lower compared to prior experiments was used to minimize mouse mortality while maintaining CDI symptoms). Following recovery from initial CDI, relapse was triggered by administering a further single dose of clindamycin (10 mg kg−1) up to 4 weeks post recovery from initial infection.
Histopathology
Histological sections of distal colon were scored blindly by an independent researcher utilizing the scoring system outlined in Theriot et al. [50]. Briefly, three criteria: edema, cellular infiltration, and epithelial damage were individually scored from 0–4 (where a score of 0 indicates no pathology) and the values summed to give an overall disease severity score.
References
Kwon JH, Olsen MA, Dubberke ER. The morbidity, mortality, and costs associated with clostridium difficile infection. Infect Dis Clin North Am. 2015;29(1):123–34.
Antibiotic Resistance Threats in the United States, 2013. http://www.cdc.gov/drugresistance/threat-report-2013/.
Britton RA, Young VB. Interaction between the intestinal microbiota and host in clostridium difficile colonization resistance. Trends Microbiol. 2012;20(7):313–9.
Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent clostridium difficile infection. Clin Infect Dis. 2011;53(10):994–1002.
Moayyedi P, Marshall JK, Yuan Y, Hunt R. Canadian association of gastroenterology position statement: fecal microbiota transplant therapy. Can J Gastroenterol Hepatol. 2014;28(2):66.
Hutton ML, Mackin KE, Chakravorty A, Lyras D. Small animal models for the study of clostridium difficile disease pathogenesis. FEMS Microbiol Lett. 2014;352(2):140–9.
Baines SD, Freeman J, Wilcox MH. Effects of piperacillin/tazobactam on clostridium difficile growth and toxin production in a human gut model. J Antimicrob Chemother. 2005;55(6):974–82.
Robinson CD, Auchtung JM, Collins J, Britton R. Epidemic clostridium difficile strains demonstrate increased competitive fitness over non-epidemic isolates. Infect Immun. 2014;82(7):2815–2825.
Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, et al. A mouse model of clostridium difficile-associated disease. Gastroenterology. 2008;135(6):1984–92.
Sun X, Wang H, Zhang Y, Chen K, Davis B, Feng H. Mouse relapse model of clostridium difficile infection. Infect Immun. 2011;79(7):2856–64.
Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing clostridium difficile disease in mice. PLoS Pathog. 2012;8(10):1002995.
Oh PL, Benson AK, Peterson DA, Patil PB, Moriyama EN, Roos S, et al. Diversification of the gut symbiont lactobacillus reuteri as a result of host-driven evolution. ISME J. 2010;4(3):377–87.
Seedorf H, Gri_n NW, Ridaura VK, Reyes A, Cheng J, Rey FE, et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell. 2014;159(2):253–66.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Small J. Fatal enterocolitis in hamsters given lincomycin hydrochloride. Lab Anim Care. 1968;18(4):411–20.
Best EL, Freeman J, Wilcox MH. Models for the study of clostridium difficile infection. Gut Microbes. 2012;3(2):145–67.
Mukherjee K, Karlsson S, Burman LG, Åkerlund T. Proteins released during high toxin production in clostridium difficile. Microbiology. 2002;148(7):2245–53.
Åkerlund T, Svenungsson B, Lagergren Å, Burman LG. Correlation of disease severity with fecal toxin levels in patients with clostridium difficile-associated diarrhea and distribution of pcr ribotypes and toxin yields in vitro of corresponding isolates. J Clin Microbiol. 2006;44(2):353–8.
Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan V, Johnson S, et al. Human hypervirulent clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol. 2010;192(19):4904–11.
Burns DA, Heeg D, Cartman ST, Minton NP. Reconsidering the sporulation characteristics of hypervirulent clostridium difficile bi/nap1/027. PLoS One. 2011;6(9):24894.
Borriello S, Welch A, Barclay FE, Davies HA. Mucosal association by clostridium difficile in the hamster gastrointestinal tract. J Med Microbiol. 1988;25(3):191–6.
Calabi E, Calabi F, Phillips AD, Fairweather NF. Binding of clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun. 2002;70(10):5770–8.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–30.
McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4):1003531.
Rolfe RD. Role of volatile fatty acids in colonization resistance to clostridium difficile. Infect Immun. 1984;45(1):185–91.
Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. 2013;51(9):2884–92.
Sorg JA, Sonenshein AL. Inhibiting the initiation of clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol. 2010;192(19):4983–90.
Ridlon JM, Kang D-J, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–59.
Giel JL, Sorg JA, Sonenshein AL, Zhu J. Metabolism of bile salts in mice influences spore germination in clostridium difficile. PLoS One. 2010;5(1):8740.
Buffe CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, et al. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to clostridium difficile-induced colitis. Infect Immun. 2012;80(1):62–73.
Pérez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, et al. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013;62(11):1591–601.
Stecher B, Maier L, Hardt W-D. ‘Blooming’ in the gut: how dysbiosis might contribute to pathogen evolution. Nat Rev Microbiol. 2013;11(4):277–84.
Schubert AM, Rogers MA, Ring C, Mogle J, Petrosino JP, Young VB, et al. Microbiome data distinguish patients with clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio. 2014;5(3):01021–14.
De Cáceres M, Legendre P, Moretti M. Improving indicator species analysis by combining groups of sites. Oikos. 2010;119(10):1674–84.
Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, et al. Butyrate-producing clostridium cluster xiva species specifically colonize mucins in an in vitro gut model. ISME J. 2013;7(5):949–61.
Buffe CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to clostridium difficile. Nature. 2015;517(7533):205–8.
Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1(1):56–66.
Eiseman B, Silen W, Bascom G, Kauvar A. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. 1958;44(5):854–9.
Alang N, Kelly CR. Weight gain after fecal microbiota transplantation. In: Open Forum Infectious Diseases, vol. 2, p. 004. Oxford: Oxford University Press; 2015.
Consortium HMP et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the illumina hiseq and miseq platforms. ISME J. 2012;6(8):1621–4.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumine sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–20.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Mothur: 454 SOP. Accessed Dec 2014. http://www.mothur.org/wiki/454_SOP.
Mothur: MiSeq SOP. Accessed Dec 2014. http://www.mothur.org/wiki/MiSeq_SOP.
R.C. Team. R: a language and environment for statistical computing. 2014.
McMurdie PJ, Holmes S. Phyloseq: an r package for reproducible interactive analysis and graphics of microbiome census data. PloS One. 2013;8(4):61217.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15(12):550.
Cáceres MD, Legendre P. Associations between species and groups of sites: indices and statistical inference. Ecology. 2009;90(12):3566–74.
Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. Cefoperazone-treated mice as an experimental platform to assess differential virulence of clostridium difficile strains. Gut Microbes. 2011;2(6):326–34.
Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, et al. The clostridium difficile spo0a gene is a persistence and transmission factor. Infect Immun. 2012;80(8):2704–11.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JC, KAE, and RAB designed the study. JC, JA, and LS carried out the experiment. JC analyzed the data. JC and RAB wrote the paper. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Clustering of humanized mouse microbiota based upon principal coordinates analysis (PCoA) of Bray-Curtis dissimilarities. A distinction along axis 1, accounting for 41.3 % dissimilarity, can be visualized between humanized mouse microbiota founder mice and their offspring. (PNG 120 kb)
Additional file 2:
Differentially abundant OTUs in pre-antibiotic vs post-antibiotic HMb mice communities. Two hundred and eighteen OTUs were significantly affected by antibiotic treatment. One hundred and ninety eight were significantly reduced while 20 OTUs were able to resist the antibiotic insult and increase in abundance. Significant differential abundances of OTUs were analyzed using DESeq2 in R. p values were corrected using the Benjamini-Hochberg false discovery rate (FDR) procedure with a corrected alpha value cutoff of 0.01. (PNG 120 kb)
Additional file 3:
Differentially abundant OTUs in pre-antibiotic vs recovered HMb mice communities. The ability of HMbmouse microbiota to return to a “normal” state following recovery from antibiotic insult and CDI was tested by comparing the two groups: pretreatment (no antibiotics, n = 38) and recovered (14–17 days post antibiotic cessation and CDI, n = 25). Significant differential abundances of OTUs were analyzed using DESeq2 in R. p values were corrected using the Benjamini-Hochberg false discovery rate (FDR) procedure with a corrected alpha value cutoff of 0.01. (PDF 260 kb)
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Collins, J., Auchtung, J.M., Schaefer, L. et al. Humanized microbiota mice as a model of recurrent Clostridium difficile disease. Microbiome 3, 35 (2015). https://doi.org/10.1186/s40168-015-0097-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-015-0097-2