
Abstract
Tumor initiation, progression, and response to therapies depend to a great extent on interactions between malignant cells and the tumor microenvironment (TME), which denotes the cancerous/non-cancerous cells, cytokines, chemokines, and various other factors around tumors. Cancer cells as well as stroma cells can not only obtain adaption to the TME but also sculpt their microenvironment through a series of signaling pathways. The post-translational modification (PTM) of eukaryotic cells by small ubiquitin-related modifier (SUMO) proteins is now recognized as a key flexible pathway. Proteins involved in tumorigenesis guiding several biological processes including chromatin organization, DNA repair, transcription, protein trafficking, and signal conduction rely on SUMOylation. The purpose of this review is to explore the role that SUMOylation plays in the TME formation and reprogramming, emphasize the importance of targeting SUMOylation to intervene in the TME and discuss the potential of SUMOylation inhibitors (SUMOi) in ameliorating tumor prognosis.
Similar content being viewed by others
Background
Over the past decade, the variety of protein PTMs has rapidly increased. PTMs alter targeted proteins’ conformation, charge state, and hydrophobicity, affecting their stability and activity. Diverse genomic and proteomic alterations are required in the process that a normal cell converts to a malignant one. Several dysregulated PTM pathways can result in cancer cell proliferation and metastasis. Nowadays, hundreds of unique PTMs in eukaryotic cells have sprung up, such as acetylation, phosphorylation, ubiquitination, and SUMOylation.
PTM with SUMO, named SUMOylation, was discovered in the middle of the 1990s [1]. Observed in numerous human pathologies, particularly in tumorigenesis and progression, SUMOylation acts in various aspects of cell biological process, such as DNA damage repair, protein trafficking, and cell cycle regulation [2, 3, 4, 5].
In our former research, we discussed the connection between epigenetic modifications and TME. For instance, distinct features of the TME tend to alter the activity of enzymes engaged in RNA methylation while each form of methylation contributes to the formation of the different TME states, including hypoxia, metabolic dysregulation, chronic inflammation, and immune escape [6]. Notably, recent studies have found that many RNA methylation-related enzymes could be SUMOylated, leading to the promotion or impression of tumor progression. The N6-Methyladenosine (m6A) methyltransferase activity of methyltransferase 3 (METTL3) is inhibited due to its SUMOylation by SUMO1 at residue K211, K177, K212, and K215, resulting in the decrease in m6A levels and increase in mRNA degradation that promotes tumorigenesis [7]. Similarly, it has been confirmed that a “reader protein” of m6A methylation named YTHDF2 (YTH N6-methyladenosine RNA binding protein 2) was SUMOylated at residue K571 according to microenvironmental hypoxia. SUMOylation of YTHDF2 increases the binding affinity to m6A sites and promotes the degradation of several tumor-related mRNAs [8]. Not only the enzymes of m6A, but also 5-methylcytosine (m5C) methylation such as NOP2/Sun RNA methyltransferase 2 (NSUN2) can be SUMOylated. The carcinogenic activity of NSUN2 is promoted by SUMOylation, mediating its stability and nuclear transport [9].
It is well known that the TME mediates interactions among proteins due to subtle changes such as oxygen content, PH, and ATP, which can potentially expose peptide motifs that are the main factor affecting interactions between SUMOylation ligase and targeted proteins, including transcription factors (TFs) and oncoproteins. To date, studies on SUMOylation have concentrated on carrying out proteomics and identifying targeted proteins with specific lysine (K) residues and motifs, but not the SUMOylation stoichiometry in the TME, which differed from each type of tumor [10]. Here, we display the crucial role of SUMOylation in various cancers and deliberate on the SUMOylation activity in distinct TME surrounding the enzyme-protein substrate and the potential ability of SUMOylation to affect the TME.
Chemical basis of SUMOylation
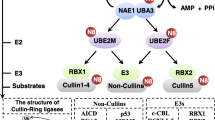
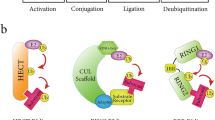
The SUMOylation process is similar to ubiquitination: it includes maturation, activation, conjugation, and deconjugation steps. (Fig. 1). In mammals, SUMO proteins begin as inactive precursors, which are firstly processed by sentrin-specific proteases (SENPs). The C-terminal di-glycine (-GG) motif of SUMO is subsequently exposed by SENPs, thus activating SUMOs. Heterodimeric E1 enzyme (SAE1/2) forms a high-energy thioester bond between its cysteine site and the C-terminal of SUMO in an ATP-dependent manner. Secondly, activated SUMOs are transferred to a cysteine residue of SUMO conjugating enzyme (UBC9, a SUMO E2 ligase). Finally, with the help of UBC9 and SUMO E3 ligase, they are conjugated to a target lysine that is usually in a consensus sequence (ψ-K-X-D/E, ψ: large hydrophobic amino acid; K: lysine; X: any amino acid; D: aspartate; E: glutamate). SUMO E3 ligases are another class of proteins in the SUMOylation cascade, mainly including members of the protein inhibitors of activated signal transducer and activator of transcription (PIAS) family [11, 12]. Although the SUMO enzymes are fewer in number than their counterparts in the ubiquitination pathway, the amount of SUMO substrates are strikingly large, affecting cells activity and disease development. Furthermore, SUMO maturation and deconjugation are both carried out by SENPs. SENP family comprises six members in mammals (SENPs 1–3 and 5–7), and SENPs 1–3 and 5 are the major executors while SENP 6–7 remove the SUMO monomers from the polymeric SUMO chains. Apart from covalent SUMO modification, non-covalent interaction between SUMOs and target protein happens thanks to the SUMO-interacting motifs (SIMs), which contain a hydrophobic core binding with the surface region of SUMO [13].

The SUMO procedure diagram and its function in various tumors. SUMO proteins are firstly processed by SENP, and after the formation of a high-energy thioester bond between the C-terminal SUMO and active site cysteine of SAE1/2, activated SUMO is then transferred to a cysteine residue in the active site of Ubc9. It is finally transferred to a target lysine with the help of Ubc9 and SUMO E3 ligase. The expression and functions in different tumors of SUMO E1, E2, E3 ligases and SENPs are also depicted in this figure
Multiple functions of SUMOylation in cancers
Neural system tumors
The SUMOylation cascade modifies targeted proteins reversibly and dynamically, and participants of SUMO circulation play distinct roles in various tumors (Fig. 1; Table 1). In glioblastoma (GBM), proteins involved in the SUMOylation cascade are upregulated, such as E1 (SAE1), E2 (UBC9) components, and SENP1, promoting tumor progression [14, 15]. Researchers found that CRMP2 SUMOylation induced by UBC9 could significantly promote GBM cell proliferation [16]. SUMOylation of promyelocytic leukemia (PML) protein promoted by prolyl-isomerase Pin1 facilitates c-Myc proteins stability, promoting glioma stem cells (GSCs) maintenance and GBM malignancy [17]. SUMO1 conjugation of FUS boosted by lncRNA RMST enhances the interaction between FUS and heterogeneous nuclear ribonucleoprotein D (hnRNPD), maintaining their expression and inhibiting tumorigenesis [18].
Respiratory tumors
The binding affinity of YTHDF2 with m6A motif can be significantly increased by SUMOylation while SUMO1-modulated METTL3 SUMOylation significantly suppresses its m6A methyltransferase activity, resulting in lung cancer progression [7, 19]. TRIM28, an uncommon protein in SUMOylation cascade, is overexpressed in lung adenocarcinoma with low immune and stromal scores, regulating the TME by SUMOlating the IRF family proteins [20]. UBC9/PIASy-mediated SUMOylation decreases sirtuin 1 (SIRT1) and increases transcriptional repression activity of SLUG, predicting more invasive types of lung cancers [21, 22]. PIAS1-induced SUMOylation of PML facilitates its degradation and thus promotes NSCLC progression, while vascular endothelial growth factor receptor 2 (VEGFR2) SUMOylation inhibits angiogenesis signaling pathway in NSCLC [23, 24]. Additionally, SAE2 was found highly expressed in small cell lung cancer (SCLC), promoting migration and invasion, and decreasing sensitivity to chemotherapy [25].
Mammary system tumors
Researchers showed that UBC9 was upregulated through Forkhead Box Protein P3 (FOXP3), a tumor-suppressing TF, which could act as a novel activator of SUMOylation in Breast Cancer (BRCA) [26]. When compared with nonmetastatic cells, metastatic breast cancer cells exhibit an upregulated SUMO2/3 modification profile, especially on MYC. SAE1/2 enzymatic activity losing facilitates MYC synthetic lethality while SENP1-driven deSUMOylation decreases its ubiquitination-mediated degradation [27, 28, 29]. SENP3-deficiency-mediated AKT1 SUMOylation leads to AKT1 hyper-phosphorylation and activation, promoting macrophages’ M2 polarization [30]. M2-type macrophages interact with tumor cells by releasing EGF, MMPs, VEGF, and TGFβ, thus promoting tumor proliferation, invasion, angiogenesis, and immune escape. In triple-negative breast cancer (TNBC), overexpressed SENP1 promotes CSN5-mediated ZEB1 protein degradation via deSUMOylation of GATA1, and ultimately enhances metastasis and invasion of tumor cells [31]. The catalytic component of the human telomerase enzyme (HTERT) was reported to be SUMOylated by SUMO1 and CBX4, and PIAS1 and TIF1γ cooperated to facilitate the SUMOylation of SnoN, both triggering the EMT program [32, 33].
Digestive system tumors
SUMO1 conjugation to SMAD4 and murine double minute 2 homolog (MDM2) increase their expression and are involved in oral squamous cell carcinoma (OSCC) aggressiveness [34, 35]. Through its PDZ domain, Regulator of G protein signaling 12 (RGS12) upregulates SUMOylation of phosphatase and tension homolog (PTEN). Moreover, specificity protein 1 (SP1) SUMOylation can activate PTEN transcription to block the AKT/mTOR signaling pathway, increasing radio sensitization of OSCC cells [36, 37]. SENP3 and SENP5 were found overexpressed and related to differentiation of OSCC [38, 39]. A germline variant of minichromosome maintenance proteins (MCMs) can increase its SUMOylation levels, facilitating Esophageal squamous cell carcinoma (ESCC) proliferation and metastasis [40]. Modified by SUMO2/3, heat shock protein 27 (HSP27) is upregulated, which increases pyruvate kinase isoenzyme M2 (PKM2) and decreases E-cadherin, enhancing the malignant extent of ESCC cells [41].
SUMOylation was found to create a positive feedback loop in gastric cancer (GC). Wang et al. found that as a result of P38α-SUMOylation, reactive oxygen species (ROS) accumulated, which facilitated p38α-SUMOylation by improving the PIASxα protein stability, creating a favorable environment for survival and metastasis [42]. PIAS1 and PIAS4 show a higher level among patients with a lower risk of mortality after second-line docetaxel-based chemotherapy [43]. SUMO-2/3 modifies NSUN2 and modulates its stability and nuclear transport, promoting its carcinogenic activity [44]. Additionally, SENP2 was suggested to stabilize N-myc downstream-regulated gene 2 (NDRG2) which functioned as a tumor suppressor gene [45]. UBC9/SUMO1-mediated insulin-like growth factor 1 receptor (IGF-1R) SUMOylation increases transcription activity of its substrate proteins, including snail family zinc finger 2 (SNAI2) [46]. SAE2 was also substantiated to play a crucial role in the aggressiveness of GC and predict a poor survival outcome [47].
SENP1 is overexpressed in colon cancer (COAD) and silencing of SENP1 inhibits cell proliferation through upregulating CDK inhibitors including p21 and p27 [48]. Besides, 46 cases of colon cancer were investigated, showing that patients who exhibited high expression of SUMO1 and SUMOylated P53 were more likely to experience metastasis [49]. Contrary to many other cancers, PIAS1, which was found associated with IRF-1 nuclear translocation and apoptosis, was downregulated in colon cancers [50].
In hepatocellular carcinoma (HCC) cells, SUMOylation reprograms the TME to affect cancer-TME crosstalk. Tumor-infiltrating macrophages, one of the most abundant stromal cell types in the HCC TME, inhibit anti-tumor immunity by inducing matrix remodeling, angiogenesis, and tumor metastasis. UBC9/SUMO1-mediated PKM2 SUMOylation induces its exosomal excretion, which triggers monocyte-to-macrophage differentiation to substantially increase the abundance of macrophages to help tumor cell invasion and metastasis [51]. RANBP2, a SUMOylation E3 ligase, mediates Fat mass- and obesity-associated gene (FTO) SUMOylation and subsequent degradation, inducing Guanine nucleotide-binding protein G (o) subunit alpha (GNAO1) instability, an m6A substrate of FTO and a tumor suppressor in HCC [52]. P53-stabilizing and activating RNA (PSTAR), a newly found lncRNA, enhances SUMO1-dependent SUMOylation of heterogeneous nuclear ribonucleoprotein K (hnRNP K) and disrupts its deSUMOylation through SENP2, facilitating transactivation of p53 [53]. SUMO1 also promotes Mesencephalic astrocyte-derived neurotrophic factor (MANF) nuclear translocation and enhances its interaction with p65, inhibiting the nuclear factor kappa B (NF-κB)/Snail signal pathway in EMT [54]. UBC9/SUMO1-mediated METTL3 SUMOylation regulated Snail expression and HCC metastasis in an m6A-dependent manner [55]. Nuclear factor erythroid-2 related factor 2 (NRF2) was found to be SUMOylated by SUMO1 at K110, promoting de novo serine synthesis and HCC tumorigenesis [56]. It was suggested that CBX4 could enhance VEGF expression in HCC cells and promote angiogenesis through enhancing hypoxia-inducible factors (HIF-1α) SUMOylation and its transcriptional activity [57]. Intriguingly, SENP1 augments the transcriptional activity of HIF-1α under hypoxic depending on deSUMOylation function. Meanwhile, a positive feedback loop was demonstrated between HIF-1α and SENP1, resulting in stemness and tumorigenesis of HCC [58].
Researchers detected coexpression of MYC and SUMO-related factors (such as SAE1, UBC9, SUMO1, SUMO2/3) in Pancreatic ductal adenocarcinoma (PDAC), and hyperactivation of MYC was associated with increased sensitivity to pharmacological SUMO inhibition, which provided a new therapeutic strategy to PDAC [59]. PIAS4 contributes to von Hippel-Lindau (VHL) SUMOylation and impairs its function, upregulating HIF1α and its targets including VEGF and STAT3 [60]. UBC9/SUMO2-regulated SUMOylation of DNA double-strand breaks (DSB) repair proteins (e.g. RNF40) sustains their stability, maintaining the DNA damage response (DDR) [61].
Urinary system tumors
SENP1 is upregulated in urinary content and is regarded as a predictor of the recurrence of bladder cancer (BLCA) [62]. While SENP2 is downregulated in BLCA. Mechanistically, SENP2 inhibits nuclear translocation of β-catenin through deSUMOylation of TBL1/TBLR1, inhibiting the MMP13 activation and BC cell metastasis [63]. UBC9 overexpression induced by lncRNA ELNAT1 catalyzes SUMOylation of hnRNPA1, promoting ELNAT1 packaged into EVs and activating SOX18 transcription to induce lymphangiogenesis [64].
In renal cell cancer (RCC), hypoxia-associated factor (HAF)-mediated HIF-2 transactivation requires SUMOylation of HAF by SUMO1 in an oxygen-sensing manner, contributing to the maximal induction of HIF-2 target genes and tumor progression [65]. RWD domain-containing protein SUMO Enhancer (RSUME) was found to SUMOylated VHL to alleviate HIF degradation, favoring RCC proliferation. Additionally, the capability of binding between HIF and VHL of RSUME was found dependent on VHL SUMOylation, specifically SUMO2 [66].
In prostate cancer (PC), UBC9 mediates SUMOylation of signal transducer and activator of transcription 4 (STAT4) on K350 which activates the immunosuppressive phenotype of tumor-associated macrophages (TAMs), while macrophage-specific UBC9 ablation can facilitate STAT4-induced interplay among TAM-CD8(+) T cells-cancer cells to curb tumor progression [67].
Female reproductive system tumors
In cervical cancer (CCA), Human Papilloma Virus (HPV) 16E6 targets hADA3 for SUMOylation-mediated degradation [68, 69]. Interaction between Forkhead box M1b (FOXM1b) and UBC9/PIAS1 can be impaired by HPV16 E7, decreasing SUMOylation of FOXM1b and sustaining its stability [70].
Dong et al. have demonstrated that UBC9 overexpression could significantly increase the proliferation of Epithelial Ovarian Cancer (EOC) through PI3K/AKT signaling pathway [71]. SENP1 was shown to upregulate HIF-1α expression through deSUMOylation, increasing cisplatin resistance in EOC cells [72]. Furthermore, PIAS4 was confirmed to induce SUMOylation of SP1, a transcriptional activator, and prevent it from binding to the SIRT1 promoter, downregulating it and impeding EMT [72].
SUMOylation takes part in the hypoxic response pathway
A hypoxic TME is a pivotal hallmark of most solid tumors. Under hypoxia, dramatic reprogramming of biological processes happens, including anaerobic energy production, lipid metabolism modulation, oxygen delivery increase, etc. Those dysregulation gene expressions in distinct events also contribute to tumor cell ethology alteration through the activation of HIFs [73, 74]. Fine-tuning of the SUMO conjugation machinery extensively occurs under various stress conditions, especially hypoxia, regarded as a homeostatic mechanism evolving in multicellular organisms to sustain cellular and tissue functions [75, 76, 77]. SUMO pathway in cancer cells is also dysregulated under the catalysis of hypoxia, facilitating proliferation, invasion, metastasis, and even resistance to chemotherapy as mentioned above.
HIF-mediated target gene activation was depicted in researches. Under normoxia, HIF is modified by pVHL, an E3 ubiquitin ligase, through hydroxylation activity of the prolyl hydroxylase domain containing proteins (PHDs) and factor inhibiting HIF (FIH), leading to subsequent proteasomal degradation. In the hypoxic TME, the decreased hydroxylation facilitates HIFα nuclear transport, dimerization with HIFβ, and recruitment of CBP/p300 coactivators. As a result, hypoxia-targeted genes are stimulated via HIF binding to hypoxia responsive elements (HRE) [78]. Effect of PTM during cellular response to hypoxia has been extensively studied and SUMO hemostasis was indispensable to plenary activation of hypoxia signaling. A significant increase in SUMO1 mRNAs and proteins was observed, and interaction between SUMO1 and HIF1α was also demonstrated under hypoxic conditions such as pulmonary hypertension [79, 80].
However, a SILAC-quantitative proteomic-based study suggested that it was not SUMO1, or SUMO2/3 increase but a massive augment in SUMOylation status that altered under hypoxia. Identically, based on comparative mass spectrometry, it was illustrated that oxygen concentration altered the activity of enzymes in the SUMO pathway, especially the SENP family (SENP1 and SENP3). Intriguingly, a few SUMO enzymes also act as hypoxia-induced SUMO1 targets that change under the TME, including RanBP2 and PIAS2 [81, 82].
Researchers have found that HIF, the most significant factor in the hypoxic response pathway, was widely modified by SUMOylation, influencing its stability and transcriptional activity (Fig. 2). HIF-1α is upregulated through SUMO1 modification at K391/K477, and overexpression of PIAS3 can maintain its stability and transcription activity [83, 84, 85]. In agreement, Li et al. discovered that E3 ligase activity of CBX4 SUMOylated HIF-1α, promoting its transcriptional activity and expression of VEGF, which facilitated tumor malignancy via angiogenesis [57, 86]. While in other studies, SUMO E3 ligase (RanBP2 and PIAS4) were found negatively regulate hypoxia-mediated HIF-1α stability and transactivation [87, 88]. In some conditions, SENP1-mediated deSUMOylation also increases HIF-1α transcriptional activity and facilitates HIF1α-dependent genes expressions such as VEGF and glucose transporter 1 (GLUT1) [89]. The SENP1/HIF-α positive feedback loop illustrated by Cui et al. mentioned above also support this scenario [58]. The reason for the controversial scenario might be the fact that several factors among the hypoxia pathway were regulated via the alteration of SUMO enzymes, resulting in distinct HIF-1α activity. Additionally, HIF-2α was found modified by SUMOylation at K394, leading to VHL and RNF4 (SUMO-targeted ubiquitin ligases)-mediated proteasomal degradation [90]. Tojo et al. elucidated that the aryl hydrocarbon receptor nuclear transporter (ARNT), also named HIF-1β, was modified by SUMO1 at K245, disturbing interaction with PML and augmenting the transcriptional activity of ARNT [91].

Crosstalk between SUMOylation related enzymes and hypoxia signaling pathway. SUMOylation related enzymes regulate HIF-1a stability and transcriptional activity through directly mediating its SUMOylation and indirectly influencing other participants involved in hypoxia signaling pathway, causing expression level alteration of critical genes that modulating cancer cells biological processes such as metastasis, angiogenesis, and glycolysis. Meanwhile, hypoxia can also affect expression of some actors via monitoring their SUMOylation state
Meanwhile, several participants apart from HIF in hypoxia signaling were suggested to be regulated by SUMOylation (Fig. 2). HIF1α E3 ligase HAF degrades HIF-1α in a SUMOylation manner, while hypoxia-induced SUMOylation of HAF enables its HIF-2α combination to enhance transcriptional activity [65]. Induced by hypoxia, RSUME enhances overall SUMO1-3 conjugation, thus promoting the stabilization of HIF-1. In addition, RSUME SUMOylates and physically interacts with pVHL, thus suppressing the complex aggregation of pVHL, Elongins and Cullins (ECV), subsequently alleviating ubiquitination-induced degradation of HIF-1/2α [92, 93]. PIAS4-induced VHL SUMOylation by SUMO1 on K171 facilitates VHL oligomerization and abrogates its inhibitory function on HIF-1α activity. Therefore, target genes of HIF-1α such as JMJD1A, VEGF, and STAT3 are activated, and tumor progression is promoted [60, 94] .PHD3 SUMOylation by SUMO2/3 was also found to repress HIF-1-dependent transcriptional activity [95]. Furthermore, p300 and CREB binding protein (CBP) act as homologous transcriptional coactivators. K1017 and K1029 of p300 are modified by SUMO1, leading to the recruitment of histone deacetylase 6 (HDAC6), and SUMOylation deficiency via SENP3 increases p300-mediated transcriptional activity [96, 97]. Hypoxia/SIRT1-mediated UBC9-K65 acetylation reduction promotes CBP SUMOylation as well as hypoxia signaling cascade [98]. On the contrary, Kuo et al. exhibited that CBP could be SUMOylated by SUMO1 at K999, K1034, and K1057, negatively regulating its translational activity through interacting with death domain-associated protein (Daxx), which recruited HDAC2 and played a role of transcriptional corepressor [99].
Moreover, the invasive ability of cancer cells is likely to be regulated by SUMOylation. Hypoxia increases Slug SUMOylation by the way of disrupting its crosstalk with SENP1 and SENP2, promoting lung cancer metastasis. Also, Xie et al. showed that Slug SUMOylation could be stabilized by p14 (ARF), promoting EMT [22, 100]. SUMOylation-dependent negative feedback between HIF-1α and CLDN6 has been elucidated by researchers. CLDN6, transcriptionally upregulated by HIF-1α, was suggested to combine β-catenin, leading to β-catenin degradation and preventing its nuclear translocation. β-catenin acts as a TF of SENP1, and degradation of it downregulates SENP1, causing HIF-1α SUMOylation and degradation [101].
SUMOylation influences metabolism signaling pathways
Metabolic stress, in addition to other stress, results in changes in endogenous synthesis and exogenous uptake of nutrients, which are fuels for various biological procedures, including protein modification. Crosstalk between SUMOylation and metabolic dysregulation can partly explain how malignant cells thrive in the TME. SUMOylation has been extensively studied in metabolic diseases, especially diabetes. SENP1 was suggested to amplify insulin exocytosis, NADPH generation, and subsequent glutathione (GSH) reduction, rescuing β cell function in type 2 diabetes [102]. In support, SENP1 can ameliorate type 1 diabetes. Mechanistically, SENP1 facilitates deSUMOylation of NEMO, in adipocytes, to alleviate NF-κB activity and subsequent inflammation [103]. However, few studies have focused on SUMOylation in metabolic reprogramming, which was indispensable for TME formation.
According to the Warburg effect, despite oxygen-rich conditions, tumor cells tend to uptake glucose more rapidly and convert it to lactate via glycolysis because aerobic glycolysis helps malignant cells to survive. Features of the TME, such as hypoxia, hypoglycemic, and acidic microenvironment are closely related to functional proteins, whose SUMO modifications are nonnegligible [104]. SUMO modification alters tumor cell metabolism either directly via metabolic enzymes or indirectly via TFs and crucial signaling pathways (Fig. 3).

The role of SUMOylation in cancer metabolism pathways. This diagram focuses on SUMO modified metabolic enzymes, closely associated signaling pathways and TFs. Noteworthy, glucose metabolism is the most important metabolic pathway regulated by SUMOylation, contributing to metabolic reprogramming such as “Warburg effect”
SUMOylation occurs on metabolic enzymes and dramatically alters the direction of metabolic flux (Fig. 3). Under hypoxic stress, SUMOylation plays a role in facilitating glycolytic pathway reprogram in that SUMO1 overexpression shows a correlation with clustering of glycolytic enzymes. In turn, the high glucose microenvironment demonstrates not only augmented SUMO pathway factors, including SUMO1-4 and SUMO ligase E3 (CBX4 and PIAS4) levels but also enhanced colocalization of HIF-1α and SUMO [19, 83]. Mo et al. suggested that SUMO2 modification promoted GLUT1 degradation, ultimately inhibiting glycolysis [105]. SUMO-defective hexokinase 2 (HK2) enters mitochondria to phosphorylate glucose to form glucose-6-phosphate, enhancing both glucose consumption and lactate production, while SUMOylated HK2 impedes its activity [106]. Long-chain saturated fatty acids starting from acetyl-CoA and malonyl-CoA are catalyzed by fatty acid synthetase (FASN), and its SUMOylation inhibits its proteasomal degradation, altering lipid metabolism and eliciting oncogene activity [107]. HSP27 SUMOylation by SUMO2/3 enhances PKM2 expression, promoting glycolysis and tumor progression [41]. In addition, K110 SUMOylation of NRF2 increases de novo serine synthesis by enhancing ROS clearance and phosphoglycerate dehydrogenase (PHGDH) upregulation. Accordingly, serine deficiency can in turn promote NRF2 SUMOylation. Moreover, NRF2 promotes glycolysis through activating PFK and MYC [56, 108].
Metabolism-associated TFs are widely SUMOylated, including sterol regulatory element-binding proteins (SREBP), peroxisome proliferator-activated receptor alpha (PPARα), and HIF-1 (Fig. 3). PTM of SREBP regulates its expression and related de novo lipogenesis procedure, a significant metabolic process in tumor cells. SUMOylation of SREBP suppresses its transcriptional activity and promotes HDAC3 recruitment, inhibiting lipid uptake and synthesis [109]. Lee et al. found that PKA-mediated SREBP1 phosphorylation facilitated PIAS4-induced SREBP1 SUMOylation, leading to degradation of SREBP1 via ubiquitination [110]. Nuclear receptor superfamily (FXR, LXR, LRH-1, and PPAR) undergo SUMOylation as well [109, 111, 112, 113]. LRH-1, one of those nuclear receptors, is SUMOylated at K289, decreasing oxysterol binding protein-like 3 (OSBPL3) expression and subsequent SREBP-1 processing [114, 115]. Another SUMOylation-mediated TF, named PPARα, is meanwhile a metabolic mediator. SENP2-induced PPARα deSUMOylation displays upregulated degradation, alleviating fatty acid oxidation and target genes transcription [116, 117]. It is well-known that HIF-1 is a crucial modulator of metabolic reprogramming and simultaneously a downstream molecule of SUMOylation. Specifically, it increases the transcription of GLUT1, lactate dehydrogenase 1 (LDH1), and pyruvate dehydrogenase kinase 1 (PDK1) (Fig. 2), turning pyruvate to acetyl-CoA and lactate for the mitochondrial tricarboxylic acid (TCA) cycle [118, 119, 120, 121].
In addition to acting on metabolic enzymes and metabolism-related TFs, various classical signaling pathways functioning in cancer metabolism undergo SUMOylation (Fig. 3). Growth factors or insulin stimulation of phosphoinositide 3-kinase (PI3K) induces glycolytic enzyme aldolase A (ALDOA) release, potentiating AKT-independent glycolytic flux. Activation of Serine-threonine kinase AKT is sufficient to stimulate tumor microenvironmental aerobic glycolysis and thus exerts its promoting effect on tumor growth and metabolism of individual cells. PI3K/AKT manages crucial phases in glycolysis through the activation of specific enzymes that mattered in cellular metabolic reprogramming, such as rapamycin (mTOR) complex 1 (mTORC1) and forkhead box O (FOXO) family [122, 123, 124, 125] p110β, which is a catalytic subunit of PI3K, can be modified by SUMO1 and SUMO2, increasing its activation of glucose metabolism in an AKT-dependent or independent manner [126]. Ong et al. displayed that SUMO E1 enzyme SAE1 was related to dysregulated cancer metabolism and became a potential target in HCC therapy. Additionally, SAE1 companied by E2 UBC9 and E3 ligase PIAS4 mediates AKT SUMOylation majorly at K276, regulating AKT activation and promoting tumor progression, which can be reversed by SENP1 [127, 128, 129]. SUMOylated AKT then promotes a series of metabolic cascade through whatever rapid or lagging courses. AKT can either directly phosphorylate ACLY or indirectly initiate ACLY via the SREBP family of TFs activation, facilitating de novo lipid synthesis. Likewise, SREBP also promotes fatty acid and sterol synthesis through many other related enzymes, such as FASN and Acetyl-coenzyme A synthetase (ACSS2) [130, 131]. AKT is activated at T308 through phosphoinositide-dependent protein kinase 1 (PDPK1) and S473 by mTORC2, leading to increased glucose uptake and glycolysis [122]. PDPK1 is also SUMOylated, and nonSUMOylated PDPK1 tethers LC3 to the endoplasmic reticulum to trigger macroautophagy/autophagy, which acts as a crucial pathway for cell metabolism [132]. mTORC1 activation was suggested to stimulate glycolysis and de novo lipid biosynthesis by influencing the transcription activity of HIF1-α and SREBP1/2 [133]. A sensor of energy status: 5’-AMP-activated protein kinase (AMPK), which comprises a catalytic α, a scaffolding β and a nucleotide binding regulator γ. Noteworthy, starvation-induced activation of AMPK results in the upregulation of catabolic metabolism and nutrient uptake, becoming a bridge that links metabolism and the SUMO pathway [134]. Dou et al. illustrated AMPKα as another pivotal substrate of SENP2, negatively regulating gluconeogenesis. SENP2-induced AMPKα deSUMOylation triggers its ubiquitination, subsequently promoting gluconeogenesis and blood glucose [135]. mTORC1 can be activated by AKT while inhibited via AMPKα, and PIAS4-induced AMPKα1 SUMOylation suppresses AMPK activation towards mTORC1 signaling [135]. Conversely, modified by PIAS4 and SUMO2, AMPKβ2 activates the α2β2γ1 AMPK complex, which might be an antagonistic mechanism relating to the ubiquitination of AMPKβ2 [136]. The significant oncogene MYC is widely known as a TF as well as an increased downstream of PI3K-AKT signaling [137]. SUMOylation and deSUMOylation mediate MYC protein stabilization and activation through the activity of SUMO-related enzymes, including PIAS1, RNF4, SENP1, and SENP7 [29, 138, 139]. However, research illustrated that SUMOylation of MYC also mediated its oncogene activity and inhibition of SUMOylation might be a possible therapy for MYC-elicited cancers [28]. Furthermore, hypoxia-mediated SENP1 inactivation alleviates basic helix-loop-helix family member e40 (BHLHE40) deSUMOylation, leading to transcriptional repression of PGC-1α, a crucial metabolic regulator, contributing to metabolic strategies inhibition, such as mitochondrial biogenesis and oxidative metabolism [82]. PTEN, a pivotal tumor suppressor factor functioning to dephosphorylate PIP3 to regenerate PIP2 and resulting in subsequent PI3K-AKT signaling inhibition, exhibits SUMO modification at K266. SUMOylated PTEN shows a low ubiquitination level, and in turn, proteasome inhibition results in PTEN SUMOylation accumulation [140, 141]. PTEN illustrates indirect regulatory activity for glucose metabolism via PI3K/AKT pathway as well as the direct mediatory potential for glycolysis through phosphoglycerate kinase 1 (PGK1), which catalyzes one of the two ATP producing reactions in the glycolytic pathway [142]. Apart from glucose metabolism, PTEN can also repress intracellular lipid accumulation by coordinating with Maf1, a TF that restrains RNA synthesis and lipid biosynthesis [142].
Recent studies have found that noncoding RNAs participated in the SUMO pathway and were associated with metabolism in a new way. Mo et al. showed that circRNF13 bound to the 3’- Untranslated Region (3’-UTR) of the SUMO2 mRNA and stabilized it, facilitating GLUT1 SUMOylation and subsequent degradation, ultimately inhibiting glycolysis and nasopharyngeal carcinoma (NPC) progression [105]. SUMO pathway mediates the tumor metabolism and the TME in many ways while the research on the regulation of SUMOylation by metabolism changes is very limited, more studies are warranted to clarify how it acts on SUMO processes.
SUMOylation involves in the inflammatory TME
There are many components of the TME, including the extracellular matrix and various structures, like vascularization and inflammatory infiltrates. Crosstalk between malignant cells and their surrounding microenvironment helps tumor cells to survive. Inflammatory or immune-related cytokines and cells play an indispensable role in regulating this interaction [143]. Critical peptide motifs functioning as PTM sites are exposed when subtle changes in the TME are triggered by infection-inflammation, such as ATP and PH [3]. Karhaosen et al. described that global SUMOylation (SUMO2/3 isoforms) increase could partly silence inflammation during metabolic stress and reinstate tissue integrity. For instance, ROR-γt SUMOylation suppresses transcription of IL-17 A, which amplifies inflammation and recruits neutrophils and monocytes via stimulating inflammatory cytokines production [144, 145].
SUMOylation participates in tumorigenesis via inflammatory pathways as well (Table 2). Recognized as an indispensable mediating factor in inflammatory pathways during carcinogenesis, NF-κB also acts as a TF, which is a nonnegligible target of SUMOylation. Simultaneous SUMOylation of Mesencephalic astrocyte-derived neurotrophic factor (MANF) and p65 by SUMO1 facilitate MANF nuclear translocation and their interaction, disrupting the NF-κB/Snail pathway and subsequent EMT in HCC [54]. TAK1 SUMOylation at K329 and K562 induced by TRIM60 suppresses MAPK/NF-κB activation via disturbing TRAF6/Table 2/TAK1 complex formation and the innate immune response [146]. In addition, IkappaBalpha (IκB) is modified by SUMO1 on K21, which acts as the site of ubiquitination as well. Therefore, SUMOylated IκB fails to enter signal-induced degradation, interfering with NF-κB signaling activation [147] . Comerford et al. also verified an analogous mechanism. Under normoxia, cAMP-response element-binding protein (CREB) and IκB are ubiquitinated, resulting in the induction of proinflammatory phenotype. While under hypoxia, modified by SUMO1, CREB, and IκB are stabilized, and inflammatory phenotype is subsequently inhibited [148]. However, the diametrical effect of NF-κB-related cascade reaction happens in that SUMO2/3-modifed IκBα is more susceptible to the proteasome [149]. Intriguingly, a regulatory loop emerges in NF-κB activity and its SUMOylation. That is PIAS3-mediated ReIA SUMOylation, a subunit of NF-κB, contributes to NF-κB inhibition, while its SUMOylation can be facilitated by NF-κB activation [150]. Furthermore, pro-apoptotic and anti-proliferative activities of interferon beta (IFNβ) were reported in BRCA. Decque et al. elucidated that SUMOylation silenced IFNβ and interferon alpha receptor 1 (IFNAR1) and thus restricted NF-κB associated cytokines production and Toll-like receptors (TLRs)-induced production of inflammatory cytokines. Therefore, SUMOylation of IFNβ tends to promote cancer cells’ malignant properties in diverse ways [151, 152, 153].
UBC9 plays a significant role in the inflammatory infiltration of TME because of its uniqueness in the SUMO pathway. Given that RSUME increases IκB contents and stabilizes HIF-1α during cellular stress like heat shock and hypoxia, leading to inhibition of NF-κB. Researchers have found that this mechanism was implemented via RSUME-enhanced Ubc9 thioester formation, favoring noncovalent binding of SUMO1 to UBC9 and SUMO polymerization [92, 154]. In bladder cancer, Xia et al. determined different SUMOylation patterns and gene clusters that shape the TME and clinicopathological features [155]. UBC9 was found overexpressed in bladder cancer, and its downregulation contributed to obvious inflammatory gene activation. A prominent marker for cancer stem cells (CD44) is mediated by interleukin-6 (IL-6), which is a hub gene in UBC9 regulatory network. Therefore, UBC9 is to eliminate inflammatory infiltration, which might be a threat to tumorigenesis [155]. Likewise, compromised UBC9 function displays decreased AKT1 SUMOylation and increased proinflammatory cytokines including RelA, cFos, and cJun [156].
In addition to the E1, E2, E3 enzymes, and the SUMO molecular, SENP family, the deSUMOylation executors, mediate inflammatory processes as well. Yang et al. found that intermittent hypoxia (IH) could not only stimulate tumor necrosis factor-α (TNF-α) and IL-6 but also NEMO SUMOylation. While SENP1 attenuates NF-κB activation via disturbing NEMO SUMOylation [103, 157]. SENP2 also efficiently deSUMOylates NEMO, inhibiting DNA-damage-induced NF-κB activation and subsequently suppressing SENP1/2 transcription in response to genotoxic stimuli [158]. K277 of NEMO/Ikkγ is SUMOylated by SUMO2/3, which hinders the binding of CYLD/NEMO and therefore enhances the inhibitor of κB kinase (Ikk) activation. While deSUMOylation of NEMO by SENP6 can inhibit Ikk activation and subsequent IκB degradation, impairing NF-κB activation and proinflammatory genes expression [153]. Besides, CD45 SUMOylation via SENP1-deficiency facilitates STAT3 dephosphorylation, suppressing tumor development by myeloid-derived suppressor cells (MDSC) infiltration [159]. In the TME, various stimuli including chemotherapy or radiotherapy can trigger the release of pro-inflammatory mediators, turning “cold” tumors “hot”. Through SUMO modifications, tumor cells change the function of various pathways such as the NF-κB signaling pathway, which provides a chance for cancer treatment to target SUMOylation.
SUMOylation participates in immune cells maturation and activation
Most tumor cells can be recognized by host CD8(+) T cells, and cancers that grow progressively must have escaped the antitumor attack. Recent research has illustrated two categories of tumor immune features in the TME, including innate immune activation which comprised immune cell infiltration and chemokine/IFN profile, and immune cell deficient phenotype. Tumor cells resist immune attack through the dominant immune-suppressive pathways and immune ignorance respectively [160]. Activation and inhibition of immune cells in the TME are critical for tumor cells to achieve immune escape, and molecules that regulate immune cells’ function usually undergo SUMOylation (Table 3).
Macrophage plasticity leads to either antitumor or protumor function in different conditions. SUMOylation of PKM2 induces monocyte-to-macrophage differentiation and TME remodeling by enhancing its exosomal excretion. Meanwhile, chemokines secreted by macrophages activate the CCL1-CCR8 axis, which promotes the PKM2-ARRDC1 combination and PKM2 excretion [51]. FOXP3-expressing regulatory T (Treg) cells eliminate aberrant immune response including anti-tumor immune response. Removal of Treg cells can restore anti-tumor immune function while Treg cell depletion may concurrently elicit autoimmunity. As a result, it is crucial to care about the exogenous adjustment of Treg cells among tumors. TCR stimulation enhances UBC9-mediated IRF4 SUMOylation, which augments Treg cell proliferation and regulates the downstream of TCR signals [161]. BTB domain and CNC homolog 2 (BACH2) functions as a mediator in primary adaptive immune response and immune deficiency. Yu and his colleagues suggested that SENP3 not only inhibited the nuclear export of BACH2 via deSUMOylation, maintaining Treg cell stability but also regulated ROS-induced immune tolerance [162]. NFATc1, a TF-regulating antigen receptor-mediated gene expression, is SUMOylated by UBC9 and PIAS1. Researchers constructed an NFATc1 SUMOylation deletion transgenic mouse and suggested that it exhibited increased IL-2 secretion, which promoted Treg expansion and impaired IL-17 and IFN-γ expression through STAT5 and Blimp-1 induction [163]. Intriguingly, Liu and his colleagues found that PIAS1 inhibited Treg cell differentiation independent of its SUMO E3 ligase activity. Mechanistically, PIAS1 binds to the Foxp3 promoter and increases histone H3 methylation, alleviating protein accessibility [164].
Apart from adoptive T cell therapy (ACT) whose function is instantaneous, endogenous T cells are more potent because of their ability to secure long-term memory with a broad repertoire of antigen specificity. SUMOylation was suggested to affect the development, activation, and function of T cells and B cells, thereby regulating the TME. Wang et al. selectively deleted UBC9 in T cells and found IL-7 signaling loss with increased apoptosis and attenuated proliferation during initial positive selection, resulting in defective late-stage maturation [165]. Immune adaptor SH2 domain containing leukocyte phosphoprotein of 76 kDa (SLP-76) is a substrate for SUMOylation cascade at K266/K284, and TCR stimulation facilitates SLP-76-UBC9 association, increasing the NFAT mediated IL-2 transcription [166]. JunB is another indispensable transcriptional activator of IL-2 and IL-4, regulating T cell function, and its SUMOylation on K237 also favors resting and activated primary T cells [167]. RORγt was found to mediate TH17 cell differentiation which regulated thymocyte development and lymph node genesis. While RORγt SUMOylation at K31 by SUMO3 and PIAS4 enhances its transcriptional activity, sustaining TH17 differentiation and CD8(+) T cell immature single-positive cells (ISPs) activity [168]. K54 of phospholipase C-γ1 (PLC-γ1) exhibits PIASxβ/PIAS3/SUMO1-regulated modification upon TCR stimulation, promoting PLC-γ1-microclusters assembly which favors T cell activation [169]. Likewise, kinase PKC-θ is SUMOylated via PIASxβ upon TCR, and its deSUMOylation impairs the coaccumulation of PKC-θ and CD28, inhibiting the formation of a mature immune synapse and T cell activation [170]. Moreover, SENP1-mediated SUMO2-STAT5 deSUMOylation facilitates its acetylation and downstream signaling, impelling the development of early T and B cells [171].
In plasma cells, SUMO1/UBC9-mediated Daxx SUMOylation was reported to not only impair the transcriptional potential of TFs but also facilitated type I IFN-regulated suppression of B cell development. Muromoto et al. constructed a SUMOylation-defective Daxx K630/631A mutant and found that the mutation transferred Daxx from the nucleus to cytoplasm and decreased the interaction with PML [172, 173]. Also, B lymphocyte-induced maturation protein-1 (Blimp-1) plays a crucial role during plasma cell differentiation that depends on PIAS1-induced Blimp-1 SUMOylation on K816, which increases the interaction with HDAC2 [174].
When present in the TME, SUMOylation influences tumor development via mediating immune cells’ activity, and this gives us a therapeutic hint that simultaneously targets SUMOylation and immune checkpoints. For example, irradiation (IR) + ATR inhibitors (ATRi) like berzosertib boosts the STING signaling and triggers strong innate immune activation by increasing SUMOylation at K127 of SHP1 [175]. Based on these, we suspect that whether SUMOylation could turn the “cold” TME “hot” to facilitate immune checkpoint inhibitor (ICI) therapy.
SUMOylation participates in the exosomes-dependent dialog in the TME
The exosomes, extracellular vesicles secreted from all cells, largely contribute to the communication between the TME and cancer cells as well. Jena et al. illustrated that not only tumor cells secreted exosomes containing various cytokines, chemokines, and miRNAs that affected stromal cells’ maturation and differentiation but also stromal cell-derived exosomes had an influence on tumor cells [176]. Intriguingly, SUMOylation is also involved in intracellular communication through exosomes. SUMOylation of hnRNPA1 in BLCA facilitates the recognition of the lncRNA ELNAT1 via the endosomal sorting complex required for transport (ESCRT). ELNAT1 in the exosome is then transmitted into human lymphatic endothelial cells (HLECs), enhancing lymphangiogenesis by transcriptionally promoting SRY-box transcription factor 18 (SOX18) [64]. Analogously, SAE1/SUMO2-mediated hnRNPA1 SUMOylation in PDAC is elevated via KRASG12D mutation-induced hyperactivation of SUMOylation, and SUMOylated hnRNPA1 tends to be packaged into exosomes and transmitted to lymphatic endothelial cells, stabilizing prospero homeodomain protein 1 (PROX1) and promoting lymph node (LN) metastasis [177]. Furthermore, SUMOylation of hnRNPA1 in small extracellular vesicles (sEVs) was verified to steer sEV-miRNAs loading, which was suggested to promote communication between malignant cells and the TME, and thus enhance proliferation and metastasis [178]. It is noteworthy that the exact mechanism by which SUMOylation regulates extracellular vesicles is limited to hnRNPA1. Since a large number of proteins can be modified by SUMO, more SUMOylation-mediated communication by exosomes in the TME remains to be discovered.
The future of SUMOylation
Orchestrating SUMOylation and the TME to resolve drug resistance
Nowadays, therapeutic resistance has become a thorny problem during cancer treatment. There is evidence that SUMOylation plays a pleiotropic role in the TME remodeling which dramatically affect drug resistance. In the hypoxic microenvironment of the EOC, SENP1 alleviates tumor cells’ sensitivity to chemotherapy via deSUMOylating HIF-1α and upregulating its expression [72]. The TME reprogramming in terms of metabolic reprogramming is also affected by SUMOylation, leading to chemotherapy resistance in PC. SENP1-mediated deSUMOylation of HK2 helps its mitochondria binding and consequently augments glucose consumption as well as lactate production [106]. In Irinotecan (CPT-11) resistant COAD cells, researchers also observed a dramatic increase of SUMO pathway members, accumulation of SENP1 and HIF-1α, and upregulation of glycolysis-relative protein markers, indicating the alterations of the metabolic microenvironment [179]. Demel and his colleagues found that SUMOylation activation inhibited MHC class I (MHC-I) antigen presentation, contributing to immune evasion from CD8(+) T cells and subsequent resistance to immunotherapies. Therefore, SUMOi is expected to restore antigen presentation machinery and augment the killing effect of CD8(+) T cells in the TME [180]. Moreover, a low SUMOylation level of SP1 contributes to SP1 and SNHG17 upregulation, which results in drug resistance through activating the Notch2 pathway in GC [181]. Although not mentioned by the authors, recent studies have shown the crucial role of the Notch2 pathway in TME remodeling in terms of regulating the anti-tumor infiltrate, which is likely to cause resistance [182]. Additionally, the SUMOylation-loss of PML triggers the abnormal NF-kB activation and is responsible for gemcitabine/oxaliplatin resistance in PDAC [183]. The evidence presented thus far supports that the SUMO pathway has the potential to modulate therapy resistance via TME remodeling, providing a window of opportunity for the application of the SUMOi to re-sensitize resistant individuals.
Applying SUMOi to clinical practice
The critical functions of SUMOylation in the TME provide opportunities for drug development and clinical trials. Recent research has displayed the potential of SUMOi for anticancer therapy. It has been confirmed that UBC9 or SAE1/2 depletion impaired cell survival through chromatin and non-chromatin-related manner, which has been identified through a proteomics mode as well [184, 185, 186]. As previously mentioned, SUMOylation cascade participants (E1, E2, E3-ligases, and SENPs) are upregulated in several tumors. Concurrently, they modify some proteins closely associated with the TME. These findings pave the way for the development of SUMOi that can be used to reverse the TME, thereby facilitating cancer treatment. Inhibitors targeting distinct parts of the SUMO circulation are shown in Fig. 4.

Functional sites of SUMOylation inhibitors. Ginkgolic acid, anacardic acid, and kerriamycin B blocks SAE1/2, while Davidiin and tannic acid impairs formation of the SAE1/2-SUMO intermediate. ML-792, TAK981, COH-000, and ML-93 inhibits SUMO E1 as well. Spectomycin B and GSK145, 2-D08, and SUBINS bind to UBC9, disturbing its interaction with SUMO. Triptolide, Momordin Ic and streptonigrin respectively inhibit SENP1 and disrupt SENP1-SUMO1 interaction. GN6958 and Ebselen suppress SENP1 and SENP2 respectively. There is no E3 inhibitor under research. Green inhibitors are natural compound while blue ones are synthetic product
-
a.
E1 inhibitors.
Natural compounds blocking SAE1/2 were first reported in 2009, including ginkgolic acid, anacardic acid, and kerriamycin B [187, 188]. From 2014 to 2015, two other compounds appeared, that is, Davidiin and tannic acid. SUMO conjugations to target proteins are blocked by these compounds since they impair the formation of the SAE1/2-SUMO intermediate [189, 190]. A phenol in Ginkgo biloba L. named Ginkgolic acid was known to have antibacterial and antitumor properties through its targeting of proinflammatory factors such as prostaglandins and leukotrienes, suppressing the pro-tumor inflammatory microenvironment formation [191]. Given the poor specificity and side effects of these natural products, synthetic inhibitors of the SUMO E1 were reported one by one from 2017 to 2021, including ML-792, its derivative TAK981, COH-000, and ML-93. Their high specificity avoids additional effects on other PTMs, such as ubiquitylation and neddylation. It was reported that COH-000 bound to Cys30 of SAE2 in an allosteric site, and ML-792, TAK-981, and ML-93 disrupted SAE1/2 activity through forming an adduct with SUMO [59, 192, 193, 194]. There was also evidence that hyperactivation of MYC sensitized PDAC cells to ML-93-mediated SUMO inhibition. Moreover, ML-93 impedes metabolic reprogramming in the TME because MYC-driven metabolic signals on which malignancies depended are susceptible to ML-93 [137]. Crowl et al. found that SUMO2/3 perturbed IFN induction, which prevented the spread of inflammation [195]. Interestingly, the anti-lymphoma activity of TAK-981 relies on IFNAR as well. Furthermore, TAK-981 can weaken the immune escape ability of tumor cells by enhancing the presentation of exogenous antigens released by dying cancer cells and facilitating subsequent cytotoxic T cell initiation [196]. Identically, Kumar et al. confirmed that TAK-981 increased the proportions of activated CD8(+) T cells and natural killer (NK) cells through activating STAT1 and IFN target genes, strengthening immune surveillance of tumor cells [197]. Noteworthy, three clinical trials (NCT03648372, NCT04065555, and NCT04074330) concentrated on TAK-981 in patients respectively with metastatic solid tumors or lymphomas, head and neck cancer, and non-Hodgkin lymphoma. These trials extensively probed TAK-981 about its safety, tolerability, pharmacokinetics, efficiency in combination with cetuximab or avelumab, or rituximab, and its biological influences within the TME.
-
b.
E2 inhibitors.
Natural product Spectomycin B and synthetic small molecular (GSK145A, 2-D08, and SUBINS) have emerged to inhibit SUMOylation through binding to and blocking UBC9-SUMO coalition [198, 199, 200, 201] 2′,3′,4′, -trihydroxyflavone (2-D08) that disrupts SUMO from transferring to substrates has been widely utilized in research. It was suggested that 2-D08 could induce apoptosis in AML cells and inhibit metastasis in PDAC via NOX2 and KRAS deSUMOylation respectively [202, 203]. Nevertheless, although various E3 ligases are liable to transfer SUMO to target proteins, there are no molecule inhibitors for E3 yet, which becomes a potential opportunity for exploration.
-
c.
SENPs inhibitors.
Upregulation of SENP1 and SUMOylation disorder are favorable for PC progression. Natural product small molecules such as triptolide and Momordin Ic can optimize PC prognosis through inhibiting SENP1 [204, 205]. Streptonigrin, another natural product, can also disrupt SENP1-SUMO1 interaction, and thus alleviates HIF1α expression [206]. The synthesized product GN6958 and Ebselen keep SUMOylation to a certain stoichiometric level by suppressing the activity of SENP1 and SENP2 respectively [207, 208]. As a result, SENPs inhibitors are also expected to achieve better efficacy in vitro, which would pave the way for further preclinical studies.
Challenges of future research on SUMOylation
Combined, studies concentrating on SUMOylation have unraveled the possible mechanism of its role in TME reprogramming and tumorigenesis, and discovered some potential inhibitors targeting SUMO circulation to hinder tumor development (Fig. 5). However, massive future work is required to reveal the functions of SUMOylation more comprehensively in the TME and expand its applications.

A summary of SUMOylation in the TME and therapeutic implications
Firstly, to resolve the lack of natural protease sites in the C-terminal tail of SUMO proteins, a method using α-lytic protease, WaLP, was reported [209]. This method can generate peptides containing SUMO-remnant diglycyllysine (KGG) and detect endogenous SUMO modification at a proteome-wide and site level. However, this method fails to distinguish SUMO1-4, and it can’t differentiate between SUMOylated, Fatylated, or Fublylated proteins yet.
The second issue deserving attention is that the networks and pathways activated or inhibited by the “SUMO switch” lack more comprehensive interpretations. Additionally, whether global SUMO proteome alterations upon stress induce distinct biological processes via common signaling pathways hasn’t been clarified.
The third issue is that most potential medical intervention targeting SUMOylation is confined to TAK-981. Given that TAK-981 promotes the functions of T cells, we wonder if its combined effect with ICIs is more beneficial. Additionally, although family members of PIAS E3 enzymes can influence hypoxia and metabolic state in the TME and even activate immune cells as described above, there is no molecular inhibitor targeting E3 ligases yet. This defect might be attributed to the diversity of E3 enzymes. As a result, we expect an inhibitor that specifically targets E3 ligase-mediated processes. In short, much remains to be learned before we gain an insight into the reversible switch of the SUMO cascade and its role in the TME reprogramming.
Conclusion
As one of the most prevalent modifications, SUMOylation is increasingly implicated in tumor initiation and progression. Recently, massive SUMOylated or deSUMOylated proteins were found to act as the executor of hypoxia adaption, metabolic reprogramming, inflammatory, and immune responses during the formation of the TME. Based on this, SUMOylation may serve as a promising diagnostic as well as therapeutic strategy benefiting from its availability of highly specific lyase and high-throughput proteomics.
Data Availability
Not applicable.
Abbreviations
- ACSS:
-
Acetyl-coenzyme A synthetase
- AMPK:
-
5’-AMP-activated protein kinase
- ARNT:
-
Aryl hydrocarbon receptor nuclear transporter
- ARRDC1:
-
Arrestin domain containing 1
- BACH2:
-
BTB domain and CNC homolog 2
- BHLHE40:
-
Basic helix-loop-helix family member e40
- BLCA:
-
Bladder cancer
- Blimp-1:
-
B lymphocyte-induced maturation protein-1
- BRCA:
-
Breast cancer
- CBP:
-
CREB binding protein
- CBX4:
-
chromobox 4
- CCA:
-
Cervical cancer
- CCL1:
-
C-C motif chemokine ligand 1
- CCR8:
-
C-C motif chemokine receptor 8
- CLDN6:
-
Claudin 6
- COAD:
-
Colon cancer
- CREB:
-
cAMP-response element-binding protein
- CRMP2:
-
Cysteine repeat modular protein 2
- CSN5:
-
COP9 signalosome subunit 5
- Daxx:
-
Death domain-associated protein
- DDR:
-
DNA damage response
- DSB:
-
DNA double-strand breaks
- ECV:
-
Elongins and Cullins
- EMT:
-
Epithelial-mesenchymal transition
- EOC:
-
Epithelial Ovarian Cancer
- ESCC:
-
Esophageal squamous cell carcinoma
- ESCRT:
-
Endosomal sorting complex required for transport
- FASN:
-
Fatty acid synthetase
- FIH:
-
Factor inhibiting HIF
- FOXM1b:
-
Forkhead box M1b
- FOXO:
-
Forkhead box O
- FOXP3:
-
Forkhead Box Protein P3
- FTO:
-
Fat mass- and obesity-associated gene
- GATA1:
-
GATA binding protein 1
- GBM:
-
Glioblastoma
- GC:
-
Gastric cancer
- GLUT1:
-
Glucose transporter 1
- GNAO1:
-
Guanine nucleotide-binding protein G (o) subunit alpha
- GSCs:
-
Glioma stem cells
- GSH:
-
Glutathione
- HAF:
-
Hypoxia-associated factor
- HDAC6:
-
Histone deacetylase 6
- HIF-1:
-
Hypoxia inducible factor 1
- HK2:
-
Hexokinase 2
- HLECs:
-
Human lymphatic endothelial cells
- HnRNPK:
-
Heterogeneous nuclear ribonucleoprotein K
- hnRNPD:
-
Heterogeneous nuclear ribonucleoprotein D
- HPV:
-
Human Papilloma Virus
- HRE:
-
Hypoxia responsive elements
- HSP27:
-
Heat shock protein 27
- HTERT:
-
Human telomerase reverse transcriptase
- ICI:
-
Immune checkpoint inhibitor
- IFNβ:
-
Interferon beta
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- IκB:
-
IkappaBalpha
- IL-2:
-
Interleukin 2
- IRF-1:
-
Interferon regulatory factor 1
- ISPs:
-
Immature single-positive cells
- LDH1:
-
Lactate dehydrogenase 1
- LN:
-
Lymph node
- m5C:
-
5-methylcytosine
- m6A:
-
N6-Methyladenosine
- MANF:
-
Mesencephalic astrocyte-derived neurotrophic factor
- MANF:
-
Mesencephalic astrocyte-derived neurotrophic factor
- MCM:
-
Minichromosome maintenance
- MDM2:
-
Murine double minute 2 homologue
- MDSC:
-
Myeloid-derived suppressor cells
- METTL3:
-
Methyltransferase 3
- mTORC1:
-
Rapamycin (mTOR) complex 1
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NDRG2:
-
N-myc downstream regulated gene 2
- NEMO:
-
Nuclear factor (NF)-kappaB essential modulator
- NFATC1:
-
Nuclear factor of activated T cells 1
- NF-κBNF-κB:
-
Nuclear Factor Kappa-Β
- NPC:
-
Nasopharyngeal carcinoma
- NRF2:
-
Nuclear factor erythroid-2 related factor 2
- NSUN2:
-
NOP2/Sun RNA methyltransferase 2
- OSBPL3:
-
Oxysterol binding protein-like 3
- OSCC:
-
Oral squamous cell carcinoma
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- PDPK1:
-
Phosphoinositide-dependent protein kinase 1
- PGK1:
-
Phosphoglycerate kinase 1
- PHDs:
-
Prolyl hydroxylase domain containing proteins
- PI3K:
-
Phosphoinositide 3-kinase
- PIAS:
-
Protein inhibitor of activated STAT
- PKC:
-
Protein kinase C
- PKM2:
-
Pyruvate kinase isoenzyme M2
- PLC-γ1:
-
Phospholipase C-γ1
- PML:
-
Promyelocytic leukemia
- PPARα:
-
Peroxisome proliferator-activated receptor alpha
- PROX1:
-
Prospero homeodomain protein 1 (PROX1)
- PC:
-
Prostate cancer
- PSTAR:
-
P53-stabilizing and activating RNA
- PTEN:
-
Phosphatase and tension homolog
- PTM:
-
Post-translational modification
- RanBP2:
-
RAN binding protein 2
- RCC:
-
Renal cell cancer
- RGS12:
-
Regulator of G protein signaling 12
- RNF4:
-
Ring finger protein 4
- ROS:
-
Reactive oxygen species
- RSUME:
-
RWD domain-containing protein SUMO Enhancer
- SCLC:
-
Small cell lung cancer
- SENPs:
-
Sentrin-specific Proteases
- sEV:
-
Small extracellular vesicle
- SIMs:
-
SUMO-interacting motifs
- SIRT1:
-
Sirtuin 1
- SNAI2:
-
Snail family zinc finger 2
- SnoN:
-
SKI like proto-oncogene
- SOX18:
-
SRY-box transcription factor 18
- SP1:
-
Specificity protein 1
- SREBP:
-
Sterol regulatory element-binding proteins
- STAT:
-
Signal transducer and activator of transcription
- SUMO:
-
Small ubiquitin-related modifier
- TCA:
-
Tricarboxylic acid
- TLRs:
-
Toll-like receptors
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- TRIM28:
-
Tripartite motif containing 28
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- VHL:
-
Von Hippel-Lindau
- YTHDF2:
-
YTH N6-methyladenosine RNA binding protein 2
- ZEB1:
-
Zinc finger E-box binding homeobox 1
References
Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88(1):97–107.
Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805.
Liu J, Qian C, Cao X. Post-Translational Modif Control Innate Immun Immun. 2016;45(1):15–30.
Rabellino A, Khanna KK. The implication of the SUMOylation pathway in breast cancer pathogenesis and treatment. Crit Rev Biochem Mol Biol. 2020;55(1):54–70.
Zamaraev AV, Kopeina GS, Prokhorova EA, Zhivotovsky B, Lavrik IN. Post-translational modification of caspases: the other side of apoptosis regulation. Trends Cell Biol. 2017;27(5):322–39.
Gu Y, Wu X, Zhang J, Fang Y, Pan Y, Shu Y, et al. The evolving landscape of N(6)-methyladenosine modification in the tumor microenvironment. Mol Ther. 2021;29(5):1703–15.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46(10):5195–208.
Hou G, Zhao X, Li L, Yang Q, Liu X, Huang C, et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Res. 2021;49(5):2859–77.
Hu Y, Chen C, Tong X, Chen S, Hu X, Pan B, et al. NSUN2 modified by SUMO-2/3 promotes gastric cancer progression and regulates mRNA m5C methylation. Cell Death Dis. 2021;12(9):842.
Hendriks IA, Vertegaal AC. A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol. 2016;17(9):581–95.
Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108(3):345–56.
Chang HM, Yeh ETH. SUMO: from bench to Bedside. Physiol Rev. 2020;100(4):1599–619.
Kerscher O. SUMO junction-what’s your function? New insights through SUMO-interacting motifs. EMBO Rep. 2007;8(6):550–5.
Kunadis E, Lakiotaki E, Korkolopoulou P, Piperi C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol Ther. 2021;220:107721.
Zhang QS, Zhang M, Huang XJ, Liu XJ, Li WP. Downregulation of SENP1 inhibits cell proliferation, migration and promotes apoptosis in human glioma cells. Oncol Lett. 2016;12(1):217–21.
Wang L, Ji S. Inhibition of Ubc9-Induced CRMP2 SUMOylation disrupts Glioblastoma Cell Proliferation. J Mol Neurosci. 2019;69(3):391–8.
Zhang A, Tao W, Zhai K, Fang X, Huang Z, Yu JS, et al. Protein sumoylation with SUMO1 promoted by Pin1 in glioma stem cells augments glioblastoma malignancy. Neuro Oncol. 2020;22(12):1809–21.
Liu C, Peng Z, Li P, Fu H, Feng J, Zhang Y, et al. lncRNA RMST suppressed GBM Cell Mitophagy through Enhancing FUS SUMOylation. Mol Ther Nucleic Acids. 2020;19:1198–208.
Agbor TA, Cheong A, Comerford KM, Scholz CC, Bruning U, Clarke A, et al. Small ubiquitin-related modifier (SUMO)-1 promotes glycolysis in hypoxia. J Biol Chem. 2011;286(6):4718–26.
Liu J, Han X, Chen L, Han D, Mu X, Hu X, et al. TRIM28 is a distinct prognostic biomarker that worsens the tumor immune microenvironment in lung adenocarcinoma. Aging. 2020;12(20):20308–31.
Sun L, Li H, Chen J, Dehennaut V, Zhao Y, Yang Y, et al. A SUMOylation-dependent pathway regulates SIRT1 transcription and lung cancer metastasis. J Natl Cancer Inst. 2013;105(12):887–98.
Hung PF, Hong TM, Chang CC, Hung CL, Hsu YL, Chang YL, et al. Hypoxia-induced slug SUMOylation enhances lung cancer metastasis. J Exp Clin Cancer Res. 2019;38(1):5.
Rabellino A, Carter B, Konstantinidou G, Wu SY, Rimessi A, Byers LA, et al. The SUMO E3-ligase PIAS1 regulates the tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer Res. 2012;72(9):2275–84.
Wang M, Jiang X. SUMOylation of vascular endothelial growth factor receptor 2 inhibits the proliferation, migration, and angiogenesis signaling pathway in non-small cell lung cancer. Anticancer Drugs. 2020;31(5):492–9.
Liu X, Xu Y, Pang Z, Guo F, Qin Q, Yin T, et al. Knockdown of SUMO-activating enzyme subunit 2 (SAE2) suppresses cancer malignancy and enhances chemotherapy sensitivity in small cell lung cancer. J Hematol Oncol. 2015;8:67.
Wang CM, Yang WH, Liu R, Wang L, Yang WH. FOXP3 activates SUMO-Conjugating UBC9 gene in MCF7 breast Cancer cells. Int J Mol Sci. 2018;19(7).
Subramonian D, Raghunayakula S, Olsen JV, Beningo KA, Paschen W, Zhang XD. Analysis of changes in SUMO-2/3 modification during breast cancer progression and metastasis. J Proteome Res. 2014;13(9):3905–18.
Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-dependent transcriptional subprogram is required for myc-driven tumorigenesis. Science. 2012;335(6066):348–53.
Sun XX, Chen Y, Su Y, Wang X, Chauhan KM, Liang J, et al. SUMO protease SENP1 deSUMOylates and stabilizes c-Myc. Proc Natl Acad Sci U S A. 2018;115(43):10983–8.
Xiao M, Bian Q, Lao Y, Yi J, Sun X, Sun X, et al. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol Oncol. 2022;16(4):1026–44.
Gao Y, Wang R, Liu J, Zhao K, Qian X, He X, et al. SENP1 promotes triple-negative breast cancer invasion and metastasis via enhancing CSN5 transcription mediated by GATA1 deSUMOylation. Int J Biol Sci. 2022;18(5):2186–201.
Sanyal S, Mondal P, Sen S, Sengupta Bandyopadhyay S, Das C. SUMO E3 ligase CBX4 regulates hTERT-mediated transcription of CDH1 and promotes breast cancer cell migration and invasion. Biochem J. 2020;477(19):3803–18.
Chanda A, Ikeuchi Y, Karve K, Sarkar A, Chandhoke AS, Deng L, et al. PIAS1 and TIF1gamma collaborate to promote SnoN SUMOylation and suppression of epithelial-mesenchymal transition. Cell Death Differ. 2021;28(1):267–82.
Liu K, Wang X, Li D, Xu D, Li D, Lv Z, et al. Ginkgolic Acid, a SUMO-1 inhibitor, inhibits the progression of oral squamous cell carcinoma by alleviating SUMOylation of SMAD4. Mol Ther Oncolytics. 2020;16:86–99.
Katayama A, Ogino T, Bandoh N, Takahara M, Kishibe K, Nonaka S, et al. Overexpression of small ubiquitin-related modifier-1 and sumoylated Mdm2 in oral squamous cell carcinoma: possible involvement in tumor proliferation and prognosis. Int J Oncol. 2007;31(3):517–24.
Fu C, Yuan G, Yang ST, Zhang D, Yang S. RGS12 represses oral Cancer via the phosphorylation and SUMOylation of PTEN. J Dent Res. 2021;100(5):522–31.
Yuan DY, Meng Z, Xu K, Li QF, Chen C, Li KY, et al. Betulinic acid increases radiosensitization of oral squamous cell carcinoma through inducing Sp1 sumoylation and PTEN expression. Oncol Rep. 2017;38(4):2360–8.
Ding X, Sun J, Wang L, Li G, Shen Y, Zhou X, et al. Overexpression of SENP5 in oral squamous cell carcinoma and its association with differentiation. Oncol Rep. 2008;20(5):1041–5.
Sun Z, Hu S, Luo Q, Ye D, Hu D, Chen F. Overexpression of SENP3 in oral squamous cell carcinoma and its association with differentiation. Oncol Rep. 2013;29(5):1701–6.
Tian J, Lu Z, Niu S, Zhang S, Ying P, Wang L, et al. Aberrant MCM10 SUMOylation induces genomic instability mediated by a genetic variant associated with survival of esophageal squamous cell carcinoma. Clin Transl Med. 2021;11(6):e485.
Zhang X, Liu T, Zheng S, Liu Q, Shen T, Han X, et al. SUMOylation of HSP27 regulates PKM2 to promote esophageal squamous cell carcinoma progression. Oncol Rep. 2020;44(4):1355–64.
Wang Q, Xu C, Fan Q, Yuan H, Zhang X, Chen B, et al. Positive feedback between ROS and cis-axis of PIASxalpha/p38alpha-SUMOylation/MK2 facilitates gastric cancer metastasis. Cell Death Dis. 2021;12(11):986.
Wei J, Costa C, Ding Y, Zou Z, Yu L, Sanchez JJ, et al. mRNA expression of BRCA1, PIAS1, and PIAS4 and survival after second-line docetaxel in advanced gastric cancer. J Natl Cancer Inst. 2011;103(20):1552–6.
He H, Qiao B, Guo S, Cui H, Li N, Liu H, et al. Induction of T helper 17 cell response by interleukin-7 in patients with primary cutaneous melanoma. Melanoma Res. 2021;31(4):328–37.
Hu XY, Liu Z, Zhang KL, Feng J, Liu XF, Wang LY, et al. SUMO-specific protease 2-mediated deSUMOylation is required for NDRG2 stabilization in gastric cancer cells. Cancer Biomark. 2017;21(1):195–201.
Izawa Y, Mori S, Tretter JT, Quintessenza JA, Toh H, Toba T, et al. Normative aortic valvar measurements in adults using Cardiac computed Tomography- A potential guide to further sophisticate aortic valve-sparing surgery. Circ J. 2021;85(7):1059–67.
Shao DF, Wang XH, Li ZY, Xing XF, Cheng XJ, Guo T, et al. High-level SAE2 promotes malignant phenotype and predicts outcome in gastric cancer. Am J Cancer Res. 2015;5(2):589–602.
Xu Y, Li J, Zuo Y, Deng J, Wang LS, Chen GQ. SUMO-specific protease 1 regulates the in vitro and in vivo growth of colon cancer cells with the upregulated expression of CDK inhibitors. Cancer Lett. 2011;309(1):78–84.
Zhang H, Kuai X, Ji Z, Li Z, Shi R. Over-expression of small ubiquitin-related modifier-1 and sumoylated p53 in colon cancer. Cell Biochem Biophys. 2013;67(3):1081–7.
Coppola D, Parikh V, Boulware D, Blanck G. Substantially reduced expression of PIAS1 is associated with colon cancer development. J Cancer Res Clin Oncol. 2009;135(9):1287–91.
Hou PP, Luo LJ, Chen HZ, Chen QT, Bian XL, Wu SF, et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the Tumor Microenvironment. Mol Cell. 2020;78(6):1192–1206e10.
Liu X, Liu J, Xiao W, Zeng Q, Bo H, Zhu Y, et al. SIRT1 regulates N(6) -Methyladenosine RNA modification in Hepatocarcinogenesis by Inducing RANBP2-Dependent FTO SUMOylation. Hepatology. 2020;72(6):2029–50.
Qin G, Tu X, Li H, Cao P, Chen X, Song J, et al. Long noncoding RNA p53-Stabilizing and activating RNA promotes p53 signaling by inhibiting Heterogeneous Nuclear Ribonucleoprotein K deSUMOylation and suppresses Hepatocellular Carcinoma. Hepatology. 2020;71(1):112–29.
Liu J, Wu Z, Han D, Wei C, Liang Y, Jiang T, et al. Mesencephalic astrocyte-derived neurotrophic factor inhibits Liver Cancer through small ubiquitin-related modifier (SUMO)ylation-Related suppression of NF-kappaB/Snail signaling pathway and epithelial-mesenchymal transition. Hepatology. 2020;71(4):1262–78.
Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W, et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10(13):5671–86.
Guo H, Xu J, Zheng Q, He J, Zhou W, Wang K, et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett. 2019;466:39–48.
Li J, Xu Y, Long XD, Wang W, Jiao HK, Mei Z, et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell. 2014;25(1):118–31.
Cui CP, Wong CC, Kai AK, Ho DW, Lau EY, Tsui YM, et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1alpha deSUMOylation and SENP1/HIF-1alpha positive feedback loop. Gut. 2017;66(12):2149–59.
Biederstadt A, Hassan Z, Schneeweis C, Schick M, Schneider L, Muckenhuber A, et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut. 2020;69(8):1472–82.
Chien W, Lee KL, Ding LW, Wuensche P, Kato H, Doan NB, et al. PIAS4 is an activator of hypoxia signalling via VHL suppression during growth of pancreatic cancer cells. Br J Cancer. 2013;109(7):1795–804.
Arnold F, Gout J, Wiese H, Weissinger SE, Roger E, Perkhofer L, et al. RINT1 regulates SUMOylation and the DNA damage response to preserve Cellular Homeostasis in Pancreatic Cancer. Cancer Res. 2021;81(7):1758–74.
Brems-Eskildsen AS, Zieger K, Toldbod H, Holcomb C, Higuchi R, Mansilla F, et al. Prediction and diagnosis of bladder cancer recurrence based on urinary content of hTERT, SENP1, PPP1CA, and MCM5 transcripts. BMC Cancer. 2010;10:646.
Tan M, Gong H, Wang J, Tao L, Xu D, Bao E, et al. SENP2 regulates MMP13 expression in a bladder cancer cell line through SUMOylation of TBL1/TBLR1. Sci Rep. 2015;5:13996.
Chen C, Zheng H, Luo Y, Kong Y, An M, Li Y et al. SUMOylation promotes extracellular vesicle-mediated transmission of lncRNA ELNAT1 and lymph node metastasis in bladder cancer. J Clin Invest. 2021;131(8).
Koh MY, Nguyen V, Lemos R Jr, Darnay BG, Kiriakova G, Abdelmelek M, et al. Hypoxia-induced SUMOylation of E3 ligase HAF determines specific activation of HIF2 in clear-cell renal cell carcinoma. Cancer Res. 2015;75(2):316–29.
Tedesco L, Elguero B, Pacin DG, Senin S, Pollak C, Garcia Marchinena PA, et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis. 2019;10(4):266.
Xiao J, Sun F, Wang YN, Liu B, Zhou P, Wang FX et al. UBC9 deficiency enhances immunostimulatory macrophage activation and subsequent antitumor T cell response in prostate cancer. J Clin Invest. 2023;133(4).
Chand V, Kapoor A, Kundu S, Nag A. Identification of a peptide that disrupts hADA3-E6 interaction with implications in HPV induced cancer therapy. Life Sci. 2022;288:120157.
Chand V, John R, Jaiswal N, Johar SS, Nag A. High-risk HPV16E6 stimulates hADA3 degradation by enhancing its SUMOylation. Carcinogenesis. 2014;35(8):1830–9.
Jaiswal N, John R, Chand V, Nag A. Oncogenic human papillomavirus 16E7 modulates SUMOylation of FoxM1b. Int J Biochem Cell Biol. 2015;58:28–36.
Dong M, Pang X, Xu Y, Wen F, Zhang Y. Ubiquitin-conjugating enzyme 9 promotes epithelial ovarian cancer cell proliferation in vitro. Int J Mol Sci. 2013;14(6):11061–71.
Ao Q, Su W, Guo S, Cai L, Huang L. SENP1 desensitizes hypoxic ovarian cancer cells to cisplatin by up-regulating HIF-1alpha. Sci Rep. 2015;5:16396.
Riera-Domingo C, Audige A, Granja S, Cheng WC, Ho PC, Baltazar F, et al. Immunity, Hypoxia, and metabolism-the Menage a Trois of Cancer: implications for Immunotherapy. Physiol Rev. 2020;100(1):1–102.
Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157.
Zheng XT, Wang C, Lin W, Lin C, Han D, Xie Q, et al. Importation of chloroplast proteins under heat stress is facilitated by their SUMO conjugations. New Phytol. 2022;235(1):173–87.
Wang L, Ma Q, Yang W, Mackensen GB, Paschen W. Moderate hypothermia induces marked increase in levels and nuclear accumulation of SUMO2/3-conjugated proteins in neurons. J Neurochem. 2012;123(3):349–59.
Keiten-Schmitz J, Wagner K, Piller T, Kaulich M, Alberti S. Muller. The nuclear SUMO-Targeted Ubiquitin Quality Control Network regulates the Dynamics of cytoplasmic stress granules. Mol Cell. 2020;79(1):54–67. e7.
Nunez-O’Mara A, Berra E. Deciphering the emerging role of SUMO conjugation in the hypoxia-signaling cascade. Biol Chem. 2013;394(4):459–69.
Jiang Y, Wang J, Tian H, Li G, Zhu H, Liu L, et al. Increased SUMO-1 expression in response to hypoxia: Interaction with HIF-1alpha in hypoxic pulmonary hypertension. Int J Mol Med. 2015;36(1):271–81.
Shao R, Zhang FP, Tian F, Anders Friberg P, Wang X, Sjoland H, et al. Increase of SUMO-1 expression in response to hypoxia: direct interaction with HIF-1alpha in adult mouse brain and heart in vivo. FEBS Lett. 2004;569(1–3):293–300.
Chachami G, Stankovic-Valentin N, Karagiota A, Basagianni A, Plessmann U, Urlaub H, et al. Hypoxia-induced changes in SUMO conjugation affect Transcriptional Regulation under Low Oxygen. Mol Cell Proteomics. 2019;18(6):1197–209.
Kunz K, Wagner K, Mendler L, Holper S, Dehne N. Muller. SUMO signaling by hypoxic inactivation of SUMO-Specific Isopeptidases. Cell Rep. 2016;16(11):3075–86.
Han X, Wang XL, Li Q, Dong XX, Zhang JS, Yan QC. HIF-1alpha SUMOylation affects the stability and transcriptional activity of HIF-1alpha in human lens epithelial cells. Graefes Arch Clin Exp Ophthalmol. 2015;253(8):1279–90.
Bae SH, Jeong JW, Park JA, Kim SH, Bae MK, Choi SJ, et al. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem Biophys Res Commun. 2004;324(1):394–400.
Nakagawa K, Kohara T, Uehata Y, Miyakawa Y, Sato-Ueshima M, Okubo N, et al. PIAS3 enhances the transcriptional activity of HIF-1alpha by increasing its protein stability. Biochem Biophys Res Commun. 2016;469(3):470–6.
Li J, Xu Y, Jiao H, Wang W, Mei Z, Chen G. Sumoylation of hypoxia inducible factor-1alpha and its significance in cancer. Sci China Life Sci. 2014;57(7):657–64.
Berta MA, Mazure N, Hattab M, Pouyssegur J. Brahimi-Horn. SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem Biophys Res Commun. 2007;360(3):646–52.
Kang X, Li J, Zou Y, Yi J, Zhang H, Cao M, et al. PIASy stimulates HIF1alpha SUMOylation and negatively regulates HIF1alpha activity in response to hypoxia. Oncogene. 2010;29(41):5568–78.
Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131(3):584–95.
van Hagen M, Overmeer RM, Abolvardi SS, Vertegaal AC. RNF4 and VHL regulate the proteasomal degradation of SUMO-conjugated hypoxia-inducible Factor-2alpha. Nucleic Acids Res. 2010;38(6):1922–31.
Tojo M, Matsuzaki K, Minami T, Honda Y, Yasuda H, Chiba T, et al. The aryl hydrocarbon receptor nuclear transporter is modulated by the SUMO-1 conjugation system. J Biol Chem. 2002;277(48):46576–85.
Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, et al. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131(2):309–23.
Gerez J, Tedesco L, Bonfiglio JJ, Fuertes M, Barontini M, Silberstein S, et al. RSUME inhibits VHL and regulates its tumor suppressor function. Oncogene. 2015;34(37):4855–66.
Cai Q, Verma SC, Kumar P, Ma M, Robertson ES. Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modification. PLoS ONE. 2010;5(3):e9720.
Nunez-O’Mara A, Gerpe-Pita A, Pozo S, Carlevaris O, Urzelai B, Lopitz-Otsoa F, et al. PHD3-SUMO conjugation represses HIF1 transcriptional activity independently of PHD3 catalytic activity. J Cell Sci. 2015;128(1):40–9.
Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, et al. P300 transcriptional repression is mediated by SUMO modification. Mol Cell. 2003;11(4):1043–54.
Huang C, Han Y, Wang Y, Sun X, Yan S, Yeh ET, et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009;28(18):2748–62.
Hsieh YL, Kuo HY, Chang CC, Naik MT, Liao PH, Ho CC, et al. Ubc9 acetylation modulates distinct SUMO target modification and hypoxia response. EMBO J. 2013;32(6):791–804.
Kuo HY, Chang CC, Jeng JC, Hu HM, Lin DY, Maul GG, et al. SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of daxx. Proc Natl Acad Sci U S A. 2005;102(47):16973–8.
Xie Y, Liu S, Lu W, Yang Q, Williams KD, Binhazim AA, et al. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol Oncol. 2014;8(7):1355–64.
Jia Y, Guo Y, Jin Q, Qu H, Qi D, Song P, et al. A SUMOylation-dependent HIF-1alpha/CLDN6 negative feedback mitigates hypoxia-induced breast cancer metastasis. J Exp Clin Cancer Res. 2020;39(1):42.
Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional beta cells. J Clin Invest. 2015;125(10):3847–60.
Shao L, Zhou HJ, Zhang H, Qin L, Hwa J, Yun Z, et al. SENP1-mediated NEMO deSUMOylation in adipocytes limits inflammatory responses and type-1 diabetes progression. Nat Commun. 2015;6:8917.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37.
Mo Y, Wang Y, Zhang S, Xiong F, Yan Q, Jiang X, et al. Circular RNA circRNF13 inhibits proliferation and metastasis of nasopharyngeal carcinoma via SUMO2. Mol Cancer. 2021;20(1):112.
Shangguan X, He J, Ma Z, Zhang W, Ji Y, Shen K, et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat Commun. 2021;12(1):1812.
Floris A, Mazarei M, Yang X, Robinson AE, Zhou J, Barberis A et al. SUMOylation protects FASN against proteasomal degradation in breast Cancer cells treated with grape Leaf Extract. Biomolecules. 2020;10(4).
Kuosmanen SM, Kansanen E, Kaikkonen MU, Sihvola V, Pulkkinen K, Jyrkkanen HK, et al. NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. 2018;46(3):1124–38.
Arito M, Horiba T, Hachimura S, Inoue J, Sato R. Growth factor-induced phosphorylation of sterol regulatory element-binding proteins inhibits sumoylation, thereby stimulating the expression of their target genes, low density lipoprotein uptake, and lipid synthesis. J Biol Chem. 2008;283(22):15224–31.
Lee GY, Jang H, Lee JH, Huh JY, Choi S, Chung J, et al. PIASy-mediated sumoylation of SREBP1c regulates hepatic lipid metabolism upon fasting signaling. Mol Cell Biol. 2014;34(6):926–38.
Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J Biol Chem. 2003;278(19):16809–19.
Balasubramaniyan N, Luo Y, Sun AQ, Suchy FJ. SUMOylation of the farnesoid X receptor (FXR) regulates the expression of FXR target genes. J Biol Chem. 2013;288(19):13850–62.
Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010;24(4):381–95.
Stein S, Lemos V, Xu P, Demagny H, Wang X, Ryu D, et al. Impaired SUMOylation of nuclear receptor LRH-1 promotes nonalcoholic fatty liver disease. J Clin Invest. 2017;127(2):583–92.
Priest C. Tontonoz. SUMOylation places LRH-1 in PROXimity to lipid metabolism. Cell Metab. 2014;20(4):558–9.
Liu Y, Dou X, Zhou WY, Ding M, Liu L, Du RQ, et al. Hepatic small ubiquitin-related modifier (SUMO)-Specific protease 2 controls systemic metabolism through SUMOylation-Dependent regulation of liver-adipose tissue crosstalk. Hepatology. 2021;74(4):1864–83.
Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62(3):720–33.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–32.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85.
Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer Cell Metabolism in Hypoxia: role of HIF-1 as Key Regulator and Therapeutic Target. Int J Mol Sci. 2021;22(11).
Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–6.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20(2):74–88.
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–9.
Hosios AM. Manning. Cancer Signaling drives Cancer Metabolism: AKT and the Warburg Effect. Cancer Res. 2021;81(19):4896–8.
Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, et al. Phosphoinositide 3-Kinase regulates glycolysis through mobilization of Aldolase from the actin cytoskeleton. Cell. 2016;164(3):433–46.
El Motiam A, de la Cruz-Herrera CF, Vidal S, Seoane R, Baz-Martinez M, Bouzaher YH, et al. SUMOylation modulates the stability and function of PI3K-p110beta. Cell Mol Life Sci. 2021;78(8):4053–65.
Yang Y, Liang Z, Xia Z, Wang X, Ma Y, Sheng Z, et al. SAE1 promotes human glioma progression through activating AKT SUMOylation-mediated signaling pathways. Cell Commun Signal. 2019;17(1):82.
Ong JR, Bamodu OA, Khang NV, Lin YK, Yeh CT, Lee WH et al. SUMO-Activating enzyme subunit 1 (SAE1) is a Promising Diagnostic Cancer Metabolism Biomarker of Hepatocellular Carcinoma. Cells. 2021;10(1).
Li R, Wei J, Jiang C, Liu D, Deng L, Zhang K, et al. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 2013;73(18):5742–53.
Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (akt) substrate in primary adipocytes. J Biol Chem. 2002;277(37):33895–900.
Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24(43):6465–81.
Hu B, Zhang Y, Deng T, Gu J, Liu J, Yang H, et al. PDPK1 regulates autophagosome biogenesis by binding to PIK3C3. Autophagy. 2021;17(9):2166–83.
Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–83.
Sanz P, Rubio T, Garcia-Gimeno MA. AMPKbeta subunits: more than just a scaffold in the formation of AMPK complex. FEBS J. 2013;280(16):3723–33.
Dou X, Zhou WY, Ding M, Ma YJ, Yang QQ, Qian SW, et al. The protease SENP2 controls hepatic gluconeogenesis by regulating the SUMOylation of the fuel sensor AMPKalpha. J Biol Chem. 2022;298(2):101544.
Rubio T, Vernia S, Sanz P. Sumoylation of AMPKbeta2 subunit enhances AMP-activated protein kinase activity. Mol Biol Cell. 2013;24(11):1801.
Stine ZE, Walton ZE, Altman BJ, Hsieh AL. Dang. MYC, metabolism, and Cancer. Cancer Discov. 2015;5(10):1024–39.
Chen Y, Sun XX, Sears RC, Dai MS. Writing and erasing MYC ubiquitination and SUMOylation. Genes Dis. 2019;6(4):359–71.
Gonzalez-Prieto R, Cuijpers SA, Kumar R, Hendriks IA. Vertegaal. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle. 2015;14(12):1859–72.
Ho J, Bassi C, Stambolic V. Characterization of nuclear PTEN and its post translational modifications. Methods. 2015;77–8:104 – 11.
Gonzalez-Santamaria J, Campagna M, Ortega-Molina A, Marcos-Villar L, de la Cruz-Herrera CF, Gonzalez D, et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 2012;3:e393.
Qian X, Li X, Shi Z, Xia Y, Cai Q, Xu D, et al. PTEN suppresses glycolysis by Dephosphorylating and Inhibiting Autophosphorylated PGK1. Mol Cell. 2019;76(3):516–27. e7.
Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014;15(11):e493–503.
Karhausen J, Ulloa L, Yang W. SUMOylation connects cell stress responses and inflammatory control: Lessons from the gut as a Model Organ. Front Immunol. 2021;12:646633.
Singh AK, Khare P, Obaid A, Conlon KP, Basrur V, DePinho RA, et al. SUMOylation of ROR-gammat inhibits IL-17 expression and inflammation via HDAC2. Nat Commun. 2018;9(1):4515.
Gu Z, Chen X, Yang W, Qi Y, Yu H, Wang X, et al. The SUMOylation of Table 2 mediated by TRIM60 inhibits MAPK/NF-kappaB activation and the innate immune response. Cell Mol Immunol. 2021;18(8):1981–94.
Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2(2):233–9.
Comerford KM, Leonard MO, Karhausen J, Carey R, Colgan SP, Taylor CT. Small ubiquitin-related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proc Natl Acad Sci U S A. 2003;100(3):986–91.
Aillet F, Lopitz-Otsoa F, Egana I, Hjerpe R, Fraser P, Hay RT, et al. Heterologous SUMO-2/3-ubiquitin chains optimize IkappaBalpha degradation and NF-kappaB activity. PLoS ONE. 2012;7(12):e51672.
Liu Y, Bridges R, Wortham A, Kulesz-Martin M. NF-kappaB repression by PIAS3 mediated RelA SUMOylation. PLoS ONE. 2012;7(5):e37636.
Decque A, Joffre O, Magalhaes JG, Cossec JC, Blecher-Gonen R, Lapaquette P, et al. Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat Immunol. 2016;17(2):140–9.
Ambjorn M, Ejlerskov P, Liu Y, Lees M, Jaattela M. Issazadeh-Navikas. IFNB1/interferon-beta-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy. 2013;9(3):287–302.
Liu X, Chen W, Wang Q, Li L, Wang C. Negative regulation of TLR inflammatory signaling by the SUMO-deconjugating enzyme SENP6. PLoS Pathog. 2013;9(6):e1003480.
Antico Arciuch VG, Tedesco L, Fuertes M, Arzt E. Role of RSUME in inflammation and cancer. FEBS Lett. 2015;589(22):3330–5.
Xia QD, Sun JX, Xun Y, Xiao J, Liu CQ, Xu JZ, et al. SUMOylation Pattern predicts prognosis and indicates Tumor Microenvironment Infiltration characterization in bladder Cancer. Front Immunol. 2022;13:864156.
Mustfa SA, Singh M, Suhail A, Mohapatra G, Verma S, Chakravorty D et al. SUMOylation pathway alteration coupled with downregulation of SUMO E2 enzyme at mucosal epithelium modulates inflammation in inflammatory bowel disease. Open Biol. 2017;7(6).
Yang T, Sun J, Wei B, Liu S. SENP1-mediated NEMO de-SUMOylation inhibits intermittent hypoxia induced inflammatory response of microglia in vitro. J Cell Physiol. 2020;235(4):3529–38.
Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell. 2011;43(2):180–91.
Huang X, Zuo Y, Wang X, Wu X, Tan H, Fan Q, et al. SUMO-Specific protease 1 is critical for myeloid-derived suppressor cell development and function. Cancer Res. 2019;79(15):3891–902.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22.
Ding X, Wang A, Ma X, Demarque M, Jin W, Xin H, et al. Protein SUMOylation is required for Regulatory T cell expansion and function. Cell Rep. 2016;16(4):1055–66.
Yu X, Lao Y, Teng XL, Li S, Zhou Y, Wang F, et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat Commun. 2018;9(1):3157.
Xiao Y, Qureischi M, Dietz L, Vaeth M, Vallabhapurapu SD, Klein-Hessling S et al. Lack of NFATc1 SUMOylation prevents autoimmunity and alloreactivity. J Exp Med. 2021;218(1).
Liu B, Tahk S, Yee KM, Fan G. Shuai. The ligase PIAS1 restricts natural regulatory T cell differentiation by epigenetic repression. Science. 2010;330(6003):521–5.
Wang A, Ding X, Demarque M, Liu X, Pan D, Xin H, et al. Ubc9 is required for positive selection and late-stage maturation of thymocytes. J Immunol. 2017;198(9):3461–70.
Xiong Y, Yi Y, Wang Y, Yang N, Rudd CE, Liu H. Ubc9 interacts with and SUMOylates the TCR adaptor SLP-76 for NFAT transcription in T cells. J Immunol. 2019;203(11):3023–36.
Garaude J, Farras R, Bossis G, Charni S, Piechaczyk M, Hipskind RA, et al. SUMOylation regulates the transcriptional activity of JunB in T lymphocytes. J Immunol. 2008;180(9):5983–90.
He Z, Zhang J, Huang Z, Du Q, Li N, Zhang Q, et al. Sumoylation of RORgammat regulates TH17 differentiation and thymocyte development. Nat Commun. 2018;9(1):4870.
Wang QL, Liang JQ, Gong BN, Xie JJ, Yi YT, Lan X, et al. T cell receptor (TCR)-Induced PLC-gamma1 Sumoylation via PIASxbeta and PIAS3 SUMO E3 Ligases regulates the Microcluster Assembly and physiological function of PLC-gamma1. Front Immunol. 2019;10:314.
Wang XD, Gong Y, Chen ZL, Gong BN, Xie JJ, Zhong CQ, et al. TCR-induced sumoylation of the kinase PKC-theta controls T cell synapse organization and T cell activation. Nat Immunol. 2015;16(11):1195–203.
Van Nguyen T, Angkasekwinai P, Dou H, Lin FM, Lu LS, Cheng J, et al. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol Cell. 2012;45(2):210–21.
Muromoto R, Ishida M, Sugiyama K, Sekine Y, Oritani K, Shimoda K, et al. Sumoylation of daxx regulates IFN-induced growth suppression of B lymphocytes and the hormone receptor-mediated transactivation. J Immunol. 2006;177(2):1160–70.
Lin DY, Huang YS, Jeng JC, Kuo HY, Chang CC, Chao TT, et al. Role of SUMO-interacting motif in daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol Cell. 2006;24(3):341–54.
Ying HY, Su ST, Hsu PH, Chang CC, Lin IY, Tseng YH, et al. SUMOylation of Blimp-1 is critical for plasma cell differentiation. EMBO Rep. 2012;13(7):631–7.
Liu C, Wang X, Qin W, Tu J, Li C, Zhao W et al. Combining radiation and the ATR inhibitor berzosertib activates STING signaling and enhances immunotherapy via inhibiting SHP1 function in colorectal cancer. Cancer Commun (Lond). 2023.
Jena BC, Mandal M. The emerging roles of exosomes in anti-cancer drug resistance and tumor progression: an insight towards tumor-microenvironment interaction. Biochim Biophys Acta Rev Cancer. 2021;1875(1):188488.
Luo Y, Li Z, Kong Y, He W, Zheng H, An M et al. KRAS mutant-driven SUMOylation controls extracellular vesicle transmission to trigger lymphangiogenesis in pancreatic cancer. J Clin Invest. 2022;132(14).
Li Y, Zhang J, Li S, Guo C, Li Q, Zhang X, et al. Heterogeneous nuclear ribonucleoprotein A1 loads batched Tumor-Promoting MicroRNAs into small extracellular vesicles with the assist of Caveolin-1 in A549 cells. Front Cell Dev Biol. 2021;9:687912.
Chen MC, Nhan DC, Hsu CH, Wang TF, Li CC, Ho TJ, et al. SENP1 participates in Irinotecan resistance in human colon cancer cells. J Cell Biochem. 2021;122(10):1277–94.
Demel UM, Boger M, Yousefian S, Grunert C, Zhang L, Hotz PW et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J Clin Invest. 2022;132(9).
Huang G, Cai G, Hu D, Li J, Xu Q, Chen Z, et al. Low SP1 SUMOylation-dependent SNHG17 upregulation promotes drug resistance of gastric cancer through impairing hsa-miR-23b-3p-induced Notch2 inhibition. Cell Oncol (Dordr). 2022;45(6):1329–46.
Meurette O, Mehlen P. Notch Signaling in the Tumor Microenvironment. Cancer Cell. 2018;34(4):536–48.
Swayden M, Alzeeb G, Masoud R, Berthois Y, Audebert S, Camoin L, et al. PML hyposumoylation is responsible for the resistance of pancreatic cancer. FASEB J. 2019;33(11):12447–63.
Hayashi T, Seki M, Maeda D, Wang W, Kawabe Y, Seki T, et al. Ubc9 is essential for viability of higher eukaryotic cells. Exp Cell Res. 2002;280(2):212–21.
Neyret-Kahn H, Benhamed M, Ye T, Le Gras S, Cossec JC, Lapaquette P, et al. Sumoylation at chromatin governs coordinated repression of a transcriptional program essential for cell growth and proliferation. Genome Res. 2013;23(10):1563–79.
Tu J, Chen Y, Cai L, Xu C, Zhang Y, Chen Y, et al. Functional proteomics study reveals SUMOylation of TFII-I is involved in Liver Cancer Cell Proliferation. J Proteome Res. 2015;14(6):2385–97.
Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, et al. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol. 2009;16(2):133–40.
Fukuda I, Ito A, Uramoto M, Saitoh H, Kawasaki H, Osada H, et al. Kerriamycin B inhibits protein SUMOylation. J Antibiot (Tokyo). 2009;62(4):221–4.
Takemoto M, Kawamura Y, Hirohama M, Yamaguchi Y, Handa H, Saitoh H, et al. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. J Antibiot (Tokyo). 2014;67(4):335–8.
Suzawa M, Miranda DA, Ramos KA, Ang KK, Faivre EJ, Wilson CG et al. A gene-expression screen identifies a non-toxic sumoylation inhibitor that mimics SUMO-less human LRH-1 in liver. Elife. 2015;4.
Gerstmeier J, Seegers J, Witt F, Waltenberger B, Temml V, Rollinger JM, et al. Ginkgolic Acid is a Multi-Target inhibitor of key enzymes in pro-inflammatory lipid mediator biosynthesis. Front Pharmacol. 2019;10:797.
He X, Riceberg J, Soucy T, Koenig E, Minissale J, Gallery M, et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol. 2017;13(11):1164–71.
Lightcap ES, Yu P, Grossman S, Song K, Khattar M, Xega K, et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci Transl Med. 2021;13(611):eaba7791.
Lv Z, Yuan L, Atkison JH, Williams KM, Vega R, Sessions EH, et al. Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat Commun. 2018;9(1):5145.
Crowl JT, Stetson DB. SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. Proc Natl Acad Sci U S A. 2018;115(26):6798–803.
Nakamura A, Grossman S, Song K, Xega K, Zhang Y, Cvet D, et al. The SUMOylation inhibitor subasumstat potentiates rituximab activity by IFN1-dependent macrophage and NK cell stimulation. Blood. 2022;139(18):2770–81.
Kumar S, Schoonderwoerd MJA, Kroonen JS, de Graaf IJ, Sluijter M, Ruano D, et al. Targeting pancreatic cancer by TAK-981: a SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut. 2022;71(11):2266–83.
Brandt M, Szewczuk LM, Zhang H, Hong X, McCormick PM, Lewis TS, et al. Development of a high-throughput screen to detect inhibitors of TRPS1 sumoylation. Assay Drug Dev Technol. 2013;11(5):308–25.
Wiechmann S, Gartner A, Kniss A, Stengl A, Behrends C, Rogov VV, et al. Site-specific inhibition of the small ubiquitin-like modifier (SUMO)-conjugating enzyme Ubc9 selectively impairs SUMO chain formation. J Biol Chem. 2017;292(37):15340–51.
Hirohama M, Kumar A, Fukuda I, Matsuoka S, Igarashi Y, Saitoh H, et al. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem Biol. 2013;8(12):2635–42.
Kim YS, Nagy K, Keyser S, Schneekloth JS. Jr. An electrophoretic mobility shift assay identifies a mechanistically unique inhibitor of protein sumoylation. Chem Biol. 2013;20(4):604–13.
Zhou P, Chen X, Li M, Tan J, Zhang Y, Yuan W, et al. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem Biophys Res Commun. 2019;513(4):1063–9.
Choi BH, Philips MR, Chen Y, Lu L. Dai. K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J Biol Chem. 2018;293(45):17574–81.
Huang W, He T, Chai C, Yang Y, Zheng Y, Zhou P, et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS ONE. 2012;7(5):e37693.
Wu J, Lei H, Zhang J, Chen X, Tang C, Wang W, et al. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget. 2016;7(37):58995–9005.
Ambaye N, Chen CH, Khanna S, Li YJ, Chen Y. Streptonigrin inhibits SENP1 and reduces the protein level of Hypoxia-Inducible factor 1alpha (HIF1alpha) in cells. Biochemistry. 2018;57(11):1807–13.
Uno M, Koma Y, Ban HS, Nakamura H. Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP)1 inhibitors. Bioorg Med Chem Lett. 2012;22(16):5169–73.
Bernstock JD, Ye D, Smith JA, Lee YJ, Gessler FA, Yasgar A, et al. Quantitative high-throughput screening identifies cytoprotective molecules that enhance SUMO conjugation via the inhibition of SUMO-specific protease (SENP)2. FASEB J. 2018;32(3):1677–91.
Lumpkin RJ, Gu H, Zhu Y, Leonard M, Ahmad AS, Clauser KR, et al. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat Commun. 2017;8(1):1171.
Acknowledgments and funding
This work was supported by grants from the National Natural Science Foundation of China (No.82173347), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801).
Author information
Authors and Affiliations
Contributions
YQ Shu, P Ma, and YR Gu designed the study. YR Gu, Y Fang, X Wu, TT Xu, drafted the manuscript. Q Wang, T Hu, and YY Xu critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, Y., Fang, Y., Wu, X. et al. The emerging roles of SUMOylation in the tumor microenvironment and therapeutic implications. Exp Hematol Oncol 12, 58 (2023). https://doi.org/10.1186/s40164-023-00420-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-023-00420-3