
Abstract
Uterine and ovarian cancers are the most common gynecologic cancers. N6−methyladenosine (m6A), an important internal RNA modification in higher eukaryotes, has recently become a hot topic in epigenetic studies. Numerous studies have revealed that the m6A-related regulatory factors regulate the occurrence and metastasis of tumors and drug resistance through various mechanisms. The m6A-related regulatory factors can also be used as therapeutic targets and biomarkers for the early diagnosis of cancers, including gynecologic cancers. This review discusses the role of m6A in gynecologic cancers and summarizes the recent advancements in m6A modification in gynecologic cancers to improve the understanding of the occurrence, diagnosis, treatment, and prognosis of gynecologic cancers.
Similar content being viewed by others

Background
The main types of gynecologic cancers, which seriously damage the female reproductive organs, include vulvar cancer, vaginal cancer, cervical cancer (CC), endometrial cancer (EC), uterine cancer, and ovarian cancer (OC). CC and OC are the most frequent types of gynecologic cancer, accounting for 6.5% and 3.4%, respectively, of all the new cancers in women [1]. A population-based study conducted on the epidemiological trends of gynecologic cancer from 1990 to 2019 indicated that the incidence and mortality of gynecologic cancers might have geographical variations and changes along with sociodemographic index value [2]. Most gynecologic cancer patients have no distinct symptoms or physical signs in the early stages. In addition, the specific biomarkers for the early diagnosis of gynecologic cancer are also lacking. Moreover, most of the cases are in advanced stages at the time of diagnosis. Therefore, understanding the pathogenesis of gynecologic cancer is particularly important. This might identify specific markers for early diagnosis and therapeutic targets for the related therapeutic drugs, thereby ultimately improving the prognosis and quality of patients [3,4,5].
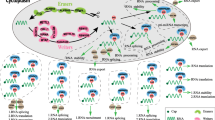
N6-methyladenosine (m6A) was first discovered in 1974 as an internal chemical modification, which was widely observed in the messenger RNAs (mRNAs) and non-coding RNAs (ncRNAs) [6]. The m6A plays important role in numerous aspects of RNA metabolism, such as pre-mRNA splicing, processing of 3′-untranslated region (UTR), export, translation, and degradation of mRNA, and processing of non-coding RNA [7,8,9,10]. Recent studies have shown that the m6A regulatory proteins act as writers, erasers, and readers, thereby modulating the dynamic deposition of mRNAs and other nuclear RNAs [11, 12]. These findings strongly suggest the dynamic regulatory role of m6A modification is similar to the other well-known chromosomal reversible epigenetic modifications. This reversible RNA methylation provides a new dimension in the post-transcriptional regulation of gene expression [11].
In addition to mRNAs, m6A is also reported in a variety of ncRNAs, such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), and ribosomal RNAs (rRNAs) and has been indicated to be crucial for their metabolism and function [13,14,15]. In addition, abnormal m6A modifications in ncRNA by some m6A regulatory proteins participate in the proliferation, invasion, and drug resistance of cancer cells, thereby indicating their potential association with cancer [16, 17]. Therefore, this new field in cancer pathogenesis might provide new opportunities for the diagnosis and treatment of cancer.
This review summarizes the recent studies on m6A modifications in OC, CC, and EC, particularly focusing on the regulatory mechanism of m6A regulatory proteins in promoting the proliferation, invasion, and metastasis of these three gynecologic cancers. Finally, the current knowledge and prospects of m6A modifications in the tumor immune microenvironment, diagnosis, and prognosis of gynecologic cancer are also discussed.
m6A modification
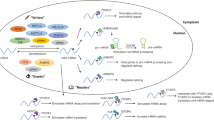
The m6A methylase complex consists of at least five methyltransferases (writers) with methyltransferase-like 3 (METTL3) as its catalytic core. METTL14 is present as the structural support for METTL3; this core complex is stabilized by wilm’s tumor-associated protein (WTAP). The RNA binding pattern protein 15 (RBM15) helps the recruitment of the complex to its target site. Another component of this complex is vir-like m6A methyltransferase-associated protein (VIRMA), which is also known as KIAA1429; its molecular function is uncertain [18]. On the other hand, a demethylase (eraser), consisting of fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5), removes the m6A modifications, thereby reducing its modification rate [19, 20]. The functional interaction between the methyltransferases and demethylase of m6A might contribute to the dynamic regulation of the m6A modifications.
Recent studies have identified the m6A binding proteins (readers) for mRNA, containing YT521-B homology (YTH) domains, such as YTHDF1-3, YTHDC1, and YTHDC2, which have shown a greater affinity for the methylated mRNAs as compared to the non-methylated mRNA [21,22,23,24,25]. If induced by the different cellular environments, the YTH proteins, belonging to the same YTH domain family, can bind to the different subsets of the m6A site and regulate different genes [21]. For instance, the YTHDF2 protein colocalizes with the decay factor and enhances the m6A-modified mRNAs expression [26, 27]. On the other hand, YTHDF1 can bind to the m6A site near the stop codon, thereby subsequently activating the translation by interacting with eukaryotic translation initiation factor 3 (eIF3) [28]. Another YTH protein, YTHDF3 can decay mRNA when working with the YTHDF2 while enhancing the translation of m6A-modified RNA when working with YTHDF1 [10, 29].
m6A and OC
Numerous studies elucidated that the m6A regulators could participate in many functions in OC, such as dysregulation of signaling pathways and anti-tumor drug resistance. The role and mechanism of m6A regulators in OC are summarized in Fig. 1 and Table 1.

In OC, m6A regulatory proteins contribute to tumorigenesis and metastasis by interacting with various RNAs. METTL3 and METTL14 stimulate the progression of OC by promoting the expression levels of FZD10, CSF-1, EIF3C, AXL, RHPN-AS1, miR-125-5p, and TROAP. HOXA10 forms a loop with ALKBH5 and jointly activates the JAK2/STAT3 signaling pathway by mediating JAK2 m6A methylation and promoting the OC resistance to cisplatin. The activated NF-κB up-regulates ALKBH5 expression and increases m6A level and NANOG expression, contributing to ovarian carcinogenesis. FTO and ALKBH5 stimulate/inhibit the progression of OC by affecting the expression levels of ATG5, ATG7, PDE1C, PDE4B, and FZD10.YTHDF1, YTHDF2 and IGF2BP1 stimulate/inhibit the progression of OC by affecting the expression of BMF, TRIM29, EIF3C, SRF, and UBA6. In addition, FBW7 and miR-145 inhibit the expression of YTHDF, leading to OC suppression
Role of m6A regulators in the progression of OC
m6A writer and OC
Studies on OC have mainly focused on the regulatory factor METTL3 [30,31,32,33]. Numerous studies have shown that METTL3 plays an important role in the tumorigeneses of lung and hepatocellular cancer (HCC) [14, 34, 35]. However, METTL3 has also been identified as a tumor suppressor in renal cell carcinoma (RCC) and inhibits the proliferation, migration, and progression of cancer [36]. Ma et al. first reported that METTL3 could promote m6A methylation in the OC without interacting with METTL14 and WTAP [31]. A previous study also showed that in human cancer, METTL3 could directly regulate the specific mRNA translation by recruiting eIF3 without coordinating with METTL14 and WTAP [34]. Studies suggested that METTL3 had a novel m6A regulatory mechanism, which might play an important function in the occurrence and development of OC. Currently, numerous studies indicate that METTL3 can promote the occurrence of OC by affecting the maturation and stability of multiple RNAs. Hua et al. first confirmed the role of METTL3 in tumor progression in the OC cells [30]. In-vivo and in-vitro studies reported that METTL3 could promote the epithelial-to-mesenchymal transition (EMT) by stimulating the mRNA translation of AXL receptor tyrosine kinase (AXL), which might play an important role in the occurrence and/or invasion of OC [30]. Further studies showed that there were no significant changes in the expression levels of AXL in YTHDF1-silenced cells [30]. A study suggested that METTL3 might act as an m6A reader, thereby directly promoting the translation of mRNA; these results were consistent with a previous study, which reported that METTL3 could promote the translation of mRNA in the human lung cancer cells, containing m6A regulatory proteins in their cytoplasm [30, 34]. However, the exact mechanism of the role of m6A requires further investigation.
Liang et al. reported that knocking down the METTL3 gene decreased the expression levels of phosphorylated AKT and its downstream p70S6K and cyclin D1, indicating a decrease in the activation of the AKT pathway in the absence of METTL3 [32]. Bi et al. confirmed that METTL3 could regulate the m6A level of miR-126-5p, promoting its maturation which activated the phosphoinositide 3-kinase (PI3K)/Protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) signaling pathway [33]. In addition, in a xenograft experiment, the knockdown of phosphatase and tensin homolog (PTEN) gene could reverse the METTL3 knockdown-induced decrease in the expression levels of PI3K, p-AKT, and p-4EBP1. The results further confirmed that the knockdown of the METTL3 gene inhibited the PI3K/AKT/mTOR signaling pathway by inhibiting the expression of miR-126-5p and upregulating that of PTEN in-vivo. These results showed that silencing the METTL3 gene could decrease tumor growth and PTEN gene silencing [33]. METTL3 could improve the m6A modification levels in RHPN1-antisense RNA 1(RHPN1-AS1) and increase its RNA stability, which might partially contribute to the up-regulation of RHPN1-AS1 in OC [37]. RHPN1-AS1 absorbs miR-596, which increases the expression of LETM1 and activates the FAK/PI3K/AKT signaling pathway, thereby contributing to the occurrence and development of cancer [37]. These results were similar to Luo’s study, which indicated that YTHDF1 could promote the HCC progression by activating the PI3K/AKT/mTOR signaling pathway and inducing EMT [38].
In most tumors, METTL14 downregulates the m6A levels in cancer cells by acting as m6A methyltransferase to inhibit the occurrence and development of tumors, thereby playing an anti-tumor role. In breast cancer (BC), the low METTL14 level was correlated with a poor prognosis, and its abnormal expression could promote the invasion of BC by affecting the tumor progression-related pathways and mediating the immunosuppression [39]. The studies on OC have shown similar results. Liang et al. reported a considerable copy number variation (CNV) in the METTL14 gene in the OC tissues and a decrease in its expression level as well as low m6A methylation level [40]. Further investigation showed that the trophinin-associated protein (TROAP) was a downstream target of METTL14 [40]. METTL14 reduced the mRNA stability of TROAP, inhibiting the proliferation of OC cells at the G1 phase [40].
In addition, WTAP, an m6A writer, has been reported as a classic biomarker for the progression and metastasis of OC, thereby contributing to the diagnosis and prognosis of OC [41, 42]. Fu et al. reported that the overexpression of WTAP in the OC cells resulted in a high malignancy and low survival rate and promoted aberrant methylation of mRNA, thereby regulating the growth and migration of tumor cells [43]. Wang et al. reported a positive correlation of WTAP expression with two genes, including a family with sequence similarity 76 member A (FAM76A) and HBS1-like translational GTPase (HBS1L) [44]. However, the mechanism of the WTAP-HBS1L/ FAM76A axis, playing a functional role in the progression of OC, remains unclear.
m6A reader and OC
In OC, numerous studies have elucidated the different functional roles of YTHDF1, YTHDF2, and Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) [45,46,47,48,49]. YTHDF1, a YTH domain family member, can identify the m6A post-transcriptional modification by the conserved aromatic cages in its YTH domain [50]. All the YTH domain-containing proteins can bind to the m6A sites in mRNA; however, they recognize different target mRNAs and play different functional roles. Studies have shown an increase in the recruitment of tripartite motif-containing 29 (TRIM29) mRNA by YTHDF1 in the cisplatin-resistant OC cells, thereby promoting the translation of TRIM29 transcripts [49]. Knocking down the YTHDF1gene inhibited the cancer stem cell (CSC)-like characteristics in the cisplatin-resistant OC cells, which were rescued by the overexpression of TRIM29 gene, thereby suggesting its oncogenic role in an m6A-YTHDF1-dependent manner [49]. Based on the multi-group analysis of OC, researchers have identified a new mechanism, involving EIF3C, a subunit of the translation initiation factor [47]. YTHDF1 could bind to the m6A-modified mRNA of EIF3C, stimulate the EIF3C translation, and promote its overall translational output, ultimately leading to the progression and metastasis of OC [47]. Recent studies have reported that the IGF2BPs in m6A “reader” also plays a similar role. Müller et al. found that IGF2BP1 could downregulate the mRNA expression of serum response factor (SRF) mediated by the miRNA production, thereby promoting the expression of SRF in an m6A-dependent manner [46]. Wang et al. also identified IGF2BP1 as an m6A reader of ubiquitin-like modifier activating enzyme 6 antisense RNA 1 (UBA6-AS1) -RBM15, which mediated the m6A mRNA level of UBA6, thereby enhancing its stability; this ultimately inhibited the proliferation, migration, and invasion of OC cells via UBA6 [51]. These studies suggested that YTHDF1 and IGF2BPs were involved in the OC progression by increasing the translation of target mRNA.
Recent studies demonstrated a novel tumor-promoting effect of YTHDF2 by analyzing its upstream signal in the OC and showed that the chemical modification of m6A as a signal center could interact with important metabolic pathways [45, 48]. Li et al. reported a close correlation between an increase in the YTHDF2 protein levels and the OC tissues in a clinical setting [48]. In addition, the investigation showed a key crosstalk between the miR-145 and YTHDF2 via a double-negative feedback loop [48]. Researchers have also reported similar double-negative feedback circuits in the HCC. The miR-145, which was down-regulated in the HCC patients, could directly target the 3′-UTR of YTHDF2 mRNA, thereby inhibiting its expression [52]. Additionally, Xu et al. demonstrated the E3-ubiquitin ligase F-box and WD repeat domain-containing 7 (FBW7), a tumor suppressor, could degrade YTHDF2 in the OC [45]. This study depicted the mechanism of YTHDF2 and FBW7 in OC; FBW7 could decrease the YTHDF2-mediated m6A-dependent mRNA decay for stabilizing the pro-apoptotic BMF mRNA [45]. In particular, Xu et al. demonstrated the role of the FBW7-YTHDF2-BMF axis in the occurrence and development of OC and described how the m6A-related regulatory factors in OC were activated, which provided new insights into the mechanism of OC.
m6A eraser and OC
In OC, studies on m6A erasers have focused on FTO and ALKBH5 [53,54,55,56,57]. The FTO gene is associated with obesity in children and adults [58] and regulates energy homeostasis by controlling food intake and fine-tuning nutritional sensing at the cellular level [59]. FTO is the first recognized nucleic acid demethylase, which physiologically targets the m6A residues in mRNA [19, 60]. Studies have reported its high expression in acute myeloid leukemia (AML), promoting cellular transformation and proliferation by the post-transcriptional regulation of ASB2, RARA, MYC, and CEBPA [61, 62]. CSCs have the abilities of self-renewal, spherical growth, differentiation and tumor formation, which are correlated with the initiation, metastasis, and recurrence of high-grade serous OC (HGSOC) after chemotherapy [63, 64]. Interestingly, Huang et al. recently reported a tumor-inhibitory effect of FTO in HGSOC, which was contradictory to the tumor-promoting role of the FTO previously reported in other types of cancer. Huang et al. demonstrated that FTO targeted two phosphodiesterase genes (PDE4B and PDE1C), thereby regulating the cAMP signal transduction and maintaining the stemness of ovarian CSCs [53]. Their study demonstrated the inhibitory effects of FTO on solid tumors for the first time and identified the cAMP signal transduction as a key pathway for the self-renewal of CSCs regulated by m6A mRNA modification [53]. In addition, another eraser ALKHB5 was also demonstrated to upregulate the expression level of NANOG [54]. A study showed that ALKBH5 could mediate the post-transcriptional NANOG expression and enrich CSCs in BC [65]. ALKBH5 showed significantly higher expression levels in OC tissues as compared to the normal ovarian tissues; however, its expression in the OC cell lines was lower than that in the normal ovarian cells in-vitro [54]. Jiang et al. reported similar expression patterns for the Toll-like receptor 4 (TLR4) in the tumor microenvironment (TME) [54]. Further investigation verified that the highly expressed TLR4 could activate the nuclear factor kappa B (NF-κB) pathway, upregulate the ALKBH5 expression, and increase the m6A level and NANOG expression, thereby contributing to the tumorigenesis of OC [54]. Their study revealed an important functional role of mRNA m6A modification in the self-renewal of OC cells, particularly in the TME.
Autophagy is related to the status of physiological processes and is involved in many pathological conditions, including tumors and inflammation. Currently, numerous studies have confirmed the effects of circRNA on the malignant behavior of tumor cells by regulating autophagy. For example, CircDnmt1 stimulated autophagy in the BC cells to promote their proliferation [66]. CircRNA 103948 acted as competing endogenous RNA (ceRNA) and inhibited autophagy in CRC [67]. CircRNA ST3GAL6 could inhibit the malignant behavior of gastric cancer by regulating autophagy through the FOXP2/MET/mTOR axis [68]. Interestingly, Zhang et al. reported that the m6A modification was involved in autophagy [57]. They demonstrated that CircRAB11FIP1 could bind to the mRNA of FTO, promoting its expression, and thereby regulating the m6A methylation level of ATG5 and ATG7 mRNA through FTO, which ultimately promoted the autophagy and malignant behavior of OC [57].
Tumors show resistance to anti-tumor drugs by numerous mechanisms, including mutation of tumor suppressor genes, activation of oncogenes, and dysregulation of the signaling pathways [69,70,71]. In OC, the FTO and ALKBH5 are involved in drug resistance by activating or upregulating a specific signaling pathway [55, 56]. Takeshi et al. demonstrated a significant increase in m6A modification in mRNA frizzled class receptor 10 (FZD10), thereby increasing its stability and upregulating the Wnt/β-catenin pathway [56]. Further investigation showed that the downregulation of m6A demethylases FTO and ALKBH5 enhanced the m6A modification of FZD10 mRNA and reduced the sensitivity of PARP inhibitor (PARPi), which increased the activity of homologous recombination [56]. Another study reported that ALKBH5- Homeobox A10 (HOXA10) loop could mediate the demethylation of Janus kinase 2 (JAK2) m6A and cisplatin resistance in the OC [55]. Noteworthily, HOXA10 could form a loop with ALKBH5 and act as an upstream transcription factor of ALKBH5. Its overexpression could also enhance chemoresistance in the OC cells [55]. The activation of the JAK2/STAT3 signaling pathway can result in the overexpression of the ALKBH5-HOXA10 loop [55]. These studies have explained the mechanism of m6A modification in the anti-tumor drug resistance of OC. However, more detailed studies are needed in the future to study the mechanism in detail.
m6A and immunoregulation in OC
TME includes tumors, surrounding matrix, and immune components, such as tumor-associated macrophages (TAMs), CD8+ T lymphocytes, and myeloid-derived suppressor cells (MDSCs) [72]. Recent studies have demonstrated numerous important roles of TME components in various biological behaviors of cancer, such as invasion, metastasis, and immune evasion from immune surveillance [73, 74]. Currently, based on the level of tumor-infiltrating immune cells (TICs) in TME, tumors have been classified into two groups: hot tumors, containing high-density CD8+ T lymphocytes, and cold tumors, lacking T lymphocytes [75,76,77]. Despite the relatively high tumor mutation burden in OC, it is a cold tumor, generally lacking the infiltration of cytotoxic T lymphocytes, and thereby lacking the ability to recognize all the tumor antigens [78]. Numerous studies have demonstrated that the degree of immune cell infiltration and expression levels of various immune gene markers are correlated with the specific m6A regulators [54, 79,80,81]. Wang et al. reported RBM15B, Zinc finger CCCH domain-containing protein 13 (ZC3H13), YTHDF1, and IGF2BP1 as important immune cell infiltration-regulated m6A regulators in the OC [79]. Yan et al. also reported cell division cycle 42 effector protein 3 (CDC42EP3) as a possible target gene of m6A, which was downregulated by m6A regulators in the OC cells and tissues [80]. Interestingly, the expression of CDC42EP3 was reported to be correlated with the various TICs, including natural killer cells, T central memory cells, and T gamma delta cells [80]. Jiang et al. further elucidated the mechanism of m6A regulatory factors involved in immune responses [54]. The m6A eraser ALKBH5 could promote the development of OC by stimulating the NF-κB pathway in the TME [54].
Prognostic effect of m6A in OC
Numerous OC patients are diagnosed in the advanced stages due to the lack of specific biomarkers for early clinical screening as well as due to their relatively nonspecific disease symptoms. Currently, more than 70% of OC patients show < 30% overall survival after five years of cytoreductive surgery and adjuvant chemotherapies [82]. In order to better control tumorigenesis and monitor the prognosis of OC patients, new factors and biomarkers are needed to be developed.
Advancements in the studies of m6A methylation in OC might elaborate the prognostic potential of m6A regulatory factors and m6A-related genes in OC. Recently, numerous studies identified that the m6A RNA methylation regulator exhibited high frequencies of genetic changes and high prognostic potential in OC (Table 4) [83,84,85,86,87]. Han et al. reported the correlation of genetic mutations in patients with OC with the survival and m6A regulator genes [84]. Zhu et al. reported the association of YTHDC2 and KIAA1429 with the prognosis of OC patients [88]. In addition, researchers have established genetic models to predict the prognosis of OC patients. Fan et al. established a genetic model, consisting of three m6A regulatory genes, which could be used to predict the progression of OC patients [86]. Li et al. also established a risk-scoring model, consisting of three m6A RNA methylation regulators (VIRMA, IGF2BP1, and HNRNPA2B1) and a related miRNA-m6A regulator-m6A target gene network [87]. Similarly, Jiao et al. also established a genetic model, containing 12 genes (WTAP, LGR6, ZC2HC1A, SLC4A8, AP2A1, NRAS, CUX1, HDAC1, CD79A, ACE2, FLG2, and LRFN1) [89]. Moreover, several studies also have verified the reliability of these results. For example, Yu et al. reported the correlation of high WTAP expression with poor prognosis in HGSOC [90]. However, different m6A regulatory genes have shown different expression patterns in the different tumor types or independent databases, further demonstrating the complexity of the m6A post-transcriptional regulation mechanism and suggesting the tumor-specificity of m6A regulatory factors.
LncRNAs are a group of 200-nucleotides long RNA, which regulate gene expression and various physiological and pathological processes [91, 92]. Using the OC-related dataset from The Cancer Genome Atlas (TCGA), Nie et al. confirmed the prognostic potential of 21 m6A modifications in OC and identified two m6A subtypes using the m6A-related gene expression profiles [93]. Then, the authors established an OC risk model based on the differential expression pattern of lncRNAs between the m6A subtypes and lncRNAs co-expressed with the m6A-related genes [93]. The risk model not only simply evaluated the predictability of tumor prognosis using the risk score but also assessed the effectiveness of immunotherapy and developed novel and more accurate immunotherapies.
m6A and CC
Numerous studies have elucidated the participation of m6A regulators in many functions in the CC, such as aerobic glycolysis and EMT. The role and mechanism of m6A regulators in CC are summarized in Fig. 2 and Table 2.

In CC, m6A regulatory proteins contribute to tumorigenesis and metastasis by interacting with various RNAs. piRNA-14633 mimic increases the mRNA stability of METTL14 and CYP1B1 expression levels, leading to the malignancy of CC. METTL3, which is up-regulated by TBP, regulates the glycolysis of CC via the regulation of PDK4. METTL3 promotes/inhibits the tumorigenesis and metastasis of CC by interacting with various RNAs, including E2F1, circ0000069, ZFAS1, miR-193b, FOXD2-AS1, and HK2. FTO and ALKBH5 promote/inhibit the tumorigenesis and metastasis of CC by regulating the expression of β-catenin, HOXC13-AS, E2F1, MYC, and GAS5. circARHGAP12 interacts with IGF2BP2 to enhance the mRNA stability of FOXM1, thereby promoting the proliferation and migration of CC. E6/E7 regulates the aerobic glycolysis of CC cells through the IGF2BP2-mediated modulation of m6A-MYC mRNA. KCNMB2-AS1 and IGF2BP3 form a positive regulatory circuit, which increases the tumorigenic effect of KCNMB2-AS1 in CC. YTHDF1, YTHDF2, and IGF2BP3 promote/inhibit the tumorigenesis and metastasis of CC by regulating the expression levels of GAS5, CTNNB1, ACIN1, PDK4, HK2, and RANBP2
Role of m6A regulators in the progression of CC
m6A writers and CC
miRNA is a small non-coding RNA (18–24 nucleotides long), which negatively regulates the gene expression by binding to the 3′-UTR of its target mRNA at the post-transcriptional stage [94]. The correlations of miRNA activity with numerous physiological and pathophysiological processes, including carcinogenesis, have been reported [95, 96]. It was reported that the miRNAs might regulate over 60% of the human mRNAs and participate in almost all the biological processes in mammalian systems [97]. The functional characteristics of the miRNA target network and identification of miRNA dysregulation emphasized their importance in malignant tumors [98]. In CC, studies have shown that the m6A methylation modification might regulate the maturation of miRNA, thereby affecting the progression of tumors. Huang et al. reported that METTL3 could modulate the maturation of miR-193b by increasing the methylation level of pri-miR-193b m6A [99]. In addition, miR-193b might play a tumor suppressor role in the CC by inhibiting the cell cycle at the G1 phase as well as inhibiting the proliferation of cells [99]. Recently, Su et al. reported that METTL3 could stimulate the proliferation of CC by regulating mRNA stability of apoptosis chromatin condensation inducer 1 (ACIN1) mRNA [100]. Further investigations showed that the overexpression of IGF2BP3 could reverse the mRNA and protein levels of ACIN1 in the METTL3-downregulated CC cells, suggesting that the METTL3 could affect the mRNA stability of ACIN1 through IGF2BP3 [100].
In the previous section, it was elucidated that METTL3 could promote EMT in the OC by stimulating the mRNA translation of AXL [30]. Li et al. reported that METTL3 could negatively regulate the expression and membrane localization of β-catenin (encoded by CTNNB1) in the CC, thereby forming a complex with E-Cad to participate in the EMT and promoting the development of CC [101]. Further studies indicated that METTL3 could negatively regulate mRNA transcription, decay, and translation of CTNNB1 through different m6A regulators and mechanisms [101]. METTL3 indirectly inhibited the CTNNB1 transcription by upregulating the expression of its inhibitor E2F1 and recruiting YTHDF2 to the 5'-UTR m6A of CTNNB1 [101]. Additionally, METTL3 regulated both the classical and non-classical mRNA translation of CTNNB1 through YTHDF1 [101].
Piwi-interacting RNA (piRNA) was first discovered in 2006 as a small non-coding RNA, consisting of about 30 nucleotides, which could specifically bind to the PIWI family proteins (highly conserved RNA binding proteins) in the testicular germ cells [102, 103]. A recent study demonstrated that it was highly expressed both in the normal and cancer cells [104]. Their presence in the cancer cells might affect cancer growth by directly binding to the PIWI proteins [105]. Xie et al. reported the piRNA-14633-METTL14-CYP1B1 signaling cascade and showed the interaction of piRNA-14633 with the 3'-UTR of METTL14, which increased its mRNA stability, thereby enhancing the METTL14 methylase activity and promoting tumorigenesis by enhancing the CYP1B1 expression [106].
In OC, circRNA might act as a pro-oncogene and participate in the occurrence and development of tumors by promoting autophagy [57]. In CC, Chen et al. reported that the m6A regulator METTL3 could enhance the stability of circ0000069 transcripts, thereby showing the carcinogenesis effects of circRNA [107]. This resulted in the production of low levels of miR-4426 in the CC cells due to the enhanced circ000006 9[107]. Therefore, through m6A modification, miR-4426 was indirectly inhibited, which promoted CC development [107].
The methylation modifications of lncRNA and m6A play a key role in human cancer. The interaction mechanism between the lncRNA and m6A methylation modifications has been elucidated in other tumors, such as non-small cell lung cancer and CRC; however, little is known about their interaction in CC [108, 109]. Therefore, researchers have attempted to explore the internal mechanism of methylation modification of lncRNA and m6A, regulating the tumorigenesis of CC. Ji et al. reported a significantly upregulated expression of METTL3-induced FOXD2-AS1 in the CC cells and tissues, which could stabilize its mRNA [110]. FOXD2-AS1 could bind to the promoter region of p21 and recruit lysine-specific demethylase 1(LSD1) to silence its transcription, thereby accelerating the progression of CC [110]. In conclusion, they suggested that the METTL3/FOXD2-AS/LSD1/P21 axis could accelerate the CC progression in an m6A-dependent manner [110]. Yang et al. reported the role of m6A modification between miRNA and lncRNA and showed that the m6A modification significantly enriched the lncRNA ZFAS1 [111]. Further investigations showed the correlation of METTL3 with ZFAS1, interacting with the m6A level of ZFAS1 without affecting its expression level [111]. The knockdown of METTL3 abolished the miR-647-mediated suppression of ZFAS1, indicating that ZFAS1 could affect the miR-647 in an m6A-dependent manner [111]. These studies revealed that the lncRNA could promote the occurrence of CC in an m6A-dependent manner, and the lncRNA blocked the miRNA through the m6A modification-related proteins, thereby establishing a link between them and suggesting the potential mechanism of m6A between miRNA and lncRNA in CC.
Warburg effect, also known as aerobic glycolysis, is a typical metabolic marker of cancer metabolism [112,113,114]. Despite the abundance of intracellular oxygen, tumor cells persist to generate energy through aerobic glycolysis rather than oxidative phosphorylation through mitochondria [115]. This characteristic energy supplementation is known as the Warburg effect. Inhibiting the Warburg effect is considered an effective treatment for CC. The effects of m6A methylation modification on aerobic glycolysis in cancer cells have rarely been studied. Recent studies have investigated the molecular mechanism of m6A methylation modification in the energy metabolism in tumor cells. Li et al. reported the upregulated expression of pyruvate dehydrogenase kinase 4 (PDK4) induced by m6A, which was reversed by the METTL3 deficiency-induced inhibition of glycolysis and ATP production in the tumor cells [116]. A previous study reported that METTL3 was highly expressed in the metastatic tissues of CRC and inhibited the mRNA degradation of SRY-box transcription factor 2 (SOX2) by specifically interacting with m6A reader IGF2BP2 [117]. Similarly, Li et al. found that m6A modification in the 5′-UTR region rather than 3′-UTR of PDK4 mRNA could positively regulate its translation and stability by binding to the YTHDF1/eEF-2 complex and IGF2BP3 [116]. In addition, the TATA-binding protein (TBP) could enhance the expression of METTL3 in the CC cells [116]. The in-vivo and clinical analyses demonstrated that the m6A could regulate the glycolysis of cancer cells by regulation of PDK4 [116]. In another study, Wang et al. elucidated that METTL3 could promote the occurrence of CC in-vivo and in-vitro by promoting cellular glycolysis [118]. Meanwhile, the authors also showed that the m6A readers could stabilize mRNA, playing a carcinogenic role in the development of CC [118]. YTHDF1 could recognize HK2 m6A and enhance its stability, thereby regulating the HK2 mRNA [118]. METTL3 stabilized the HK2 mRNA by recruiting YTHDF1 and exerted an oncogenic effect via the YTHDF1/HK2 axis by accelerating glycolysis [118].
m6A reader and CC
In CC, studies on the m6A readers have mainly focused on the YTHDF1, YTHDF2, and IGFBPs. Previous studies showed that m6A readers could promote the progression of tumors by regulating mRNA translation [119]. Similarly, Wang et al. reported an elevated expression level of YTHDF1, which was closely related to the poor prognosis of CC patients [120]. RANBP2 was identified as a crucial target of YTHDF1 in the CC cells; YTHDF1 affected the RANBP2 translation in an m6A-dependent manner without affecting its transcription level [120]. Consequently, the overexpression of RANBP2 enhanced the progression of CC [120].
The lncRNAs have several functional roles, such as the organization of nuclear architecture, regulation of translation, mRNA stability, translation, and post-translational modifications [121, 122]. Currently, lncRNAs are reported to be deregulated in non-small cell lung cancer and CRC and play a tumor suppressor or oncogene function by inhibiting the miRNAs [123, 124]. Studies have also demonstrated the interaction of lncRNA with the m6A-related factors, affecting the occurrence and development of tumors. In CC, m6A readers promoted the growth and proliferation of the CC cells by affecting the fate of lncRNA [125, 126]. Zhang et al. reported that KCNMB2-AS1 was predominantly located in the cytoplasm and served as a ceRNA to inhibit the binding of miR-130b-5p and miR-4294 to the IGF2BP3 mRNA, resulting in its upregulation; therefore, it is a well-documented oncogene in CC [125]. Moreover, IGF2BP3 could bind to KCNMB2-AS1 through its three m6A modification sites acting as an m6A reader and stabilizing KCNMB2-AS1 [125]. The KCNMB2-AS1 and IGF2BP3 formed a positive regulatory circuit, which enhanced the tumorigenic effects of KCNMB2-AS1 in the CC [125]. Similarly, another study showed that the degradation of m6A-mediated GAS5 RNA relied on the YTHDF2 [126].
The circRAB11FIP1 might promote tumor autophagy in OC by regulating the m6A methylation of the autophagy-related proteins via FTO [57]. Similarly, Ji et al. showed that IGF2BP2 could interact with circARHGAP12, enhancing the mRNA stability of FOXM1 and forming a circARHGAP12/IGF2BP2/FOXM1 complex to promote the proliferation and migration of CC cells [127].
Human Papillomavirus (HPV) exists in several types, each of which, contains a circular double-stranded DNA genome. HPV primarily infects the basal keratinocytes of the squamous epithelium, which is poorly differentiated. The most common genotypes of HPV are HPV16/18, which are the high-risk types [128]. Generally, CC results from the persistent infection of high-risk HPV [129]. The HPV infection alters the metabolism of tumor cells, causing immune suppression and immune evasion, which lead to the occurrence of CC [130]. The alteration in metabolic phenotypes after HPV infection, contributing to the progression of malignant CC is a critical factor [131, 132]. Hu et al. elucidated the roles and mechanisms, underlying the biological effects of HPV E6/E7 and IGF2BP2 on the CC progression in-vitro and in-vivo [133]. They demonstrated that the E6/E7 proteins could stimulate aerobic glycolysis, proliferation, and metastasis in the CC cells by modulating the MYC mRNA via IGF2BP2 [133]. The E6/ E7 of HPV16 could enhance the expression of HK2 in glycolysis by increasing c-MYC [134]. These studies suggested complex correlations among the E6/E7, m6A methylation modification, promotion of aerobic glycolysis, and CC progression, the mechanisms of which require further studies.
m6A eraser and CC
In the CC, studies on the m6A eraser have focused on the FTO and ALKBH5 [126, 135,136,137]. The m6A demethylases play a critical role in the CC, including the interaction of FTO with the transcription factors E2F1 and MYC, which significantly reduces their translational efficiency [137]. The overexpression of E2F1 or MYC can compensate for the lack of FTO, which negatively impacts the proliferation and migration of cells, suggesting that these two genes might have mutual regulatory functions in CC cells [137].
Researchers elucidated that the lncRNA GAS5-AS1 could increase the expression and stability of GAS5 through YTHDF2 [126]. They reported that GAS5-AS1 could reduce the m6A level of GAS5 by interacting with m6A eraser ALKBH5, thereby increasing the stability of GAS5 [126]. Wang et al. suggested that FTO could regulate the fate of lncRNA to promote the development of CC [135]. They demonstrated that the reduction of the m6A level could improve the stability of HOXC13-AS in the CC cells [135]. Further investigations in the same study revealed that the HOXC13-AS promoted the epigenetically-mediated upregulation of FZD6 and activation of Wnt/β-catenin for promoting CC proliferation, invasion, and EMT [135].
Chemoradiotherapy is a major therapeutic option in CC treatment [138,139,140]. However, both the acquired and primary resistances to chemoradiotherapy cause treatment failure [140, 141]. Therefore, the mechanism of resistance to chemoradiotherapy in CC is needed to be investigated. Numerous studies showed that EMT was involved in drug resistance in cancer [142,143,144,145]. FTO could positively regulate the expression levels of β-catenin by reducing the m6A methylation level of its mRNA in EMT [136]. In the OC, the upregulation of the Wnt/β-catenin signaling pathway in EMT involved m6A modification [135, 146]. Zhou et al. also screened the markers of the Wnt/β-catenin pathway and demonstrated that the canonical Wnt/β-catenin pathway was not involved in the FTO-induced upregulation of β-catenin [136]. However, the excision repair cross-complementation group 1 (ERCC1) was determined as a downstream regulator of the FTO-induced up-regulation of β-catenin, confirming that FTO/β-catenin/ERCC1 axis might play an important role in developing resistance to the chemotherapeutic drugs in CC [136].
m6A and immunoregulators in CC
The immunosuppressive cells in the TME, such as regulatory cells and MDSCs, affect each other as well as the development of tumors [147, 148]. Tumor-infiltrating MDSCs usually induce anti-tumor immune tolerance by inhibiting the proliferation and function of T cells, such as blocking the antigen presentation by the antigen-presenting cells [149]. Ni et al. reported that METTL3 could directly induce the differentiation of MDSCs and tumor-associated MDSCs in-vitro, suggesting that METTL3 might play an important role in the TME of CC [150].
Programmed death-1 (PD-1) is present in apoptotic T-cell hybridomas. It is predominantly present on the surface of activated T cells and B cells as a surface receptor for the activation of T cells. There are two ligands for PD-1, including PD-ligand 1 (PD-L1) and PD-ligand 2 (PD-L2) [151, 152]. TME enhances the expression levels of PD-1 molecules in the infiltrating T cells as well as those of PD-L1 and PD-L2 molecules in the tumor cells, thereby leading to constant activation of the PD-1 pathway within the TME. Zhang et al. investigated the effects of m6A-related lncRNA modifications on the immune response of CC patients [153]. The results showed a significant increase in the expression levels of several immune checkpoints in the high-risk subgroups associated with m6A, including PD-1 and PD-L1, which suggested the potential responses to PD-1 [153].
Prognostic effects of m6A in CC
The diagnosis and treatment of cancer have been greatly improved over the past decades. However, the 5-year survival rate of the patients remains low. Therefore, accurate prognostic indicators are needed to establish an individualized treatment strategy for CC patients (Table 4). Wang et al. reported that a decrease in the m6A methylation level was closely related to the cancer progression and low survival rate, suggesting its potential as a target for the prognosis and treatment of CC [154]. Ma et al. reported that four genes had the prognostic potential for CC, including HNRNPC, KIAA1429, and ZC3H13, and a protective gene YTHDF1 [155]. Pan et al. established a characteristic model, consisting of three genes (ZC3H13, YTHDC1, and YTHDF1), and showed good performance in predicting the survival rates of cervical squamous cell carcinoma (CESC) patients [156]. Moreover, the results were validated using bioinformatics analysis in clinical cohort of CESC [156]. The protein and mRNA expression levels of ZC3H13, YTHDC1, and YTHDF1 were detected in further experiments [156]. The experimental results were consistent with the in-silico results, confirming their prognostic potential in CESC [156].
m6A and EC
Numerous studies have explored the participation of m6A regulators in many functions in the EC, such as cell cycle regulation and self-renewal of CSCs. The role and mechanism of m6A regulators in EC are summarized in Fig. 3 and Table 3.

In EC, m6A regulatory proteins contribute to tumorigenesis and metastasis by interacting with various RNAs. METTL14 mutation or reduced expression of METTL3 increases the proliferation and tumorigenicity of EC by activating the AKT pathway. WTAP downregulates CAV‐1 expression to activate the NF‐κB signaling pathway in EC, promoting EC progression. HIF-1α and HIF-2α activate the expression of ALKBH5 under hypoxic conditions, facilitating the SOX2 expression by demethylating the SOX2 mRNA, leading to the tumorigenesis of EC. ALKBH5 demethylates the target transcript IGF1R and enhances its mRNA stability to promote tumorigenesis and metastasis of EC. FTO promotes HOXB13 protein expression, activates the WNT signaling pathway, and promotes EC invasion and metastasis. PADI2 activates the IGF2BP1 expression and helps in maintaining the mRNA stability and expression of SOX2, thereby supporting the malignancy state of EC. IGF2BP1 recruits PABPC1 to promote PEG10 protein expression, contributing to the tumorigenesis of EC. YTHDF2 inhibits the expression of IRS1 and inhibits IRS1/AKT signaling pathway, consequently inhibiting the tumorigenicity of EC. YTHDF2-mediated LncRNA FENDRR degradation promotes cellular proliferation by elevating the SOX4 expression in EC
Role of m6A in the progression of EC
m6A writer and EC
The signal transduction network is a communication line among cells, and is used for perceiving signals, including those from the extracellular environment, and transmitting them to the downstream targets for the proper functioning and maintenance of cells. The abnormal changes in the signal transduction pathways are the important biological characteristics of the tumor cells. Current studies have shown that the alternation in the signal transduction pathways affects the cellular metabolism and immune response in the tumor cells [157, 158]. In EC, the mechanism of m6A methylation effects on tumor growth through signaling pathways has been elucidated. Liu et al. reported higher expression levels of WTAP in the EC tissues as compared to the adjacent normal tissues [159]. Their further investigations confirmed that the expression of CAV-1 was regulated by WTAP in an m6A-dependent manner [159]. They also showed that, after being regulated by WTAP, the CAV-1 could activate the downstream NF-κB pathway [159].
PI3K/AKT pathway also plays an important role in various biological processes. The dysregulation of the AKT signaling pathway has shown crucial roles in the proliferation and apoptosis of numerous tumor cells [160,161,162]. Some studies showed that the stem cells and cancer cells proliferated with the reduction in the m6A methylation [36, 163, 164]. However, other studies reported that some cancers were related to the high expression of METTL3 and increased m6A methylation, which might involve different mechanisms and require more in-depth and detailed studies [35, 165, 166].
m6A reader and EC
Researchers have shown that the dynamic m6A-modification in mRNAs, particularly in the key transcripts, might alter the physiology of cells [167]. For instance, the decreased m6A mRNA methylation could stimulate cellular proliferation by modulating the expression of the critical enzymes, which were involved in the AKT signaling pathway [168]. Hong et al. showed that the overexpressed YTHDF2 could bind to the m6A-modified insulin receptor substrate 1 (IRS1), which reduced its translation, thereby blocking the IRS1/AKT pathway [169]. In the same study, the results indicated that several vital proteins could regulate the cellular activities of EC cells through the AKT pathway; these proteins were also responsible for regulating the dynamic equilibrium of EC cells [169].
On the other hand, studies showed that YTHDF2 could also regulate the proliferation of EC cells by affecting the metabolism of lncRNA. According to Shen et al., the expression levels of lncRNA FENDRR in the EC tissues were reduced; however, its m6A methylation levels showed a negative effect trend [170]. The subsequent in-vitro experiments in the same study demonstrated that YTHDF2 could recognize the abundance of m6A-modified lncRNA FENDRR in the EC cells and degrade it [170]. After expressing the YTHDF2 gene, the expression levels of lncRNA FENDRR were restored, thereby inhibiting proliferation and stimulating the apoptosis of EC cells [170]. Furthermore, they reported that overexpressing the lncRNA FENDRR could reduce SOX4 translation and result in inhibiting EC cell proliferation and promoting cellular apoptosis [170]. These results were consistent with those of the previous study by Liu et al., which reported an adverse effect of the lncRNA FENDRR on the SOX4 expression in CRC [171].
Among the molecular mechanisms of cellular proliferation, cell cycle acceleration is of great importance, which is regulated by the CDK-cyclin complexes and cyclin-dependent kinase inhibitors [172]. A recent study reported that paternally expressed gene 10 (PEG10), a critical factor, which directly modulates the key proteins, was involved in cell cycle proteins [173]. Numerous studies have demonstrated the contributions of PEG10 to cellular proliferation; however, its mechanism has rarely been studied [174, 175]. The knockdown of the PEG10 increased the p21 and p27 expression levels [173]. Zhang et al. reported that IGF2BP1 could recognize the m6A site in the 3′-UTR of the PEG10 mRNA in EC and recruit the polyadenylate binding protein 1 (PABPC1) to stabilize PEG10 mRNA, thereby increasing its protein expression levels and accelerating the cell cycle [176].
The peptide arginine deaminases (PADIs) family contains five members, including PADI1-4 and PADI6. Except for the PADI6 which has no enzymatic activity and is expressed only in the ovary [177], other PADIs can deaminate positively charged arginine residues in substrate proteins into the neutral non-coding residues called citrulline [178, 179]. The expression of PADIs was higher in various malignant tumor tissues as compared to healthy tissues [180, 181]. Numerous studies showed that the PADIs-catalyzed protein citrullination could alter signal transduction, cell differentiation, and EMT in a variety of human cancer cells [180, 182]. Xue et al., for the first time, reported that m6A reader IGF2BP1 could be used as a downstream factor of PADI2 and could regulate it to promote tumor progression in EC [183]. They further showed that PADI2 could interact with MEK1 kinase in the MAPK pathway and catalyze the citrullination, which was beneficial for the phosphorylation of ERK1/2 by MEK1, thereby activating the expression of IGF2BP1. In addition, IGF2BP1 could also bind to the m6A site in the 3′-UTR of SOX mRNA to prevent its degradation [183]. This study revealed that the PADI2/MEK1/ERK/IGF2BP1 pathway could promote the characteristics of carcinogenic tumor cells in EC, and the combination of specific PADI2 and MEK1 inhibitors might provide a novel therapeutic target site for the treatment of the MEK inhibitor-resistant EC patients [183].
m6A eraser and EC
The insulin-like growth factor (IGF) is involved in many functions in most organs [184,185,186]. IGF1 and IGF2 can affect EC, as observed in both clinical and experimental data [187]. Both the IGF1 and IGF2 ligands can activate insulin-like growth factor 1 receptor (IGF1R), a tyrosine kinase receptor present on the cell surface, which is coupled with several intracellular secondary messenger pathways, including Ras/Raf/MAPK and PI3K/AKT signaling pathways, especially in regulating the normal uterine physiology [188]. Pu et al. reported that ALKBH5 enhanced the stability and translation of IGF1R mRNA by the demethylation of m6A and increased the protein levels of COL1A1 and MMP9, thereby promoting the tumorigenesis of EC cells [189]. However, the possibility of involving other signaling pathways cannot be excluded. Other signaling pathways may alter either directly or indirectly due to the changes in ALKBH5 and require further investigation.
The role of m6A methylation, participating in the Wnt signaling pathway by regulating the related RNAs or proteins, has been elucidated in the CC [56, 135, 136]. FTO could remove the m6A modification of HOXB13 mRNA, abolish the degradation of HOXB13 mRNA mediated by YTHDF2, promote the expression of HOXB13 protein, activate the Wnt signaling pathway, and promote the invasion and metastasis of EC [146].
Hypoxia is an important niche feature of the CSCs, positively affecting the growth of stem cells and tumor progression [190, 191]. Hypoxia-inducible factors (HIFs), including HIF-1α and HIF-2α, are the main media of hypoxia and indispensable for the activation and self-renewal of CSCs; they are strongly associated with tumors [192]. The ability of CSCs to tolerate hypoxia can be attributed to the rearrangement of genes involved in cellular multipotency and differentiation [193]. All these studies further deepen the understanding of the correlations among hypoxia, HIFs, and SOX2 [194,195,196]. Chen et al. demonstrated that the hypoxia and high levels of ALKBH5 could restore the stemness of differentiated endometrial CSCs (ECSCs) and increase the ECSC-like phenotype [197]. In addition, a recent study showed that the changes in mRNA stability were negatively correlated with the expression of these multipotency factors [65]. The m6A reader IGF2BP1 could stabilize the degradation of SOX2 mRNA in EC, thereby promoting tumor progression [183]. Similarly, Chen et al. verified that ALKBH5 could stimulate SOX2 mRNA expression by reducing its m6A methylation level, thereby increasing the stemness and carcinogenicity of ECSCs [197]. These studies revealed that the decrease in the m6A mRNA methylation in the key mRNAs might be a potential mechanism of most EC. These studies also confirmed that m6A methylation was a regulatory factor for cell growth.
m6A and immunoregulation in EC
In EC, the correlation between m6A methylation modification and TME has rarely been reported. Recently, Ma et al. analyzed the EC patients’ data from TCGA and identified the genetic changes in the m6A regulatory genes. The results showed a significant correlation between the negative changes in the m6A levels and adverse prognostic outcomes [198]. The study identified ZC3H13, METTL14, and YTHDC1 as independent prognostic factors for EC patients [198]. Noteworthy, the expression, mutation, and somatic copy number alterations (SCNAs) of these genes were associated with immune cell infiltration [198].
Prognostic effect of m6A in EC
The incidence of EC has increased over the past few decades, making it one of the most prevalent gynecologic cancers [199]. In 2020, 417,367 new cases of EC were diagnosed, causing 97,370 deaths [1]. The global incidence rate of EC is continuously increasing, while those of several other types of cancer have decreased over the past two decades [200,201,202,203,204]. Despite the better prognosis of EC than that of CC and OC, screening for the high-risk parts of EC patients is imperative due to more likeliness of developing advanced cancer and early death. With the advancements in the studies on m6A methylation modification in EC, numerous research groups have reported the prognostic potential of m6A regulatory factors and m6A-related genes in EC (Table 4). In a recent study, Zhai et al. analyzed the TCGA dataset of EC patients and established a risk model based on the m6A regulators, particularly FTO, RBM15, and YTHDF1, and revealed their crucial roles in the development and prognosis of EC [205]. Further analysis of the datasets showed that FTO and RBM15 could affect the survival of EC patients by regulating the m6A-associated genes involved in the development of connective tissue, catabolism, RNA stability, oxidative demethylation, temperature balance, and energetic metabolism [205]. Zhang et al. established a reliable protective model based on seven significant CpG sites located in the m6A regulators. The model could effectively predict the prognosis of EC, indicating that the CpG sites might be advantageous in predicting the EC [206]. Three m6A-associated lncRNAs revealed by Shi et al. that, in contrast to the TRAF3IP2-AS1, AL133243.2, the patients with the high SCARNA9 expression tended to have a worse prognosis [207]. These lncRNAs were verified to participate in the development of endometriosis by modulating m6A-related enzymes, suggesting that these RNAs might be associated with the diagnosis and treatment of EC [207].
Conclusions and perspectives
In this review, the studies on the m6A methylation modification in OC, CC, and EC were summarized from the aspects of tumor development, immune microenvironment, and prognosis. From the existing studies, it was concluded that the abnormal expression of the m6A regulator in gynecological tumors might lead to an increase or decrease in the m6A modification level of RNA. The m6A modifications of RNA might affect the fate of various RNAs and lead to the proliferation, invasion, and metastasis of tumors as well as also alter the tumor immune microenvironment of the patients, thereby participating in the occurrence and development of tumors.
The advancements in medical technology have greatly improved the survival rate of gynecologic cancer patients than before. However, due to the lack of specific biomarkers, early diagnosis is still challenging. At the same time, the resistance to the anti-tumor drugs in some patients also urges researchers to deepen the understanding of tumorigenesis and identify new immune targets for the development of anti-tumor drugs. This review summarized the results of numerous studies, which showed that the m6A regulator and related genes could be used as potential biomarkers or prognostic indicators for the early diagnosis of gynecologic cancers. Studying the mechanism of the m6A regulator in tumor development also supported this view. The role of m6A modification in the drug resistance mechanism has also been reported, which showed that the inhibitors of m6A regulators might have the potential of being used as anti-tumor drugs in drug-resistant patients.
At present, numerous studies have reported the mechanism of m6A-promoting effects on the development of gynecologic cancer; however, the knowledge is still insufficient for a deeper understanding of tumorigenesis. The mechanisms, explaining the upregulation of the m6A regulator in gynecologic cancer and their relationship with oncogenes, are still unclear. Therefore, further studies are needed in the future to explain these mechanisms in order to develop effective therapeutic strategies.
Availability of data and materials
Not applicable.
Abbreviations
- 3′-UTR:
-
3′-Untrasnlated region
- 5′-UTR:
-
5′-Untranslated region
- ACIN1:
-
Apoptosis chromatin condensation inducer 1
- ALKBH5:
-
AlkB homolog 5
- AML:
-
Acute myeloid leukemia
- AXL:
-
AXL receptor tyrosine kinase
- CC:
-
Cervical cancer
- CDC42EP3:
-
Cell division cycle 42 effector protein 3
- ceRNA:
-
Competing endogenous RNA
- CESC:
-
Cervical squamous cell carcinoma
- circRNAs:
-
Circular RNAs
- CNV:
-
Copy number variation
- CRC:
-
Colorectal cancer
- CSC:
-
Cancer stem cell
- EC:
-
Endometrial cancer
- ECSCs:
-
Endometrial cancer stem cells
- EMT:
-
Epithelial–mesenchymal transition
- ERCC1:
-
Excision repair cross-complementation group 1
- FAM76A:
-
Family with sequence similarity 76 member A
- FBW7:
-
F-box and WD repeat domain-containing 7
- FTO:
-
Fat mass and obesity-associated protein
- FZD10:
-
Frizzled class receptor 10
- HBS1L:
-
HBS1 like translational GTPase
- HCC:
-
Hepatocellular carcinoma
- HGSOC:
-
High-grade serous ovarian cancer
- HIFs:
-
Hypoxia-inducible factors
- HOXA10:
-
Homeobox A10
- HPV:
-
Human papillomavirus
- IGF:
-
Insulin like growth factor
- IGF1R:
-
Insulin-like growth factor 1 receptor
- IGF2BPs:
-
Insulin-like growth factor 2 mRNA-binding proteins
- IRS1:
-
Insulin receptor substrate 1
- JAK2:
-
Janus kinase 2
- lncRNAs:
-
Long noncoding RNAs
- LSD1:
-
Lysine-specific demethylase 1
- m6A:
-
N6-methyladenosine
- MDSCs:
-
Myeloid-derived suppressor cells
- METTL14:
-
Methyltransferase-like 14
- METTL3:
-
Methyltransferase-like 3
- miRNAs:
-
MicroRNAs
- mRNA:
-
Messenger RNAs
- mTOR:
-
Mammalian target of the rapamycin
- NANOG:
-
Nanog homeobox
- ncRNA:
-
Non-coding RNAs
- OC:
-
Ovarian cancer
- PABPC1:
-
Polyadenylate binding protein 1
- PADIs:
-
Peptide arginine deaminases
- PARPi:
-
PARP Inhibitor
- PD-1:
-
Programmed death-1
- PDK4:
-
Pyruvate dehydrogenase kinase 4
- PD-L1:
-
Programmed death ligand 1
- PD-L2:
-
Programmed death ligand 2
- PEG10:
-
Paternally expressed gene 10
- PI3K:
-
Phosphoinositide 3-kinase
- piRNA:
-
Piwi-interacting RNA
- PTEN:
-
Phosphatase and tensin homolog
- RBM15:
-
RNA binding pattern protein 15
- rRNA:
-
Ribosomal RNAs
- SCNAs:
-
Somatic copy number changes
- SOX2:
-
SRY-box transcription factor 2
- SOX4:
-
SRY-related HMG box transcription factor 4
- STAT3:
-
Signal transducer and activator of transcription 3
- TCGA:
-
The cancer genome atlas
- TICs:
-
Tumor-infiltrating immune cells
- TLR4:
-
Toll-like receptor 4
- TME:
-
Tumor microenvironment
- TRIM29:
-
Tripartite motif containing 29
- TROAP:
-
Trophinin associated protein
- VIRMA/KIAA1429:
-
Vir-like m6A methyltransferase-associated protein
- WTAP:
-
Wilm’s tumor-associated protein
- YTHDC1:
-
YTH domain containing 1
- YTHDF1:
-
YTH N6-methyladenosine RNA binding protein 1
- YTHDF2:
-
YTH N6-methyladenosine RNA binding protein 2
- ZC3H13:
-
Zinc finger CCCH domain-containing protein 13
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Yi M, Li TY, Niu MK, Luo SX, Chu Q, Wu KM. Epidemiological trends of women’s cancers from 1990 to 2019 at the global, regional, and national levels: a population-based study. Biomark Res. 2021;9(1):12.
Cao K, Du Y, Bao X, Han M, Su R, Pang J, et al. Glutathione-bioimprinted nanoparticles targeting of N6-methyladenosine FTO demethylase as a strategy against leukemic stem cells. Small. 2022;18(13): e2106558.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593(7860):597–601.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38(1):79-96.e11.
Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1(1):37–42.
Gilbert Wendy V, Bell Tristan A, Schaening C. Messenger RNA modifications: form, distribution, and function. Science. 2016;352(6292):1408–12.
Lipshitz HD, Claycomb JM, Smibert CA. Post-transcriptional regulation of gene expression. Methods. 2017;126:1–2.
Roignant J-Y, Soller M. m6A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. 2017;33(6):380–90.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15(5):293–306.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Coker H, Wei G, Brockdorff N. m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim Biophys Acta. 2019;1862(3):310–8.
Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue Q, et al. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. 2017;482(4):582–9.
Wu J, Guo X, Wen Y, Huang S, Yuan X, Tang L, et al. N6-methyladenosine modification opens a new chapter in circular RNA biology. Front Cell Dev Biol. 2021. https://doi.org/10.3389/fcell.2021.709299.
Ma S, Chen C, Ji X, Liu JB, Zhou QB, Wang GX, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):15.
Huang W, Chen T-Q, Fang K, Zeng Z-C, Ye H, Chen Y-Q. N6-methyladenosine methyltransferases: functions, regulation, and clinical potential. J Hematol Oncol. 2021. https://doi.org/10.1186/s13045-021-01129-8.
Ear J, Lin S. RNA methylation regulates hematopoietic stem and progenitor cell development. J Genet Genomics. 2017;44(10):473–4.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, et al. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540(7632):301–4.
Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42(22):13911–9.
Wang X, He C. Reading RNA methylation codes through methyl-specific binding proteins. RNA Biol. 2014;11(6):669–72.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–9.
Zhu T, Roundtree IA, Wang P, Wang X, Wang L, Sun C, et al. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014;24(12):1493–6.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–7.
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151(2):356–65.
Ma Z, Li Q, Liu P, Dong W, Zuo Y. METTL3 regulates m6A in endometrioid epithelial ovarian cancer independently of METTl14 and WTAP. Cell Biol Int. 2020;44(12):2524–31.
Liang S, Guan H, Lin X, Li N, Geng F, Li J. METTL3 serves an oncogenic role in human ovarian cancer cells partially via the AKT signaling pathway. Oncol Lett. 2020;19(4):3197–204.
Bi X, Lv X, Liu D, Guo H, Yao G, Wang L, et al. METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther. 2021;28(3–4):335–49.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–45.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Li X, Tang J, Huang W, Wang F, Li P, Qin C, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8(56):96103–16.
Wang J, Ding W, Xu Y, Tao E, Mo M, Xu W, et al. Long non-coding RNA RHPN1-AS1 promotes tumorigenesis and metastasis of ovarian cancer by acting as a ceRNA against miR-596 and upregulating LETM1. Aging. 2020;12(5):4558–72.
Luo XY, Cao MD, Gao F, He XX. YTHDF1 promotes hepatocellular carcinoma progression via activating PI3K/AKT/mTOR signaling pathway and inducing epithelial-mesenchymal transition. Exp Hematol Oncol. 2021;10(1):14.
Gong PJ, Shao YC, Yang Y, Song WJ, He X, Zeng YF, et al. Analysis of N6-methyladenosine methyltransferase reveals METTL14 and ZC3H13 as tumor suppressor genes in breast cancer. Front Oncol. 2020. https://doi.org/10.3389/fonc.2020.578963.
Li Y, Peng H, Jiang P, Zhang J, Zhao Y, Feng X, et al. Downregulation of methyltransferase-like 14 promotes ovarian cancer cell proliferation through stabilizing TROAP mRNA. Front Oncol. 2022;12: 824258.
Wu LS, Qian JY, Wang M, Yang H. Identifying the role of Wilms tumor 1 associated protein in cancer prediction using integrative genomic analyses. Mol Med Rep. 2016;14(3):2823–31.
Barbolina MV, Adley BP, Shea LD, Stack MS. Wilms tumor gene protein 1 is associated with ovarian cancer metastasis and modulates cell invasion. Cancer. 2008;112(7):1632–41.
Fu Y, Jia XC. WTAP-mediated N6-methyladenosine modification on EGR3 in different types of epithelial ovarian cancer. J Biol Regul Homeost Agents. 2020;34(4):1505–12.
Wang J, Xu J, Li K, Huang Y, Dai Y, Xu C, et al. Identification of WTAP-related genes by weighted gene co-expression network analysis in ovarian cancer. J Ovarian Res. 2020. https://doi.org/10.1186/s13048-020-00710-y.
Xu F, Li J, Ni M, Cheng J, Zhao H, Wang S, et al. FBW7 suppresses ovarian cancer development by targeting the N(6)-methyladenosine binding protein YTHDF2. Mol Cancer. 2021;20(1):45.
Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47(1):375–90.
Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y, et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020;48(7):3816–31.
Li J, Wu L, Pei M, Zhang Y. YTHDF2, a protein repressed by miR-145, regulates proliferation, apoptosis, and migration in ovarian cancer cells. J Ovarian Res. 2020;13(1):111.
Hao L, Wang JM, Liu BQ, Yan J, Li C, Jiang JY, et al. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochim Biophys Acta Mol Cell Res. 2021;1868(1): 118878.
Xu C, Liu K, Ahmed H, Loppnau P, Schapira M, Min J. Structural basis for the discriminative recognition of N-6-methyladenosine RNA by the human YT521-B homology domain family of proteins. J Biol Chem. 2015;290(41):24902–13.
Wang Y, Chen Z. Long noncoding RNA UBA6-AS1 inhibits the malignancy of ovarian cancer cells via suppressing the decay of UBA6 mRNA. Bioengineered. 2022;13(1):178–89.
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, et al. MicroRNA-145 modulates N-6-methyladenosine levels by targeting the 3′-untranslated mRNA region of the N-6-methyladenosine binding YTH domain family 2 protein. J Biol Chem. 2017;292(9):3614–23.
Huang H, Wang Y, Kandpal M, Zhao G, Cardenas H, Ji Y, et al. FTO-dependent N (6)-methyladenosine modifications inhibit ovarian cancer stem cell self-renewal by blocking cAMP signaling. Cancer Res. 2020;80(16):3200–14.
Jiang Y, Wan Y, Gong M, Zhou S, Qiu J, Cheng W. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-kappaB pathway. J Cell Mol Med. 2020;24(11):6137–48.
Nie S, Zhang L, Liu J, Wan Y, Jiang Y, Yang J, et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J Exp Clin Cancer Res. 2021;40(1):284.
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, et al. N(6)-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019;79(11):2812–20.
Zhang Z, Zhu H, Hu J. CircRAB11FIP1 promoted autophagy flux of ovarian cancer through DSC1 and miR-129. Cell Death Dis. 2021;12(2):219.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–94.
Gulati P, Yeo GSH. The biology of FTO: from nucleic acid demethylase to amino acid sensor. Diabetologia. 2013;56(10):2113–21.
Gerken T, Girard CA, Tung YCL, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318(5855):1469–72.
Li ZJ, Weng HY, Su R, Weng XC, Zuo ZX, Li CY, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N-6-methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41.
Su R, Dong L, Li CY, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172(1–2):90.
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12(2):133–43.
Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–20.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113(14):E2047–56.
Du WW, Yang W, Li X, Awan FM, Yang Z, Fang L, et al. A circular RNA circ-DNMT1 enhances breast cancer progression by activating autophagy. Oncogene. 2018;37(44):5829–42.
Zhang N, Zhang X, Xu W, Zhang X, Mu Z. CircRNA_103948 inhibits autophagy in colorectal cancer in a ceRNA manner. Ann N Y Acad Sci. 2021;1503(1):88–101.
Xu P, Zhang X, Cao J, Yang J, Chen Z, Wang W, et al. The novel role of circular RNA ST3GAL6 on blocking gastric cancer malignant behaviours through autophagy regulated by the FOXP2/MET/mTOR axis. Clin Transl Med. 2022;12(1): e707.
Shen H, Wang G-C, Li X, Ge X, Wang M, Shi Z-M, et al. S6K1 blockade overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Oncogene. 2020;39(49):7181–95.
Thompson PA, Eam B, Young NP, Fish S, Chen J, Barrera M, et al. Targeting oncogene mRNA translation in B-cell malignancies with eFT226, a potent and selective inhibitor of eIF4A. Mol Cancer Ther. 2021;20(1):26–36.
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–83.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
Torphy RJ, Schulick RD, Zhu Y. Understanding the immune landscape and tumor microenvironment of pancreatic cancer to improve immunotherapy. Mol Carcinog. 2020;59(7):775–82.
Zahn LM. Effects of the tumor microenvironment. Science. 2017;355(6332):1386–8.
Pelly VS, Moeini A, Roelofsen LM, Bonavita E, Bell CR, Hutton C, et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 2021;11(10):2602–19.
Too NSH, Ho NCW, Adine C, Iyer NG, Fong ELS. Hot or cold: bioengineering immune contextures into in vitro patient-derived tumor models. Adv Drug Deliv Rev. 2021;175: 113791.
Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. 2021;11(11):5365–86.
Zhao JX, Lu LG. Interplay between RNA methylation eraser FTO and writer METTL3 in renal clear cell carcinoma patient survival. Recent Pat Anticancer Drug Discov. 2021;16(3):363–76.
Wang Q, Zhang Q, Li Q, Zhang J, Zhang J. Clinicopathological and immunological characterization of RNA m(6) A methylation regulators in ovarian cancer. Mol Genet Genomic Med. 2021;9(1): e1547.
Yan Y, Liang Q, Xu Z, Yi Q. Integrative bioinformatics and experimental analysis revealed down-regulated CDC42EP3 as a novel prognostic target for ovarian cancer and its roles in immune infiltration. PeerJ. 2021;9: e12171.
Gu J, Bi F. Significance of N6-methyladenosine RNA methylation regulators in immune infiltrates of ovarian cancer. Front Genet. 2021;12: 671179.
Prat J. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int J Gynaecol Obstet. 2014;124(1):1–5.
Wei Q, Yang D, Liu X, Zhao H, Yang Y, Xu J, et al. Exploration of the role of m(6)A RNA methylation regulators in malignant progression and clinical prognosis of ovarian cancer. Front Genet. 2021;12: 650554.
Han X, Liu J, Cheng G, Cui S. Gene signatures and prognostic values of m6A RNA methylation regulators in ovarian cancer. Cancer Control. 2020;27(1):1073274820960460.
Zhang C, Liu J, Guo H, Hong D, Ji J, Zhang Q, et al. m6A RNA methylation regulators were associated with the malignancy and prognosis of ovarian cancer. Bioengineered. 2021;12(1):3159–76.
Fan L, Lin Y, Lei H, Shu G, He L, Yan Z, et al. A newly defined risk signature, consisting of three m(6)A RNA methylation regulators, predicts the prognosis of ovarian cancer. Aging. 2020;12(18):18453–75.
Li Q, Ren CC, Chen YN, Yang L, Zhang F, Wang BJ, et al. A risk score model incorporating three m6A RNA methylation regulators and a related network of miRNAs-m6A regulators-m6A target genes to predict the prognosis of patients with ovarian cancer. Front Cell Dev Biol. 2021;9: 703969.
Zhu W, Zhao L, Kong B, Liu Y, Zou X, Han T, et al. The methylation modification of m6A regulators contributes to the prognosis of ovarian cancer. Ann Transl Med. 2022;10(2):59.
Jiao J, Jiang L, Luo Y. N6-methyladenosine-related RNA signature predicting the prognosis of ovarian cancer. Recent Pat Anticancer Drug Discov. 2021;16(3):407–16.
Yu H-L, Ma X-D, Tong J-F, Li J-Q, Guan X-J, Yang J-H. WTAP is a prognostic marker of high-grade serous ovarian cancer and regulates the progression of ovarian cancer cells. OncoTargets Ther. 2019;12:6191–201.
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–81.
St Laurent G, Wahlestedt C, Kapranov P. The landscape of long noncoding RNA classification. Trends Genet. 2015;31(5):239–51.
Nie X, Tan J. N6-methyladenosine-related lncRNAs is a potential marker for predicting prognosis and immunotherapy in ovarian cancer. Hereditas. 2022;159(1):17.
Rigoutsos I. New tricks for animal microRNAS: targeting of amino acid coding regions at conserved and nonconserved sites. Cancer Res. 2009;69(8):3245–8.
O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10(2):111–22.
Tornesello ML, Faraonio R, Buonaguro L, Annunziata C, Starita N, Cerasuolo A, et al. The role of microRNAs, long non-coding RNAs, and circular RNAs in cervical cancer. Front Oncol. 2020;10:150.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66.
Huang C, Liang J, Lin S, Wang D, Xie Q, Lin Z, et al. N(6)-methyladenosine associated silencing of miR-193b promotes cervical cancer aggressiveness by targeting CCND1. Front Oncol. 2021;11: 666597.
Su C, Zhang Y, Chen P, Yang W, Du J, Zhang D. Methyltransferase-like 3 induces the development of cervical cancer by enhancing insulin-like growth factor 2 mRNA-binding proteins 3-mediated apoptotic chromatin condensation inducer 1 mRNA stability. Bioengineered. 2022;13(3):7034–48.
Li J, Xie G, Tian Y, Li W, Wu Y, Chen F, et al. RNA m(6)A methylation regulates dissemination of cancer cells by modulating expression and membrane localization of β-catenin. Mol Ther. 2022. https://doi.org/10.1016/j.ymthe.2022.01.019.
Ninova M, Fejes TK. New players on the piRNA field. Nat Struct Mol Biol. 2020;27(9):777–9.
Wang X, Lv C, Guo Y, Yuan S. Mitochondria associated germinal structures in spermatogenesis: piRNA pathway regulation and beyond. Cells. 2020. https://doi.org/10.3390/cells9020399.
Liu Y, Dou M, Song X, Dong Y, Liu S, Liu H, et al. The emerging role of the piRNA/piwi complex in cancer. Mol Cancer. 2019;18(1):123.
Zeng Q, Wan H, Zhao S, Xu H, Tang T, Oware KA, et al. Role of PIWI-interacting RNAs on cell survival: Proliferation, apoptosis, and cycle. IUBMB Life. 2020;72(9):1870–8.
Xie Q, Li Z, Luo X, Wang D, Zhou Y, Zhao J, et al. piRNA-14633 promotes cervical cancer cell malignancy in a METTL14-dependent m6A RNA methylation manner. J Transl Med. 2022;20(1):51.
Chen Z, Ling K, Zhu Y, Deng L, Li Y, Liang Z. circ0000069 promotes cervical cancer cell proliferation and migration by inhibiting miR-4426. Biochem Biophys Res Commun. 2021;551:114–20.
Xue L, Li J, Lin Y, Liu D, Yang Q, Jian J, et al. m(6) A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J Cell Physiol. 2021;236(4):2649–58.
Guo T, Liu DF, Peng SH, Xu AM. ALKBH5 promotes colon cancer progression by decreasing methylation of the lncRNA NEAT1. Am J Transl Res. 2020;12(8):4542–9.
Ji F, Lu Y, Chen S, Lin X, Yu Y, Zhu Y, et al. m(6)A methyltransferase METTL3-mediated lncRNA FOXD2-AS1 promotes the tumorigenesis of cervical cancer. Mol Ther Oncolytics. 2021;22:574–81.
Yang Z, Ma J, Han S, Li X, Guo H, Liu D. ZFAS1 exerts an oncogenic role via suppressing miR-647 in an m(6)A-dependent manner in cervical cancer. Onco Targets Ther. 2020;13:11795–806.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Spencer NY, Stanton RC. The Warburg effect, Lactate, and nearly a century of trying to cure cancer. Semin Nephrol. 2019;39(4):380–93.
Thakur C, Chen F. Connections between metabolism and epigenetics in cancers. Semin Cancer Biol. 2019;57:52–8.
Schwartz L, Supuran CT, Alfarouk KO. The Warburg effect and the hallmarks of cancer. Anticancer Agents Med Chem. 2017;17(2):164–70.
Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J, et al. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat Commun. 2020;11(1):2578.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112.
Wang Q, Guo X, Li L, Gao Z, Su X, Ji M, et al. N(6)-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis. 2020;11(10):911.
Zhuang M, Li X, Zhu J, Zhang J, Niu F, Liang F, et al. The m6A reader YTHDF1 regulates axon guidance through translational control of Robo31 expression. Nucleic Acids Res. 2019;47(9):4765–77.
Wang H, Luo Q, Kang J, Wei Q, Yang Y, Yang D, et al. YTHDF1 aggravates the progression of cervical cancer through m(6)A-mediated up-regulation of RANBP2. Front Oncol. 2021;11: 650383.
Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393–407.
Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21(5):542–51.
Zeng F, Wang Q, Wang S, Liang S, Huang W, Guo Y, et al. Linc00173 promotes chemoresistance and progression of small cell lung cancer by sponging miR-218 to regulate Etk expression. Oncogene. 2020;39(2):293–307.
Huang Y, Sun H, Ma X, Zeng Y, Pan Y, Yu D, et al. HLA-F-AS1/miR-330-3p/PFN1 axis promotes colorectal cancer progression. Life Sci. 2020;254: 117180.
Zhang Y, Wang D, Wu D, Zhang D, Sun M. Long noncoding RNA KCNMB2-AS1 stabilized by N(6)-methyladenosine modification promotes cervical cancer growth through acting as a competing endogenous RNA. Cell Transplant. 2020;29:963689720964382.
Wang X, Zhang J, Wang Y. Long noncoding RNA GAS5-AS1 suppresses growth and metastasis of cervical cancer by increasing GAS5 stability. Am J Transl Res. 2019;11(8):4909–21.
Ji F, Lu Y, Chen S, Yu Y, Lin X, Zhu Y, et al. IGF2BP2-modified circular RNA circARHGAP12 promotes cervical cancer progression by interacting m(6)A/FOXM1 manner. Cell Death Discov. 2021;7(1):215.
Fontham ETH, Wolf AMD, Church TR, Etzioni R, Flowers CR, Herzig A, et al. Cervical cancer screening for individuals at average risk: 2020 guideline update from the American Cancer Society. CA Cancer J Clin. 2020;70(5):321–46.
Eun TJ, Perkins RB. Screening for cervical cancer. CA Cancer J Clin. 2020;70(5):347–8.
Shamseddine AA, Burman B, Lee NY, Zamarin D, Riaz N. Tumor immunity and immunotherapy for HPV-Related cancers. Cancer Discov. 2021;11(8):1896–912.
Yuan Y, Cai X, Shen F, Ma F. HPV post-infection microenvironment and cervical cancer. Cancer Lett. 2021;497:243–54.
Ilhan ZE, Łaniewski P, Thomas N, Roe DJ, Chase DM, Herbst-Kralovetz MM. Deciphering the complex interplay between microbiota, HPV, inflammation and cancer through cervicovaginal metabolic profiling. EBioMedicine. 2019;44:675–90.
Hu C, Liu T, Han C, Xuan Y, Jiang D, Sun Y, et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m(6)A-MYC expression. Int J Biol Sci. 2022;18(2):507–21.
Zeng Q, Chen J, Li Y, Werle KD, Zhao RX, Quan CS, et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene. 2017;36(9):1245–55.
Wang T, Li W, Ye B, Zhang S, Lei X, Zhang D. FTO-stabilized lncRNA HOXC13-AS epigenetically upregulated FZD6 and activated Wnt/β-catenin signaling to drive cervical cancer proliferation, invasion, and EMT. J buon. 2021;26(4):1279–91.
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol Carcinog. 2018;57(5):590–7.
Zou D, Dong L, Li C, Yin Z, Rao S, Zhou Q. The m(6)A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019;19:321.
Datta NR, Stutz E, Liu M, Rogers S, Klingbiel D, Siebenhuner A, et al. Concurrent chemoradiotherapy vs. radiotherapy alone in locally advanced cervix cancer: a systematic review and meta-analysis. Gynecol Oncol. 2017;145(2):374–85.
Petrelli F, De Stefani A, Raspagliesi F, Lorusso D, Barni S. Radiotherapy with concurrent cisplatin-based doublet or weekly cisplatin for cervical cancer: a systematic review and meta-analysis. Gynecol Oncol. 2014;134(1):166–71.
Tewari KS, Monk BJ. New strategies in advanced cervical cancer: from angiogenesis blockade to immunotherapy. Clin Cancer Res. 2014;20(21):5349–58.
Wieringa HW, van der Zee AGJ, de Vries EGE, van Vugt MATM. Breaking the DNA damage response to improve cervical cancer treatment. Cancer Treat Rev. 2016;42:30–40.
Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472.
Meng J, Li P, Zhang Q, Yang Z, Fu S. A radiosensitivity gene signature in predicting glioma prognostic via EMT pathway. Oncotarget. 2014;5(13):4683–93.
Yao Y-H, Cui Y, Qiu X-N, Zhang L-Z, Zhang W, Li H, et al. Attenuated LKB1-SIK1 signaling promotes epithelial-mesenchymal transition and radioresistance of non-small cell lung cancer cells. Chin J Cancer. 2016. https://doi.org/10.1186/s40880-016-0113-3.
Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525.
Zhang L, Wan Y, Zhang Z, Jiang Y, Lang J, Cheng W, et al. FTO demethylates m6A modifications in HOXB13 mRNA and promotes endometrial cancer metastasis by activating the WNT signalling pathway. RNA Biol. 2021;18(9):1265–78.
Wu L, Liu H, Guo H, Wu Q, Yu S, Qin Y, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in cervical carcinoma patients. Oncol Lett. 2018;15(6):9507–15.
Liu Y, Zheng P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci. 2020;41(1):4–12.
Tan Z, Liu L, Chiu MS, Cheung KW, Yan CW, Yu Z, et al. Virotherapy-recruited PMN-MDSC infiltration of mesothelioma blocks antitumor CTL by IL-10-mediated dendritic cell suppression. Oncoimmunology. 2019;8(1): e1518672.
Ni HH, Zhang L, Huang H, Dai SQ, Li J. Connecting METTL3 and intratumoural CD33(+) MDSCs in predicting clinical outcome in cervical cancer. J Transl Med. 2020;18(1):393.
Bachy E, Coiffier B. Anti-PD1 antibody: a new approach to treatment of lymphomas. Lancet Oncology. 2014;15(1):7–8.
Gunturi A, McDermott DF. Potential of new therapies like anti-PD1 in kidney cancer. Curr Treat Options Oncol. 2014;15(1):137–46.
Zhang H, Kong W, Zhao X, Han C, Liu T, Li J, et al. N6-Methyladenosine-related lncRNAs as potential biomarkers for predicting prognoses and immune responses in patients with cervical cancer. BMC Genom Data. 2022;23(1):8.
Wang X, Li Z, Kong B, Song C, Cong J, Hou J, et al. Reduced m(6)A mRNA methylation is correlated with the progression of human cervical cancer. Oncotarget. 2017;8(58):98918–30.
Ma X, Li Y, Wen J, Zhao Y. m6A RNA methylation regulators contribute to malignant development and have a clinical prognostic effect on cervical cancer. Am J Transl Res. 2020;12(12):8137–46.
Pan J, Xu L, Pan H. Development and validation of an m6A RNA methylation regulator-based signature for prognostic prediction in cervical squamous cell carcinoma. Front Oncol. 2020;10:1444.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20(2):74–88.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501.
Li Q, Wang C, Dong W, Su Y, Ma Z. WTAP facilitates progression of endometrial cancer via CAV-1/NF-κB axis. Cell Biol Int. 2021;45(6):1269–77.
Nath S, Mandal C, Chatterjee U, Mandal C. Association of cytosolic sialidase Neu2 with plasma membrane enhances Fas-mediated apoptosis by impairing PI3K-Akt/mTOR-mediated pathway in pancreatic cancer cells. Cell Death Dis. 2018;9(2):210.
Liu JS, Huo CY, Cao HH, Fan CL, Hu JY, Deng LJ, et al. Aloperine induces apoptosis and G2/M cell cycle arrest in hepatocellular carcinoma cells through the PI3K/Akt signaling pathway. Phytomedicine. 2019;61: 152843.
Liu H, Deng H, Zhao Y, Li C, Liang Y. LncRNA XIST/miR-34a axis modulates the cell proliferation and tumor growth of thyroid cancer through MET-PI3K-AKT signaling. J Exp Clin Cancer Res. 2018;37(1):279.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591-606.e6.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6)-methyladenosine-dependent primary microRNA processing. Hepatology. 2017;65(2):529–43.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22(2):191-205.e9.
Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24(12):1403–19.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20(9):1074–83.
Hong L, Pu X, Gan H, Weng L, Zheng Q. YTHDF2 inhibit the tumorigenicity of endometrial cancer via downregulating the expression of IRS1 methylated with m(6)A. J Cancer. 2021;12(13):3809–18.
Shen J, Feng XP, Hu RB, Wang H, Wang YL, Qian JH, et al. N-methyladenosine reader YTHDF2-mediated long noncoding RNA FENDRR degradation promotes cell proliferation in endometrioid endometrial carcinoma. Lab Invest. 2021;101(6):775–84.
Liu J, Du W. LncRNA FENDRR attenuates colon cancer progression by repression of SOX4 protein. Onco Targets Ther. 2019;12:4287–95.
Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34(1):9–20.
Akamatsu S, Wyatt AW, Lin D, Lysakowski S, Zhang F, Kim S, et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;12(6):922–36.
Wang D, Zhao J, Li S, Wei J, Nan L, Mallampalli RK, et al. Phosphorylated E2F1 is stabilized by nuclear USP11 to drive Peg10 gene expression and activate lung epithelial cells. J Mol Cell Biol. 2018;10(1):60–73.
Massagué J. TGFbeta in cancer. Cell. 2008;134(2):215–30.
Zhang L, Wan Y, Zhang Z, Jiang Y, Gu Z, Ma X, et al. IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics. 2021;11(3):1100–14.
Raijmakers R, Zendman AJ, Egberts WV, Vossenaar ER, Raats J, Soede-Huijbregts C, et al. Methylation of arginine residues interferes with citrullination by peptidylarginine deiminases in vitro. J Mol Biol. 2007;367(4):1118–29.
Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. BioEssays. 2003;25(11):1106–18.
Yuzhalin AE. Citrullination in cancer. Cancer Res. 2019;79(7):1274–84.
Wang L, Song G, Zhang X, Feng T, Pan J, Chen W, et al. PADI2-mediated citrullination promotes prostate cancer progression. Cancer Res. 2017;77(21):5755–68.
Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer. 2009;9:40.
Deplus R, Denis H, Putmans P, Calonne E, Fourrez M, Yamamoto K, et al. Citrullination of DNMT3A by PADI4 regulates its stability and controls DNA methylation. Nucleic Acids Res. 2014;42(13):8285–96.
Xue T, Liu X, Zhang M, Qiukai E, Liu S, Zou M, et al. PADI2-catalyzed MEK1 citrullination activates ERK1/2 and promotes IGF2BP1-mediated SOX2 mRNA stability in endometrial cancer. Adv Sci. 2021;8(6):2002831.
Forbes K, Westwood M. The IGF axis and placental function. A mini review. Horm Res. 2008;69(3):129–37.
Díaz E, Cárdenas M, Ariza AC, Larrea F, Halhali A. Placental insulin and insulin-like growth factor I receptors in normal and preeclamptic pregnancies. Clin Biochem. 2005;38(3):243–7.
Dobolyi A, Lékó AH. The insulin-like growth factor-1 system in the adult mammalian brain and its implications in central maternal adaptation. Front Neuroendocrinol. 2019;52:181–94.
Bruchim I, Sarfstein R, Werner H. The IGF hormonal network in endometrial cancer: functions, regulation, and targeting approaches. Front Endocrinol. 2014;5:76.
Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107(6):873–7.
Pu X, Gu Z, Gu Z. ALKBH5 regulates IGF1R expression to promote the proliferation and tumorigenicity of endometrial cancer. J Cancer. 2020;11(19):5612–22.
Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129(3):465–72.
Najafi M, Farhood B, Mortezaee K, Kharazinejad E, Majidpoor J, Ahadi R. Hypoxia in solid tumors: a key promoter of cancer stem cell (CSC) resistance. J Cancer Res Clin Oncol. 2020;146(1):19–31.
Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. Embo J. 2017;36(3):252–9.
Fuchs Q, Pierrevelcin M, Messe M, Lhermitte B, Blandin AF, Papin C, et al. Hypoxia inducible factors’ signaling in pediatric high-grade gliomas: role modelization and innovative targeted approaches. Cancers. 2020. https://doi.org/10.3390/cancers12040979.
Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511(7508):246–50.
Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71(13):4640–52.
Bae KM, Dai Y, Vieweg J, Siemann DW. Hypoxia regulates SOX2 expression to promote prostate cancer cell invasion and sphere formation. Am J Cancer Res. 2016;6(5):1078–88.
Chen G, Liu B, Yin S, Li S, Guo Y, Wang M, et al. Hypoxia induces an endometrial cancer stem-like cell phenotype via HIF-dependent demethylation of SOX2 mRNA. Oncogenesis. 2020;9(9):81.
Ma J, Yang D, Ma XX. Immune infiltration-related N6-methyladenosine RNA methylation regulators influence the malignancy and prognosis of endometrial cancer. Aging. 2021;13(12):16287–315.
Lu KH, Broaddus RR. Endometrial cancer. N Engl J Med. 2020;383(21):2053–64.
Zhang S, Gong TT, Liu FH, Jiang YT, Sun H, Ma XX, et al. Global, regional, and national burden of endometrial cancer, 1990–2017: results from the global burden of disease study, 2017. Front Oncol. 2019. https://doi.org/10.3389/fonc.2019.01440.
Mullins MA, Cote ML. Beyond obesity: the rising incidence and mortality rates of uterine corpus cancer. J Clin Oncol. 2019;37(22):1851.
Lortet-Tieulent J, Ferlay J, Bray F, Jemal A. International patterns and trends in endometrial cancer incidence, 1978–2013. J Natl Cancer Inst. 2018;110(4):354–61.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Sheikh MA, Althouse AD, Freese KE, Soisson S, Edwards RP, Welburn S, et al. USA endometrial cancer projections to 2030: should we be concerned? Future Oncol. 2014;10(16):2561–8.
Zhai J, Li S, Li Y, Du Y. Data mining analysis of the prognostic impact of N(6)-methyladenosine regulators in patients with endometrial adenocarcinoma. J Cancer. 2021;12(15):4729–38.
Zhang X, Pang X, Huang Y, Qian S. A seven-m6A regulator-related CpG site-based prognostic signature for endometrial carcinoma. Medicine. 2021;100(29): e26648.
Shi R, Wang Z, Zhang J, Yu Z, An L, Wei S, et al. N6-Methyladenosine-related long noncoding RNAs as potential prognosis biomarkers for endometrial cancer. Int J Gen Med. 2021;14:8249–62.
Acknowledgements
The authors would like to thank BMCSCI (www.bmcscience.com) for providing English editing services during the preparation of this manuscript.
Funding
This work was supported by the National Nature Science Foundation of China (Grant Numbers 81802586).
Author information
Authors and Affiliations
Contributions
CJ, WB, CY, and ZL conceived the review. CJ and ZL wrote the manuscript. GB, LX, ZJ, ZJ, FY, and ZS revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors give consent for the publication of the manuscript in Experimental Hematology & Oncology.
Competing interests
The authors declare that there is no potential competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, J., Guo, B., Liu, X. et al. Roles of N6-methyladenosine (m6A) modifications in gynecologic cancers: mechanisms and therapeutic targeting. Exp Hematol Oncol 11, 98 (2022). https://doi.org/10.1186/s40164-022-00357-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00357-z