
Abstract
Classical Hodgkin lymphoma (cHL) and anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma (ALK+ ALCL) are B and T cell lymphomas respectively, which express the tumour necrosis factor receptor superfamily member, CD30. Another feature shared by cHL and ALK+ ALCL is the aberrant expression of multiple members of the activator protein-1 (AP-1) family of transcription factors which includes proteins of the Jun, Fos, ATF, and Maf subfamilies. In this review, we highlight the varied roles these proteins play in the pathobiology of these lymphomas including promoting proliferation, suppressing apoptosis, and evading the host immune response. In addition, we discuss factors contributing to the elevated expression of these transcription factors in cHL and ALK+ ALCL. Finally, we examine therapeutic strategies for these lymphomas that exploit AP-1 transcriptional targets or the signalling pathways they regulate.
Similar content being viewed by others

Introduction
AP-1 proteins are a versatile family of dimeric transcription factors
The activator protein-1 (AP-1) proteins are a collection of transcription factors characterized by the presence of a basic leucine zipper (bZip) domain (Fig. 1a). This family of proteins was first described in the 1980s when v-Fos and v-Jun were identified as the oncogenic factors associated with FBJ murine osteosarcoma virus [1] and avian sarcoma virus 17 [2], respectively. Subsequent work identified cellular homologues for both genes (c-Fos and c-Jun) [1, 3] as well as related proteins within the Jun (JunB, JunD) and Fos (FRA-1, FRA-2, and FosB) subfamilies [4, 5]. The AP-1 family has further expanded to include members of the ATF and Maf subfamilies [4, 5] (Fig. 1b).

AP-1 proteins are bZip domain-containing transcription factors that comprise four subfamilies. a Cartoons illustrating the relative size and location of the basic leucine zipper (bZip) domain in representative AP-1 proteins from each of the different subfamilies. Protein size and the location of the bZip were determined using the Conserved Domain Database [152] (CDD v3.18) at the National Center for Biotechnology Information (NCBI) website. b Representative AP-1 family members from the Jun, Fos, ATF, and Maf subfamilies are shown
The AP-1 proteins function as dimers. Both homodimers and heterodimers are found; although not all proteins can homodimerize and not all heterodimers are possible [4]. Dimerization is mediated by the leucine zipper (Fig. 1a). This domain adopts an alpha helical structure where leucine side chains interact with the alpha helix of the leucine zipper of the other family member to mediate dimerization [6, 7]. The basic domain is important for interacting with DNA [7]. AP-1 proteins bind 12-O-tetradecanoylphorbol-13-acetate (TPA) responsive elements (TRE) (TGA(G/C)TCA), cAMP responsive elements (CRE) (TGACGTCA), and related sequences [8,9,10,11]. Individual dimers differ in their DNA binding and transcriptional activities. For example, c-Jun:c-Fos dimers prefer TRE sites, whereas c-Jun:ATF dimers prefer CRE sites [12]. Moreover, c-Jun:c-Fos heterodimers have higher affinity for TRE sites than c-Jun:c-Jun homodimers [8, 9], and dimers containing JunB are less transcriptionally active than those containing c-Jun [13, 14]. While these proteins are primarily thought to function as transcriptional activators, there are situations where they appear to function as repressors ([15,16,17] as examples). Thus, the AP-1 family is a diverse collection of proteins that generate an even greater collection of dimers with varied DNA binding and transcriptional activities. Not surprisingly, AP-1 family proteins regulate a wide range of cellular and biological activities. These include the cell cycle and proliferation [5, 18], programmed cell death including apoptosis [5, 18, 19] and autophagy [20], and lipid synthesis [21]. As well, AP-1 proteins regulate migration and invasion through modulation of the cytoskeleton [22], and are implicated in inflammatory diseases [23,24,25], bone development [26,27,28], the nervous system [29,30,31,32], immune cell development and activation [26, 33], and cancer.
AP-1 proteins are implicated in the development and maintenance of cancers
AP-1 proteins play important roles in multiple malignancies including cancers of the lung [34, 35], breast [25, 36], gastrointestinal tract [25, 37], brain [38,39,40], skin [41, 42], ovaries [34, 43], and bone [44]. They regulate many of the hallmarks and enabling characteristics of cancer described by Hanahan and Weinberg [45] including sustaining proliferative signalling [5, 18], resisting cell death [5, 18], inducing angiogenesis [46,47,48], activating invasion and metastasis [22], tumour-promoting inflammation [49], and avoiding immune destruction [33]. AP-1 proteins are also implicated in the pathogenesis of leukemia and lymphoma where these transcription factors can act as oncogenes [50, 51] or tumour suppressors [52,53,54]. This includes the CD30-positive lymphomas, classical Hodgkin lymphoma (cHL) and anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma (ALK+ ALCL) where AP-1 proteins perform a variety of pro-tumour functions.
cHL and ALK+ ALCL are CD30-positive lymphomas
The CD30-positive lymphomas are characterized by the expression of tumour necrosis factor receptor superfamily member 8 (TNFRSF8) which is a 120-kDa type I transmembrane glycoprotein more commonly referred to as CD30 [55]. CD30 is recognized by the Ki-1 monoclonal antibody (mAb), first described by Stein and colleagues, which stains the mononuclear Hodgkin cells and multinuclear Reed-Sternberg (HRS) cells of cHL [56]. Subsequently, CD30 was shown to be highly expressed in both ALK+ and ALK- ALCL, as well as a number of other lymphoid cancers and proliferative disorders including cutaneous ALCL, mycosis fungoides, Sézary syndrome, lymphomatoid papulosis, and a subset of diffuse large B cell lymphomas [57,58,59].
Hodgkin lymphoma, originally called Hodgkin’s disease, was first identified in 1832 by Thomas Hodgkin [60]. Hodgkin lymphoma is a mature B cell lymphoma that is subdivided into classical Hodgkin lymphoma (cHL), and nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) [61]. cHL accounts for approximately 90% of Hodgkin lymphoma cases, and is characterized morphologically by the presence of HRS cells [61]. Interestingly, HRS cells constitute only a small proportion of cells at the tumour site with infiltrating immune cells making up the majority of the tumour mass [62, 63]. The current hypothesis is that HRS cells arise from germinal centre B cells that have failed to undergo apoptosis [62]. HRS cells exhibit the aberrant activation of multiple signalling pathways including the NF-κB [64, 65], JAK/STAT [66,67,68], and PI3K/Akt [69, 70] pathways.
Anaplastic large cell lymphomas are T cell lymphomas which include ALK+ ALCL, ALK- ALCL, cutaneous ALCL, and breast implant-associated ALCL [58]. In addition to the expression of CD30, ALK+ ALCL are characterized by chromosomal translocations and inversions involving the gene encoding for the ALK tyrosine kinase [58]. The most common translocation (~ 80%) is with the gene encoding for nucleophosmin (NPM) [71]. The resulting t(2;5)(p23;q35) translocation results in a fusion protein (NPM-ALK) consisting of the N-terminal dimerization domain of NPM and the C-terminal kinase and intracellular domains of ALK [71]. This fusion protein exhibits constitutive tyrosine kinase activity and activates many signalling events including the JAK/STAT [72,73,74,75,76] and PI3K/Akt [77, 78] pathways.
The elevated expression of several AP-1 proteins including c-Jun [79, 80], JunB [79, 81,82,83], ATF3 [84], BATF [85], and BATF3 [85, 86] has also been described in CD30-positive lymphomas. In the following sub-sections we will discuss how these transcription factors, in collaboration with other signalling pathways, benefit cHL and ALK+ ALCL by promoting proliferation/growth, suppressing apoptosis, and evading the host immune response (Fig. 2). Of note, while many activities/transcriptional targets we discuss have only been described in either ALK+ ALCL or cHL, many could be common to both lymphomas. Likewise, while specific activities/transcriptional targets have been ascribed to particular AP-1 proteins, there may be overlap with other AP-1 family members.

Functions of AP-1 family proteins in cHL and/or ALK+ ALCL. The major cellular activities regulated by AP-1 family proteins, and their transcriptional targets mediating these effects, in cHL and/or ALK+ ALCL are illustrated. Specific details are described in the text
The function of AP-1 proteins in ALK+ ALCL and cHL
AP-1 proteins regulate proliferation and growth
Several studies have implicated AP-1 proteins in promoting proliferation in ALK+ ALCL and cHL. Inhibition of AP-1 activity in the L-428 cHL cell line with a dominant negative c-Fos construct, A-Fos, decreased proliferation which was associated with a decrease in cyclin D2 expression [79]. In addition, pharmacological inhibition of the c-Jun activator, c-Jun N-terminal kinase (JNK), in the SU-DHL-1 ALK+ ALCL cell line resulted in cell cycle arrest in G2/M phase [87]. This was likely due to the upregulation of the cyclin-dependent kinase (CDK) inhibitor, p21cip1, and decreased expression of Cyclin A [87]. A similar cell cycle defect was observed in cHL cell lines when JNK was inhibited and this was also associated with an up-regulation of p21cip1 [88].
A role for specific AP-1 proteins in the regulation of proliferation has been revealed by short interfering RNA (siRNA)/short hairpin RNA (shRNA) knock-down and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 knock-out studies. Several groups have reported that JunB knock-down, in most ALK+ ALCL cell lines, decreased proliferation [89,90,91]. The common cell cycle defect observed in these studies was an increased percentage of cells in G0/G1 with decreased percentages of cells in G2/M [89] or S [90, 91] phase. These defects correlated with decreased expression of CDK2 and multiple cyclins including Cyclin A2, Cyclin D2, Cyclin D3, and Cyclin E, as well as increased expression of CDK inhibitors p14ink4A, p18ink4, p21cip1 and p27kip1 [90, 91]. In contrast, differing roles for c-Jun in regulating proliferation in ALK+ ALCL have been reported. Two studies observed no effect on proliferation when c-Jun was knocked-down [89, 92], whereas another study found that siRNA-mediated knock-down of c-Jun reduced cell viability and growth which correlated with increased levels of p21cip1 and decreased levels of Cyclin A and Cyclin D3 [87]. In cHL, stable knock-down of either c-Jun or JunB with shRNA was shown to reduce proliferation. This was characterized by an increased percentage of cells in G0/G1 and a decreased percentage in S phase, and likely due to elevated p21cip1 levels [91].
There is also evidence that c-Jun and JunB have overlapping functions with respect to promoting proliferation and/or growth in these lymphomas. For example, in a mouse model of ALK+ ALCL where expression of NPM-ALK was driven in T cells by a CD4 promoter [93], tumour formation was only compromised when both c-Jun and JunB were knocked-out, and double knock-out cells exhibited impaired proliferation [93]. Furthermore, knocking down both c-Jun and JunB with siRNA resulted in a more dramatic reduction in colony formation in Karpas 299 cells compared to single knock-down cells [90].
Other members of the AP-1 family have also been suggested to regulate proliferation in ALK+ ALCL. Schleussner and colleagues demonstrated that CRISPR/Cas9-mediated knock-out of BATF or BATF3 in the Karpas 299 and SUP-M2 cell lines reduced the growth rate of these cells, and reducing the expression of both resulted in an even greater defect [85]. Similar findings were observed in a separate study when BATF was knocked-out of cHL and ALK+ ALCL cell lines [86]. This defect in proliferation is likely due to failure of BATF3, in collaboration with other AP-1 proteins, to promote the expression of c-Myc [86]. Finally, siRNA-mediated knock-down of ATF3 in the L540Cy cHL cell line resulted in decreased [3H]-thymidine incorporation consistent with a proliferation defect [84].
Several other AP-1 transcriptional targets have been implicated in the regulation of proliferation and viability. In the CD4-NPM-ALK mouse model, c-Jun and JunB were found to promote transcription of the Platelet-Derived Growth Factor Receptor β (PDGFRβ), a receptor tyrosine kinase, which was subsequently found to be highly expressed in ALK+ ALCL patient samples and some ALK+ ALCL cell lines [93]. Importantly, tumour cells isolated from the CD4-NPM-ALK transgenic mice exhibited reduced proliferation when treated with Imatinib, a tyrosine kinase inhibitor which targets PDGFRβ [93]. CD30 is also a JunB transcriptional target [83, 94, 95]. Knock-down of CD30 was demonstrated to decrease the percentage of cells in S phase and increase the percentage in G0/G1 in the SU-DHL-1 ALK+ ALCL cell line [90], and found to decrease viability in cHL cell lines [96]. In the former study, CD30 knock-down correlated with an increase in p21cip1 and p14ink4A. Likewise, knock-down of the heat shock protein 90 (Hsp90) co-chaperone, Cyclophilin 40 (Cyp40), a JunB transcriptional target in ALK+ ALCL, resulted in reduced viability in multiple ALK+ ALCL cell lines, which could be due in part to a defect in proliferation [97].
Finally, AP-1 proteins also promote proliferation in ALK+ ALCL through PI3K/Akt signalling. The Akt 1, 2, and 3 serine/threonine kinases are transcriptional targets of c-Jun and JunB [98]. Pharmacological inhibition of Akt was shown to decrease the percentage of cells in S phase, and upregulate the CDK inhibitor, p27kip1 [99]. As well, a dominant negative Akt was shown to affect in vitro colony formation and in vivo tumour development in BaF3 cells expressing NPM-ALK [78]. Several Akt substrates linked to the regulation of proliferation have been studied in ALK+ ALCL. McDonnell and colleagues demonstrated that NPM-ALK signalling, through PI3K/Akt, leads to phosphorylation and inactivation of the serine/threonine kinase, GSK3β [100]. Inactivation of GSK3β prevented the phosphorylation and degradation of the cell cycle phosphatase, CDC25A [100]. Inactivation of GSK3β in ALK+ ALCL cell lines also promotes proliferation through stabilizing the sonic hedgehog (SHH) pathway transcription factor, Gli1, which results in up-regulation of Cyclin D2 [101]. Akt-mediated activation of mammalian target of rapamycin (mTor) signalling is also important for promoting proliferation, as siRNA-mediated knock-down of mTor decreased the number of cells in S phase [102]. The FOXO3a transcription factor is another substrate of Akt in ALK+ ALCL [103]. Phosphorylation of FOXO3a by Akt prevented FOXO3a from translocating to the nucleus and promoting the transcription of p27kip1 [103]. Collectively, these studies show that multiple AP-1 proteins, and their transcriptional targets, promote proliferation and growth in cHL and ALK+ ALCL.
AP-1 proteins and their transcriptional targets influence apoptosis
Protecting cells from apoptosis is also an important function of some AP-1 proteins in cHL and ALK+ ALCL. Forced expression of dominant negative A-Fos in Karpas 299 cells increased the number of cells with condensed or fragmented nuclei, illustrating the importance of AP-1 signalling in protecting these cells from apoptosis [79]. Knock-down of BATF3 with siRNA led to increased Annexin V staining in the SUP-M2 cell line, but not in Karpas 299 cells [85]. Increased apoptosis was also observed in cHL cell lines when ATF3 was knocked-down and this may in part be due to decreased c-Myc transcription [84].
Knock-down of all AP-1 proteins is not associated with apoptosis in these lymphomas. Stable knock-down of c-Jun or JunB in multiple cHL and ALK+ ALCL cell lines was not associated with significant apoptosis as measured by TUNEL staining [91]. However, loss of both c-Jun and JunB was associated with increased apoptosis in the CD4-NPM-ALK model as was inhibition of PDGFRβ activity with Imatinib [93]. JunB knock-down in ALK+ ALCL cell lines was found to sensitize cells to etoposide-induced decreases in cell growth and colony formation; however, whether this was due to decreased proliferation and/or increased apoptosis was not explored [90].
There is also evidence that transcriptional targets of AP-1 proteins regulate apoptosis in these lymphomas. As mentioned, knock-down of CD30 [90, 96] or Cyp40 [97] was associated with decreased viability, and a dominant negative Akt decreased colony formation in BaF3 cells expressing NPM-ALK [78]. These phenotypes could be due in part to increased apoptosis. Likewise, knock-down or pharmacological inhibition of Myc was shown to reduce the viability of ALK+ ALCL cell lines [104, 105], and increase the number of sub-G0/G1 cells [105]. Inhibition of GSK3β by Akt in ALK+ ALCL is also important for preventing the GSK3β-mediated phosphorylation, and subsequent targeting for degradation, of the pro-survival Bcl-2 family member, Mcl-1 [100]. This same study found that inhibition of GSK3β decreased poly(ADP-ribose) polymerase (PARP) cleavage in cells treated with an ALK inhibitor [100]. Other Akt substrates including mTor [102] and FOXO3a [103] are also important for promoting survival in ALK+ ALCL, and inhibition of SHH/GLI1 signalling in ALK+ ALCL cell lines resulted in an increased percentage of Annexin V-positive cells [101].
On the other hand, there is evidence that AP-1 transcriptional targets may promote apoptosis in ALK+ ALCL. The serine protease, Granzyme B (GzB), is highly expressed in ALK+ ALCL [106, 107], and its transcription is promoted by NPM-ALK signalling and JunB [108]. GzB is primarily expressed by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, where it allows these cells to kill virally-infected or transformed cells [109]. While knock-down of GzB in ALK+ ALCL cell lines did not result in appreciable apoptosis on its own, knock-down cells were less sensitive to staurosporine and doxorubicin-induced apoptosis [110]. This suggests that GzB expression could be one reason why ALK+ ALCL patients are generally responsive to chemotherapy.
AP-1 proteins regulate immunomodulatory genes
There are several immunomodulatory molecules which are AP-1 transcription targets in cHL and ALK+ ALCL. Galectin-1 (Gal-1) is an immunoglycan highly expressed in cHL and ALK+ ALCL patients and its expression strongly correlates with c-Jun levels [111,112,113]. Moreover, an AP-1 site within the Gal-1 enhancer was shown to bind c-Jun and promote Gal-1 transcription [111]. In cHL, Gal-1 was shown to create an immunosuppressive tumour microenvironment by promoting the expression of T helper 2 (TH2)-promoting cytokines and increasing the number of regulatory T cells (Tregs) [111]. In addition, analysis of patient samples demonstrated that HRS cells with high Gal-1 expression had lower infiltrating CD8-positive T cells, and in vitro experiments with recombinant Gal-1 demonstrated that Gal-1 can impair CD8 proliferation and effector function [113].
The transcription of Program death-ligand 1 (PD-L1) is also mediated by AP-1 transcription factors in cHL and ALK+ ALCL [114, 115]. PD-L1 is a ligand for the immune inhibitory receptor, program death-1 (PD-1), and engagement of PD-1 by PD-L1 allows cancers expressing PD-L1 to evade killing by CTLs and NK cells [116, 117]. Both c-Jun and JunB bind to a PD-L1 enhancer region, and this was found to be important for promoting PD-L1 transcription [115]. Furthermore, BATF3, together with the IRF4 transcription factor, was found to be important for PD-L1 expression in ALK+ ALCL [114], and inhibition of the PD-1/PD-L1 signalling axis in ALK+ ALCL cell lines was found to increase the ability of these cell lines to activate T cells and be killed by NK cells [114].
Additional activities
There are also additional functions performed by the AP-1 proteins in cHL and ALK+ ALCL. JunB has been linked to genomic instability in ALK+ ALCL through directly repressing the expression of the DEAD-box helicase, DDX11, which regulates sister chromatid cohesion [118]. Finally, c-Jun and c-Fos activity were implicated in the expression of Lymphotoxin-α (LTA) in cHL [119]. LTA is a member of the TNF family and is important for lymphoid organ development, inflammation, and antiviral responses [120, 121]. LTA secreted by cHL cell lines facilitated the interaction of CD4+ T cells with human umbilical vein endothelial cells (HUVECs) through the induction of the ICAM-1, VCAM-1, and E-selectin adhesion molecules in HUVECs [119]. Thus, through secreting LTA, HRS cells may contribute to the immune cell infiltrate characteristic of cHL.
AP-1 proteins are also critical for the expression of genes that characterize ALK+ ALCL and cHL. This includes CD30 [83, 94, 95] and GzB, with the latter being a hallmark of the cytotoxic phenotype of ALK+ ALCL [106, 107]. BATF/BATF3 are important for expression of genes in ALK+ ALCL that are associated with the TH17/group 3 innate lymphoid cell gene signature observed in this lymphoma [85]. Likewise, BATF3 expression is important for both the expression and repression of genes that characterize cHL [122].
Multiple mechanisms account for elevated AP-1 protein expression
In the previous section we discussed the many important activities influenced by AP-1 proteins in cHL and ALK+ ALCL. In this section we will examine the mechanisms and signalling events that lead to their elevated expression and/or activation in these lymphomas.
Atsaves et al. reported that JunB gene amplifications are common in ALK+ ALCL, but this did not correlate with increased JunB expression [90]. JunB transcription in ALK+ ALCL is dependent on NPM-ALK [123, 124], and on signalling events initiated by CD30 through a Mek/Erk/Ets-1 pathway [89, 94, 124]. The latter pathway is also important for promoting JunB transcription in cHL [94, 124], and NF-κB has also been suggested to increase JunB transcription [79], though others have reported different findings [94]. Importantly, the fact that CD30 is both a target and regulator of JunB means that a positive feedback loop is generated that ensures high levels of CD30 and JunB in these lymphomas. Levels of BATF3 in ALK+ ALCL are regulated by NPM-ALK/STAT3 signalling [114], and JAK/STAT signalling is also important for BATF3 expression in cHL [86]. This highlights an example of cross-talk between the AP-1 and JAK/STAT signalling pathways in these lymphomas. Signalling through Sphingosine 1-phosphate receptor 1 (S1PR1) also activates BATF3 transcription in cHL through a PI3K-dependent pathway, and BATF further promotes S1PR1 transcription generating a positive feedback loop [122]. As well, the IRF5 transcription factor promotes the transcription of multiple AP-1 genes in cHL including c-Jun, JunB, and ATF3; however, whether this up-regulation was directly or indirectly mediated by IRF5 was not determined [125].
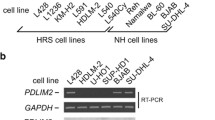
Post-transcriptional and post-translational mechanisms also influence c-Jun/JunB levels and activity. Recently, expression of miR-939 in ALK+ ALCL was found to reduce JunB levels [126], and JunB translation was promoted in ALK+ ALCL by targeting JunB mRNA to polysomes via a PI3K/Akt/mTor-dependent pathway [89]. There are several examples of the post-translational regulation of the AP-1 proteins in cHL and ALK+ ALCL. JNK, activated by NPM-ALK signalling, mediates phosphorylation and activation of c-Jun in ALK+ ALCL [87]. As well, a lack of GSK3β activity in this lymphoma has been argued to lead to a failure of JunB to be phosphorylated, and subsequently targeted for degradation, by the Fbxw7 E3 ubiquitin ligase [127]. Because Akt is a transcriptional target of JunB, and signalling mediated by Akt regulates JunB translation and stability, this illustrates cross-talk between these two pathways in ALK+ ALCL. Loss of expression of another E3 ubiquitin ligase, PDLIM2, was found to be common to both cHL and ALK+ ALCL, and reduced PDLIM2 expression increased AP-1 transcriptional activity through an unknown mechanism [128]. Thus, many factors contribute to the elevated expression of AP-1 family proteins in cHL and ALK+ ALCL.
Therapies directed at targets of AP-1 proteins in cHL and ALK+ ALCL
Frontline treatments
Frontline treatments for ALK+ ALCL are combination chemotherapy regimens. These are generally quite effective at treating the disease with event free survival ranging from 68 to 76% depending on the trial [129]. cHL is also treated successfully with combination chemotherapy in conjunction with radiotherapy [130]. Despite the success of these treatments, some patients are refractory to treatment and others relapse [129, 130]. Also, there are long-term negative consequences associated with chemotherapy and radiation [131]. Thus, precision medicine approaches that exploit specific features of these cancers have been developed. This includes ALK tyrosine kinase inhibitors, such as Crizotinib, which are currently in clinical trials to treat ALK+ ALCL [129]. Pertinent to this review, there are several therapies directed at AP-1 transcriptional targets (Fig. 3). For example, inhibition of PDGFRβ kinase activity with Imatinib was shown to successfully treat an ALK+ ALCL patient that was refractory to chemotherapy and had relapsed after autologous stem cell transplantation [93]. In addition, several therapies targeting CD30 or the interaction between PD-1/PD-L1 have been developed and we will discuss these in more detail in the next sub-section.

Therapies targeting AP-1 regulated genes in cHL and/or ALK+ ALCL. Therapies targeting the protein products of AP-1 transcriptional targets in cHL and/or ALK+ ALCL are highlighted. These include the small molecule tyrosine kinase inhibitor, Imatinib, which targets PDGFRβ, blocking antibodies that interfere with PD-1/PD-L1 interaction (Nivolumab, Pembrolizumab), a bispecfic antibody that recruits NK cells to cells expressing CD30 (AFM13), and an antibody-toxin conjugate which targets CD30-positive cells (Brentuximab Vedotin). Additional details are provided in the text
Therapies targeting CD30 and PD-1/PD-L1
CD30 therapies include several monoclonal antibodies as well as antibody conjugates [55]. Brentuximab vedotin (also known as SGN-35) is a CD30-specific monoclonal antibody (mAb) conjugated to the anti-mitotic agent, monomethylauristatin E (MMAE) via a cathepsin protease cleavable linker [132]. The anti-CD30 Ab component of brentuximab vedotin binds to CD30, which leads to the internalization of the Ab-conjugate, cleavage of the peptide linker, and release of MMAE. MMAE is a synthetic compound, related to a compound isolated from a shell-less marine mollusk, which kills cells through inhibiting tubulin polymerization [133]. Brentiximab vedotin is effective in treating relapsed or refractory cHL and ALK+ ALCL [134,135,136], and shown to be promising as a frontline treatment for these lymphomas in combination with chemotherapy [137, 138].
Other treatments targeting CD30 include bispecific antibodies such as AFM13. AFM13 consists of the heavy and light chain variable regions from the HRS-3 anti-CD30 mAb fused as a single polypeptide to the heavy and light chain variable regions of a mAb that recognizes CD16a expressed on NK cells [139]. This polypeptide dimerizes to form a tetravalent molecule with two CD30 and two CD16a binding sites, and mediates killing of cHL and ALK+ ALCL cell lines by recruiting NK cells [139, 140]. Finally, chimeric antigen receptor (CAR) T cells that specifically target CD30 are also being investigated for the treatment of CD30-positive lymphomas [141].
Interfering with the interaction between PD-L1-expressing tumour cells and T cells expressing PD-1 is an effective therapy for many cancers [116], and this includes cHL and ALK+ ALCL [142]. The PD-1 binding mAbs, Nivolumab [143, 144] and Pembrolizumab [145, 146] have been shown to be effective in treating relapsed or resistant cHL. Furthermore, two cases reports have reported a positive effect of Nivolumab treatment on individual ALK+ ALCL patients that failed other treatments [147, 148].
Concluding remarks
In this review, we discussed the key roles AP-1 proteins play in the pathobiology of cHL and ALK+ ALCL, the events that lead to the aberrant expression of these proteins, and how AP-1 transcriptional targets, or the components of pathways they function within, can be exploited as therapeutic targets. Nonetheless, there are still important questions to be addressed.
We need to know more mechanistically how AP-1 proteins regulate pro-tumour functions in these lymphomas. For example, studies have demonstrated the knock-down of AP-1 proteins results in the down-regulation of cyclins and CDKs and the up-regulation of CDK inhibitors. However, in most cases whether these changes are a direct transcriptional consequence of reduced expression of the AP-1 protein, or more likely, indirect due to dysregulation of signalling events mediated by transcriptional targets is not clear. Moreover, with regard to the latter possibility, determining which transcriptional targets are the most critical, and how they signal to regulate proliferation, or other activities, requires further investigation.
Proteomic and microarray studies have been performed to identify genome-wide dysregulated genes in knock-down cells for some family members [86, 97, 108, 122]. Extending these experiments to additional AP-1 proteins, and although technically more challenging, when multiple AP-1 proteins are knocked-down or knocked-out will provide a more comprehensive understanding of cellular activities regulated by these transcription factors. Likewise, chromatin immunoprecipitation-sequencing (ChIP-Seq) experiments will complement these studies by globally characterizing genomic sites occupied by these transcription factors and identifying those genes more likely to be direct transcriptional targets. In addition, ChIP-Seq experiments will help reveal which genes are more likely regulated by individual AP-1 proteins versus those regulated by multiple family members. Likewise, although many AP-1 proteins are aberrantly expressed in these lymphomas, little is known about the abundance and function of specific AP-1 dimers. Quantitative mass spectrometry studies and experiments utilizing defined AP-1 dimers [149] will help address these questions.
Finally, as more AP-1-regulated genes are identified, some of these, or the signalling pathways they function within, could become novel treatments. Even the AP-1 family proteins themselves could become drug targets. Small molecules and peptides that interfere with AP-1 DNA binding or dimer formation are being investigated as therapeutics [150, 151] and these could be attractive treatments for cHL and ALK+ ALCL.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- cHL:
-
Classical Hodgkin lymphoma
- ALK+ ALCL:
-
Anaplastic lymphoma kinase positive, anaplastic large cell lymphoma
- AP-1:
-
Activator protein-1
- bZip:
-
basic leucine zipper
- TPA:
-
12-O-tetradecanoylphorbol-13-acetate
- TRE:
-
12-O-tetradecanoylphorbol-13-acetate responsive elements
- CRE:
-
cAMP responsive elements
- TNFRSF8:
-
Tumour necrosis factor receptor superfamily member 8
- NLPHL:
-
Nodular lymphocyte predominant Hodgkin lymphoma
- HRS:
-
Hodgkin and Reed-Sternberg cells
- NPM:
-
Nucleophosmin
- S1P:
-
Sphingosine-1-phosphate
- JNK:
-
c-Jun N-terminal kinase
- CDK:
-
Cyclin-dependent kinase
- PDGFRβ:
-
Platelet-derived growth factor β
- HSP90:
-
Heat shock protein 90
- Cyp40:
-
Cyclophilin 40
- SHH:
-
SONIC hedgehog
- mTOR:
-
mammalian target of rapamycin
- PARP:
-
Poly(ADP-ribose) polymerase
- GzB:
-
Granzyme B
- CTLs:
-
Cytotoxic T lymphocytes
- NK:
-
Natural killer
- Gal-1:
-
Galectin-1
- TH2:
-
T-helper 2
- Tregs:
-
Regulatory T cells
- PD-L1:
-
Programmed death ligand-1
- PD-1:
-
Programmed death-1
- LTA:
-
Lymphotoxin-α
- HUVECs:
-
Human umbilical vein endothelial cells
- EFS:
-
Event free survival
- mAb:
-
Monoclonal antibody
- MMAE:
-
Monomethylauristatin E
- CAR:
-
Chimeric antigen receptor
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
References
Curran T, Peters G, Van Beveren C, Teich NM, Verma IM. FBJ murine osteosarcoma virus: identification and molecular cloning of biologically active proviral DNA. J Virol. 1982;44(2):674–82.
Maki Y, Bos TJ, Davis C, Starbuck M, Vogt PK. Avian sarcoma virus 17 carries the jun oncogene. Proc Natl Acad Sci USA. 1987;84(9):2848–52.
Bohmann D, Bos TJ, Admon A, Nishimura T, Vogt PK, Tjian R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science. 1987;238(4832):1386–92.
Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20(19):2438–52.
Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–400.
Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240(4860):1759–64. Epub 1988/06/24.
Glover JN, Harrison SC. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature. 1995;373(6511):257–61. Epub 1995/01/19.
Nakabeppu Y, Ryder K, Nathans D. DNA binding activities of three murine Jun proteins: stimulation by Fos. Cell. 1988;55(5):907–15. Epub 1988/12/02.
Rauscher FJ 3rd, Voulalas PJ, Franza BR Jr, Curran T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 1988;2(12B):1687–99.
Kataoka K, Noda M, Nishizawa M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol Cell Biol. 1994;14(1):700–12. Epub 1994/01/01.
Kerppola TK, Curran T. A conserved region adjacent to the basic domain is required for recognition of an extended DNA binding site by Maf/Nrl family proteins. Oncogene. 1994;9(11):3149–58. Epub 1994/11/01.
Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci USA. 1991;88(9):3720–4 Epub 1991/05/01.
Chiu R, Angel P, Karin M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell. 1989;59(6):979–86. Epub 1989/12/22.
Deng T, Karin M. JunB differs from c-Jun in its DNA-binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes Dev. 1993;7(3):479–90. Epub 1993/03/01.
Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, et al. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13(5):607–19. Epub 1999/03/11.
Crist SA, Griffith TS, Ratliff TL. Structure/function analysis of the murine CD95L promoter reveals the identification of a novel transcriptional repressor and functional CD28 response element. J Biol Chem. 2003;278(38):35950–8. Epub 2003/07/12.
Perez-Benavente B, Garcia JL, Rodriguez MS, Pineda-Lucena A, Piechaczyk M, Font de Mora J, et al. GSK3-SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2012;32(17):2189–99. Epub 2012/06/20.
Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4(5):E131-6.
Ameyar M, Wisniewska M, Weitzman JB. A role for AP-1 in apoptosis: the case for and against. Biochimie. 2003;85(8):747–52. Epub 2003/10/31.
Yogev O, Goldberg R, Anzi S, Shaulian E. Jun proteins are starvation-regulated inhibitors of autophagy. Cancer Res. 2010;70(6):2318–27. Epub 2010/03/04.
Caputto BL, Cardozo Gizzi AM, Gil GA. c-Fos: an AP-1 transcription factor with an additional cytoplasmic, non-genomic lipid synthesis activation capacity. Biochim Biophys Acta. 2014;1841(9):1241–6. Epub 2014/06/03.
Ozanne BW, Spence HJ, McGarry LC, Hennigan RF. Transcription factors control invasion: AP-1 the first among equals. Oncogene. 2007;26(1):1–10. Epub 2006/06/27.
Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann Rheum Dis. 2011;70:i109-12. Epub 2011/02/26.
Shiozawa S, Tsumiyama K. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle. 2009;8(10):1539–43. Epub 2009/04/28.
Trop-Steinberg S, Azar Y. AP-1 Expression and its Clinical Relevance in Immune Disorders and Cancer. Am J Med Sci. 2017;353(5):474–83. Epub 2017/05/16.
Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005;208:126–40. Epub 2005/11/30.
Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genom Hum Genet. 2008;9:183–96. Epub 2008/09/05.
Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251–64. Epub 2006/11/14.
Perez-Cadahia B, Drobic B, Davie JR. Activation and function of immediate-early genes in the nervous system. Biochemistry cell biology = Biochimie et biologie cellulaire. 2011;89(1):61–73. Epub 2011/02/18.
Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiological reviews. 2009;89(1):121–45. Epub 2009/01/08.
Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Progress in neurobiology. 2006;78(6):347–63. Epub 2006/05/24.
Herdegen T, Waetzig V. AP-1 proteins in the adult brain: facts and fiction about effectors of neuroprotection and neurodegeneration. Oncogene. 2001;20(19):2424–37. Epub 2001/06/13.
Atsaves V, Leventaki V, Rassidakis GZ, Claret FX. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers. 2019;11:7 Epub 2019/07/26.
Jiang X, Xie H, Dou Y, Yuan J, Zeng D, Xiao S. Expression and function of FRA1 protein in tumors. Mol Biol Rep. 2020;47(1):737–52 Epub 2019/10/16.
Reddy SP, Mossman BT. Role and regulation of activator protein-1 in toxicant-induced responses of the lung. Am J Physiol Lung Cell Mol Physiol. 2002;283(6):L1161-78 Epub 2002/11/09.
Rohini M, Haritha Menon A, Selvamurugan N. Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int J Biol Macromol. 2018;120(Pt A):310–7. Epub 2018/08/26.
Ashida R, Tominaga K, Sasaki E, Watanabe T, Fujiwara Y, Oshitani N, et al. AP-1 and colorectal cancer. Inflammopharmacology. 2005;13(1–3):113–25. Epub 2005/11/02.
Gil GA, Silvestre DC, Tomasini N, Bussolino DF, Caputto BL. Controlling cytoplasmic c-Fos controls tumor growth in the peripheral and central nervous system. Neurochem Res. 2012;37(6):1364–71. Epub 2012/04/06.
Debinski W, Gibo DM. Fos-related antigen 1 (Fra-1) pairing with and transactivation of JunB in GBM cells. Cancer Biol Ther. 2011;11(2):254–62. Epub 2010/11/23.
Peterziel H, Muller J, Danner A, Barbus S, Liu HK, Radlwimmer B, et al. Expression of podoplanin in human astrocytic brain tumors is controlled by the PI3K-AKT-AP-1 signaling pathway and promoter methylation. Neurooncology. 2012;14(4):426–39. Epub 2012/03/08.
Kappelmann M, Bosserhoff A, Kuphal S. AP-1/c-Jun transcription factors: regulation and function in malignant melanoma. Eur J Cell Biol. 2014;93(1–2):76–81. Epub 2013/12/10.
Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. The activator protein-1 transcription factor in respiratory epithelium carcinogenesis. Mol Cancer Res. 2007;5(2):109–20. Epub 2007/02/23.
Hein S, Mahner S, Kanowski C, Loning T, Janicke F, Milde-Langosch K. Expression of Jun and Fos proteins in ovarian tumors of different malignant potential and in ovarian cancer cell lines. Oncol Rep. 2009;22(1):177–83. Epub 2009/06/11.
Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20(19):2401–12. Epub 2001/06/13.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. Epub 2011/03/08.
Kolch W, Martiny-Baron G, Kieser A, Marme D. Regulation of the expression of the VEGF/VPS and its receptors: role in tumor angiogenesis. Breast Cancer Res Treat. 1995;36(2):139–55. Epub 1995/01/01.
Yoshitomi Y, Ikeda T, Saito H, Yoshitake Y, Ishigaki Y, Hatta T, et al. JunB regulates angiogenesis and neurovascular parallel alignment in mouse embryonic skin. J Cell Sci. 2017;130(5):916–26. Epub 2017/01/18.
Singh NK, Quyen DV, Kundumani-Sridharan V, Brooks PC, Rao GN. AP-1 (Fra-1/c-Jun)-mediated induction of expression of matrix metalloproteinase-2 is required for 15S-hydroxyeicosatetraenoic acid-induced angiogenesis. J Biol Chem. 2010;285(22):16830–43. Epub 2010/04/01.
Ji Z, He L, Regev A, Struhl K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc Natl Acad Sci USA. 2019;116(19):9453–62 Epub 2019/03/27.
Blonska M, Zhu Y, Chuang HH, You MJ, Kunkalla K, Vega F, et al. Jun-regulated genes promote interaction of diffuse large B-cell lymphoma with the microenvironment. Blood. 2015;125(6):981–91. Epub 2014/12/24.
Fan F, Bashari MH, Morelli E, Tonon G, Malvestiti S, Vallet S, et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia. 2017;31(7):1570–81. Epub 2016/11/29.
Szremska AP, Kenner L, Weisz E, Ott RG, Passegue E, Artwohl M, et al. JunB inhibits proliferation and transformation in B-lymphoid cells. Blood. 2003;102(12):4159–65.
Ott RG, Simma O, Kollmann K, Weisz E, Zebedin EM, Schorpp-Kistner M, et al. JunB is a gatekeeper for B-lymphoid leukemia. Oncogene. 2007;26(33):4863–71. Epub 2007/02/14.
Passegue E, Jochum W, Schorpp-Kistner M, Mohle-Steinlein U, Wagner EF. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001;104(1):21–32. Epub 2001/02/13.
van der Weyden CA, Pileri SA, Feldman AL, Whisstock J, Prince HM. Understanding CD30 biology and therapeutic targeting: a historical perspective providing insight into future directions. Blood Cancer J. 2017;7(9):e603 Epub 2017/09/09.
Schwab U, Stein H, Gerdes J, Lemke H, Kirchner H, Schaadt M, et al. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. 1982;299(5878):65–7. Epub 1982/09/02.
Prieto-Torres L, Rodriguez-Pinilla SM, Onaindia A, Ara M, Requena L, Piris MA. CD30-positive primary cutaneous lymphoproliferative disorders: molecular alterations and targeted therapies. Haematologica. 2019;104(2):226–35. Epub 2019/01/12.
Leventaki V, Bhattacharyya S, Lim MS. Pathology and genetics of anaplastic large cell lymphoma. Semin Diagn Pathol. 2020;37(1):57–71. Epub 2019/12/29.
Salas MQ, Climent F, Tapia G, DomingoDomenech E, Mercadal S, Oliveira AC, et al. Clinicopathologic features and prognostic significance of CD30 expression in de novo diffuse large B-cell lymphoma (DLBCL): results in a homogeneous series from a single institution. Biomarkers. 2020;25(1):69–75 Epub 2019/11/23.
Hodgkin T. On some morbid appearances of the absorbent glands and spleen. Med Chirurg Trans. 1832;17:68–114 Epub 1832/01/01.
Stein H, Pileri SA, Weiss LM, Poppema S, Gascoyne RD, Jaffe ES. Hodgkin Lymphomas: Introduction. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: International Agency for Research on Cancer (IARC); 2017.
Mathas S, Hartmann S, Kuppers R. Hodgkin lymphoma: Pathology and biology. Semin Hematol. 2016;53(3):139–47. Epub 2016/08/09.
Liu Y, Sattarzadeh A, Diepstra A, Visser L, van den Berg A. The microenvironment in classical Hodgkin lymphoma: an actively shaped and essential tumor component. Sem Cancer Biol. 2014;24:15–22. Epub 2013/07/23.
Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, et al. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood. 1996;87(10):4340–7. Epub 1996/05/15.
Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Investig. 1997;100(12):2961–9. Epub 1998/01/31.
Kube D, Holtick U, Vockerodt M, Ahmadi T, Haier B, Behrmann I, et al. STAT3 is constitutively activated in Hodgkin cell lines. Blood. 2001;98(3):762–70. Epub 2001/07/27.
Skinnider BF, Elia AJ, Gascoyne RD, Patterson B, Trumper L, Kapp U, et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2002;99(2):618–26. Epub 2002/01/10.
Cochet O, Frelin C, Peyron JF, Imbert V. Constitutive activation of STAT proteins in the HDLM-2 and L540 Hodgkin lymphoma-derived cell lines supports cell survival. Cell Signal. 2006;18(4):449–55. Epub 2005/06/22.
Morrison JA, Gulley ML, Pathmanathan R, Raab-Traub N. Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer Res. 2004;64(15):5251–60. Epub 2004/08/04.
Dutton A, Reynolds GM, Dawson CW, Young LS, Murray PG. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin’s lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol. 2005;205(4):498–506. Epub 2005/02/17.
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4.
Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002;168(1):466–74. Epub 2001/12/26.
Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21(7):1038–47. Epub 2002/02/19.
Khoury JD, Medeiros LJ, Rassidakis GZ, Yared MA, Tsioli P, Leventaki V, et al. Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK + and ALK- anaplastic large cell lymphoma. Clin Cancer Res. 2003;9(10 Pt 1):3692–9. Epub 2003/09/25.
Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003;22(35):5399–407. Epub 2003/08/23.
Lai R, Rassidakis GZ, Lin Q, Atwell C, Medeiros LJ, Amin HM. Jak3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum Pathol. 2005;36(9):939–44. Epub 2005/09/13.
Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood. 2000;96(13):4319–27. Epub 2000/12/09.
Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001;61(5):2194–9. Epub 2001/03/31.
Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. Embo J. 2002;21(15):4104–13.
Drakos E, Leventaki V, Schlette EJ, Jones D, Lin P, Medeiros LJ, et al. c-Jun expression and activation are restricted to CD30 + lymphoproliferative disorders. Am J Surg Pathol. 2007;31(3):447–53.
Rassidakis GZ, Thomaides A, Atwell C, Ford R, Jones D, Claret FX, et al. JunB expression is a common feature of CD30 + lymphomas and lymphomatoid papulosis. Mod Pathol. 2005;18(10):1365–70.
Mao X, Orchard G, Lillington DM, Russell-Jones R, Young BD, Whittaker SJ. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood. 2003;101(4):1513–9.
Watanabe M, Ogawa Y, Ito K, Higashihara M, Kadin ME, Abraham LJ, et al. AP-1 mediated relief of repressive activity of the CD30 promoter microsatellite in Hodgkin and Reed-Sternberg cells. Am J Pathol. 2003;163(2):633–41.
Janz M, Hummel M, Truss M, Wollert-Wulf B, Mathas S, Johrens K, et al. Classical Hodgkin lymphoma is characterized by high constitutive expression of activating transcription factor 3 (ATF3), which promotes viability of Hodgkin/Reed-Sternberg cells. Blood. 2006;107(6):2536–9. Epub 2005/11/03.
Schleussner N, Merkel O, Costanza M, Liang HC, Hummel F, Romagnani C, et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia. 2018;32(9):1994–2007. Epub 2018/03/29.
Lollies A, Hartmann S, Schneider M, Bracht T, Weiss AL, Arnolds J, et al. An oncogenic axis of STAT-mediated BATF3 upregulation causing MYC activity in classical Hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia. 2017;32(1):92–101 Epub 2017/07/01.
Leventaki V, Drakos E, Medeiros LJ, Lim MS, Elenitoba-Johnson KS, Claret FX, et al. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood. 2007;110(5):1621–30.
Leventaki V, Drakos E, Karanikou M, Psatha K, Lin P, Schlette E, et al. c-JUN N-terminal kinase (JNK) is activated and contributes to tumor cell proliferation in classical Hodgkin lymphoma. Hum Pathol. 2014;45(3):565–72. Epub 2014/01/25.
Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, et al. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood. 2007;110(9):3374–83.
Atsaves V, Lekakis L, Drakos E, Leventaki V, Ghaderi M, Baltatzis GE, et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK + anaplastic large cell lymphoma. Br J Haematol. 2014;167(4):514–23. Epub 2014/08/26.
Zhang J, Wu Z, Savin A, Yang M, Hsu YR, Jantuan E, et al. The c-Jun and JunB transcription factors facilitate the transit of classical Hodgkin lymphoma tumour cells through G1. Sci Rep. 2018;8(1):16019 Epub 2018/10/31.
Arechiga AF, Habib T, He Y, Zhang X, Zhang ZY, Funk A, et al. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol. 2009;182(6):3343–7. Epub 2009/03/07.
Laimer D, Dolznig H, Kollmann K, Vesely PW, Schlederer M, Merkel O, et al. PDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat Med. 2012;18(11):1699–704 Epub 2012/10/16.
Watanabe M, Sasaki M, Itoh K, Higashihara M, Umezawa K, Kadin ME, et al. JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and reed-sternberg cells of Hodgkin lymphoma. Cancer Res. 2005;65(17):7628–34.
Watanabe M, Ogawa Y, Itoh K, Koiwa T, Kadin ME, Watanabe T, et al. Hypomethylation of CD30 CpG islands with aberrant JunB expression drives CD30 induction in Hodgkin lymphoma and anaplastic large cell lymphoma. Lab Invest. 2008;88(1):48–57.
Watanabe M, Nakano K, Togano T, Nakashima M, Higashihara M, Kadin ME, et al. Targeted repression of overexpressed CD30 downregulates NF-kappaB and ERK1/2 pathway in Hodgkin lymphoma cell lines. Oncol Res. 2011;19(10–11):463–9 Epub 2011/01/01.
Pearson JD, Mohammed Z, Bacani JT, Lai R, Ingham RJ. The heat shock protein-90 co-chaperone, Cyclophilin 40, promotes ALK-positive, anaplastic large cell lymphoma viability and its expression is regulated by the NPM-ALK oncoprotein. BMC Cancer. 2012;12:229. Epub 2012/06/12.
Atsaves V, Zhang R, Ruder D, Pan Y, Leventaki V, Rassidakis GZ, et al. Constitutive control of AKT1 gene expression by JUNB/CJUN in ALK + anaplastic large-cell lymphoma: a novel crosstalk mechanism. Leukemia. 2015;29(11):2162–72. Epub 2015/05/20.
Rassidakis GZ, Feretzaki M, Atwell C, Grammatikakis I, Lin Q, Lai R, et al. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005;105(2):827–9 Epub 2004/09/18.
McDonnell SR, Hwang SR, Basrur V, Conlon KP, Fermin D, Wey E, et al. NPM-ALK signals through glycogen synthase kinase 3beta to promote oncogenesis. Oncogene. 2011;31(32):3733–40 Epub 2011/12/20.
Singh RR, Cho-Vega JH, Davuluri Y, Ma S, Kasbidi F, Milito C, et al. Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res. 2009;69(6):2550–8. Epub 2009/02/27.
Vega F, Medeiros LJ, Leventaki V, Atwell C, Cho-Vega JH, Tian L, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. 2006;66(13):6589–97. Epub 2006/07/05.
Gu TL, Tothova Z, Scheijen B, Griffin JD, Gilliland DG, Sternberg DW. NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood. 2004;103(12):4622–9. Epub 2004/02/14.
Weilemann A, Grau M, Erdmann T, Merkel O, Sobhiafshar U, Anagnostopoulos I, et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood. 2015;125(1):124–32. Epub 2014/11/02.
Wu C, Zhang HF, Gupta N, Alshareef A, Wang Q, Huang YH, et al. A positive feedback loop involving the Wnt/beta-catenin/MYC/Sox2 axis defines a highly tumorigenic cell subpopulation in ALK-positive anaplastic large cell lymphoma. J Hematol Oncol. 2016;9(1):120. Epub 2016/11/09.
Foss HD, Anagnostopoulos I, Araujo I, Assaf C, Demel G, Kummer JA, et al. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood. 1996;88(10):4005–11.
Foss HD, Demel G, Anagnostopoulos I, Araujo I, Hummel M, Stein H. Uniform expression of cytotoxic molecules in anaplastic large cell lymphoma of null/T cell phenotype and in cell lines derived from anaplastic large cell lymphoma. Pathobiology. 1997;65(2):83–90.
Pearson JD, Lee JK, Bacani JT, Lai R, Ingham RJ. NPM-ALK and the JunB transcription factor regulate the expression of cytotoxic molecules in ALK-positive, anaplastic large cell lymphoma. Int J Clin Exp Pathol. 2011;4(2):124–33. Epub 2011/02/18.
Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest. 2009;89(11):1195–220.
Pearson JD, Zhang J, Wu Z, Thew KD, Rowe KJ, Bacani JT, et al. Expression of granzyme B sensitizes ALK + ALCL tumour cells to apoptosis-inducing drugs. Mol Cancer. 2014;13(1):199. Epub 2014/08/30.
Juszczynski P, Ouyang J, Monti S, Rodig SJ, Takeyama K, Abramson J, et al. The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Natl Acad Sci USA. 2007;104(32):13134–9 Epub 2007/08/03.
Rodig SJ, Ouyang J, Juszczynski P, Currie T, Law K, Neuberg DS, et al. AP1-dependent galectin-1 expression delineates classical hodgkin and anaplastic large cell lymphomas from other lymphoid malignancies with shared molecular features. Clin Cancer Res. 2008;14(11):3338–44. Epub 2008/06/04.
Gandhi MK, Moll G, Smith C, Dua U, Lambley E, Ramuz O, et al. Galectin-1 mediated suppression of Epstein-Barr virus specific T-cell immunity in classic Hodgkin lymphoma. Blood. 2007;110(4):1326–9. Epub 2007/04/18.
Zhang JP, Song Z, Wang HB, Lang L, Yang YZ, Xiao W, et al. A novel model of controlling PD-L1 expression in ALK(+) anaplastic large cell lymphoma revealed by CRISPR screening. Blood. 2019;134(2):171–85. Epub 2019/06/04.
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18(6):1611–8. Epub 2012/01/25.
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Science translational medicine. 2016;8(328):328rv4. Epub 2016/03/05.
Mariotti FR, Quatrini L, Munari E, Vacca P, Tumino N, Pietra G, et al. Inhibitory checkpoints in human natural killer cells: IUPHAR Review 28. Br J Pharmacol. 2020;177(13):2889–903. Epub 2020/04/27.
Perez-Benavente B, Garcia JL, Rodriguez MS, Pineda-Lucena A, Piechaczyk M, Font de Mora J, et al. GSK3-SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2013;32(17):2189–99.
Fhu CW, Graham AM, Yap CT, Al-Salam S, Castella A, Chong SM, et al. Reed-Sternberg cell-derived lymphotoxin-alpha activates endothelial cells to enhance T-cell recruitment in classical Hodgkin lymphoma. Blood. 2014;124(19):2973–82. Epub 2014/08/21.
Koroleva EP, Fu YX, Tumanov AV. Lymphotoxin in physiology of lymphoid tissues: implication for antiviral defense. Cytokine. 2018;101:39–47 Epub 2016/09/14.
Hirose T, Fukuma Y, Takeshita A, Nishida K. The role of lymphotoxin-alpha in rheumatoid arthritis. Inflamm Res. 2018;67(6):495–501 Epub 2018/03/16.
Vrzalikova K, Ibrahim M, Vockerodt M, Perry T, Margielewska S, Lupino L, et al. S1PR1 drives a feedforward signalling loop to regulate BATF3 and the transcriptional programme of Hodgkin lymphoma cells. Leukemia. 2018;32(1):214–23. Epub 2017/09/08.
Hsu FY, Johnston PB, Burke KA, Zhao Y. The expression of CD30 in anaplastic large cell lymphoma is regulated by nucleophosmin-anaplastic lymphoma kinase-mediated JunB level in a cell type-specific manner. Cancer Res. 2006;66(18):9002–8.
Watanabe M, Itoh K, Togano T, Kadin ME, Watanabe T, Higashihara M, et al. Ets-1 activates overexpression of JunB and CD30 in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. Am J Pathol. 2012;180(2):831–8. Epub 2011/11/24.
Kreher S, Bouhlel MA, Cauchy P, Lamprecht B, Li S, Grau M, et al. Mapping of transcription factor motifs in active chromatin identifies IRF5 as key regulator in classical Hodgkin lymphoma. Proc Natl Acad Sci U S A. 2014;111(42):E4513-22. Epub 2014/10/08.
Garbin A, Lovisa F, Holmes AB, Damanti CC, Gallingani I, Carraro E, et al. miR-939 acts as tumor suppressor by modulating JUNB transcriptional activity in pediatric anaplastic large cell lymphoma. Haematologica. 2020;2019:241307 Epub 2020/04/18.
Perez-Benavente B, Garcia JL, Rodriguez MS, Pineda-Lucena A, Piechaczyk M, Font de Mora J, et al. GSK3-SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2012;32(17):2189–99 Epub 2012/06/20.
Wurster KD, Hummel F, Richter J, Giefing M, Hartmann S, Hansmann ML, et al. Inactivation of the putative ubiquitin-E3 ligase PDLIM2 in classical Hodgkin and anaplastic large cell lymphoma. Leukemia. 2017;31(3):602–13. Epub 2016/08/20.
Larose H, Burke GAA, Lowe EJ, Turner SD. From bench to bedside: the past, present and future of therapy for systemic paediatric ALCL, ALK. Br J Haematol. 2019;185(6):1043–54. Epub 2019/01/27.
Eichenauer DA, Aleman BMP, Andre M, Federico M, Hutchings M, Illidge T, et al. Hodgkin lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv19-29 Epub 2018/10/01.
von der Weid NX. Adult life after surviving lymphoma in childhood. Suppor Care Cancer. 2008;16(4):339–45 Epub 2008/01/16.
Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21(7):778–84. Epub 2003/06/05.
Bai R, Pettit GR, Hamel E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem Pharmacol. 1990;39(12):1941–9. Epub 1990/06/15.
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–21. Epub 2010/11/05.
Chen R, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2016;128(12):1562–6. Epub 2016/07/20.
Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190–6 Epub 2012/05/23.
Horwitz S, O’Connor OA, Pro B, Illidge T, Fanale M, Advani R, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet. 2019;393(10168):229–40. Epub 2018/12/14.
Connors JM, Jurczak W, Straus DJ, Ansell SM, Kim WS, Gallamini A, et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N Engl J Med. 2018;378(4):331–44. Epub 2017/12/12.
Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30 + tumor cells. mAbs. 2014;6(3):728–39. Epub 2014/03/29.
Pahl JHW, Koch J, Gotz JJ, Arnold A, Reusch U, Gantke T, et al. CD16A activation of NK cells promotes NK cell proliferation and memory-like cytotoxicity against cancer cells. Cancer Immunol Res. 2018;6(5):517–27 Epub 2018/03/09.
Grover NS, Savoldo B. Challenges of driving CD30-directed CAR-T cells to the clinic. BMC Cancer. 2019;19(1):203. Epub 2019/03/08.
Xie W, Medeiros LJ, Li S, Yin CC, Khoury JD, Xu J. PD-1/PD-L1 Pathway and its blockade in patients with classic Hodgkin lymphoma and non-Hodgkin large-cell lymphomas. Curr Hematol Malign Rep. 2020;15(4):372–81 Epub 2020/05/13.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9. Epub 2014/12/09.
Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016;17(9):1283–94.
Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after Brentuximab Vedotin failure. J Clin Oncol. 2016;34(31):3733–9 Epub 2016/06/30.
Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol. 2017;35(19):2125–32.
Rigaud C, Abbou S, Minard-Colin V, Geoerger B, Scoazec JY, Vassal G, et al. Efficacy of nivolumab in a patient with systemic refractory ALK + anaplastic large cell lymphoma. Pediatr Blood Cancer. 2018;65:4 Epub 2017/12/02.
Hebart H, Lang P, Woessmann W. Nivolumab for refractory anaplastic large cell lymphoma: a case report. Ann Intern Med. 2016;165(8):607–8 Epub 2016/10/18.
Bakiri L, Matsuo K, Wisniewska M, Wagner EF, Yaniv M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol. 2002;22(13):4952–64. Epub 2002/06/08.
Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. 2014;57(16):6930–48.
Brennan A, Leech JT, Kad NM, Mason JM. Selective antagonism of cJun for cancer therapy. J Exp Clin Cancer Res. 2020;39(1):184 Epub 2020/09/13.
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48(D1):D265–8 Epub 2019/11/30.
Acknowledgements
The authors would like to thank Dr. Julinor Bacani for reading and offering suggestions on the manuscript. We would also like to apologize to any colleagues whose work we were unable to discuss.
Funding
The work in the Ingham Lab is supported by operating grants from the Canadian Institutes of Health Research operating grant (MOP 77645; current PI, RJI) and Natural Sciences and Engineering Research Council of Canada (RGPIN-2017-05743; PI, RJI).
Author information
Authors and Affiliations
Contributions
ZW, MN and RJI were involved in drafting and revising this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
not applicable.
Consent for publication
not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, Z., Nicoll, M. & Ingham, R.J. AP-1 family transcription factors: a diverse family of proteins that regulate varied cellular activities in classical hodgkin lymphoma and ALK+ ALCL. Exp Hematol Oncol 10, 4 (2021). https://doi.org/10.1186/s40164-020-00197-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-020-00197-9