
Abstract
Background
Sand is often considered the preferred bedding material for dairy cows as it is thought to have lower bacterial counts than organic bedding materials and cows bedded on sand experience fewer cases of lameness and disease. Sand can also be efficiently recycled and reused, making it cost-effective. However, some studies have suggested that the residual organic material present in recycled sand can serve as a reservoir for commensal and pathogenic bacteria, although no studies have yet characterized the total bacterial community composition. Here we sought to characterize the bacterial community composition of a Wisconsin dairy farm bedding sand recycling system and its dynamics across several stages of the recycling process during both summer and winter using 16S rRNA gene amplicon sequencing.
Results
Bacterial community compositions of the sand recycling system differed by both seasons and stage. Summer samples had higher richness and distinct community compositions, relative to winter samples. In both summer and winter samples, the diversity of recycled sand decreased with time drying in the recycling room. Compositionally, summer sand 14 d post-recycling was enriched in operational taxonomic units (OTUs) belonging to the genera Acinetobacter and Pseudomonas, relative to freshly washed sand and sand from cow pens. In contrast, no OTUs were found to be enriched in winter sand. The sand recycling system contained an overall core microbiota of 141 OTUs representing 68.45% ± 10.33% SD of the total bacterial relative abundance at each sampled stage. The 4 most abundant genera in this core microbiota included Acinetobacter, Psychrobacter, Corynebacterium, and Pseudomonas. Acinetobacter was present in greater abundance in summer samples, whereas Psychrobacter and Corynebacterium had higher relative abundances in winter samples. Pseudomonas had consistent relative abundances across both seasons.
Conclusions
These findings highlight the potential of recycled bedding sand as a bacterial reservoir that warrants further study.
Similar content being viewed by others


Background
Providing suitable housing and bedding is critical for maintaining quality of life for dairy cattle. Cattle that are comfortable and well cared produce more milk and are more profitable [1,2,3]. One method to increase cow comfort is to use appropriate bedding material. There are several bedding options available, including rubber mats, straw, wood shavings, dried manure solids, newspaper clippings, and sand. Many consider sand as the ideal bedding type because it improves cow comfort and resting time, and studies comparing sand to other bedding types show decreases in lameness and incidences of hock lesions [4,5,6,7]. Moreover, studies have also shown that total bacterial counts in sand bedding are lower than in other organic bedding types [8,9,10,11]. Given that organic bedding is often implicated in the spread of bacterial pathogens responsible for diseases such as mastitis [12], the use of sand bedding may help in mitigating such diseases.
In addition to the health and comfort benefits of sand bedding, it is also cost effective as sand can be recycled and reused. The process involves mechanically removing used sand from free stalls and then pushing the sand using recycled water (grey water) to a central drain which leads to a separate sand recycling room. In the recycling room, sand is separated from organic material, cleaned with a disinfectant, and left to dry in the recycling room. This method typically leads to a sand recovery rate of > 90% and several studies have shown recycled sand to have no significant difference in bacterial load, relative to clean sand [8, 13, 14]. However, these results contrast with a more recent survey of 169 U.S. dairy herds, which found unused recycled bedding sand to have increased total bacterial counts when compared to unused fresh sand [11]. Additionally, recycling bedding sand does not remove all organic material and some bacteria (e.g., coliforms, Klebsiella pneumoniae) are known to utilize the available and residual organic nutrients to proliferate [13]. As a result, it has been suggested that recycled bedding sand could serve as a potential reservoir for bacterial pathogens [11, 15].
To date, there is limited information regarding the microbiota of recycled bedding sand. Most prior studies have only utilized culture-based surveys to quantify bacterial colonies or counts of pathogens commonly implicated in mastitis [9, 11, 13, 14]. As a result, these studies may lack the ability to identify less common or unculturable environmental pathogens. Moreover, no prior work has been done to examine the broader bacterial communities of a recycled bedding sand system. Here, we hypothesized that the microbiota of bedding sand is dynamic across both season (summer vs. winter) and stage of the recycling process, and that a core microbiota exists and persists across all recycling stages and seasons. To test this, we characterized the microbiota of recycled bedding sand collected in both summer and winter on a dairy farm in South-central Wisconsin, USA, across different stages of the recycling process using 16S rRNA gene amplicon sequencing. We report the bacterial taxa conserved throughout the recycling process and determined whether the diversity of these bacterial communities is affected by season and recycling stage.
Methods
Recycled sand collection
Recycled bedding sand was collected at various points in the recycling process from a dairy farm (approximately 1,500 Jersey cows and heifers) in South-central Wisconsin, USA. In brief, the recycling process involves removal of the top layer of sand from free stalls; thereafter, it is mechanically pushed into the aisle and flushed into a central drain using recycled water (“grey water”). Grey water is water stored in the sand recycling room used exclusively to move sand through the recycling system and is itself constantly reused across many recycling productions. The sand and grey water then travel through a drain into a central reception pit located in a dedicated recycling room in a separate building on the farm. The sand is allowed to settle in the reception pit and filled with grey water. Organic material is siphoned off and sent for composting. Sand is removed from the bottom of the pit using a commercial rotating auger. As the sand moves upward through the auger, it is doused with a disinfectant containing hypochlorous acid prior to being deposited into a large “wet recycled sand” pile. The wet recycled sand piles are approximately 2 m tall and 5–6 m wide. Once deposited, these piles remain stationary and are allowed to air-dry indoors in the recycling room for 11–14 d; they are then redistributed to re-bed free stalls on an as need basis. A full diagram of this process is presented in Supplementary Fig. S1. At the time of this study, the farm did not utilize commercial sand dryers, nor did they implement any process to hasten the sand drying process.
Sand samples were collected using sterile wooden tongue depressors (McKesson Medical-Surgical Inc., Richmond VA) and placed into sterile 50-mL conical tubes (Falcon, Fisher Bioscience, Waltham MA). At each location of the recycling process, five separate 5 mL sand samples were collected and used to create a single composite 25 mL sand sample. This was done in triplicate at all sand sampling locations except for cow pens where a total of 5 composite samples were collected from randomly chosen locations in the free stall cow pens. Sand was collected as available once in May 2018 (“summer”) and again in January 2019 (“winter”). Summer sand samples were collected from cow pens, from a pile of freshly washed wet recycled sand, a sand pile 4 d post-recycling (“4-d”) (from the surface of the pile and 7 in subsurface), a sand pile 14 d post-recycling (“14-d”) (from the surface and 7 in subsurface), and a pile of new sand that had not yet been used or recycled. In addition, approximately 40 mL of grey water was collected directly into a 50-mL conical tube, in triplicate, from the central holding tank of the sand recycling system. Winter samples were collected from a pile of freshly washed wet recycled sand, a sand pile 2 d post-recycling (“2-d”) (from the surface and 7 in subsurface), and a sand pile 7 d post-recycling (“7-d”) (from the surface and 7 in subsurface). Approximately 40 mL of grey water was also collected directly into 50-mL conical tubes, in triplicate, from the reception pit used to separate sand from organic material. In the winter we were unable to collect sand samples at the same timepoints as summer (4-d and 14-d post recycling) as the sand is processed for recycling on an as-needed basis. Sand was not collected from free stalls during the winter due to logistical constraints. After each collection, samples were transported on ice to our laboratory and stored at − 80 °C.
DNA extraction, amplification, and sequencing
All samples were thawed to room temperature and processed individually. For sand samples, 3 mL of sand was vortexed in 4 mL DNA extraction buffer and 1 mL of the resultant liquid was used for DNA extraction [16]. For grey water, DNA was extracted directly from 1 mL of sample. Total genomic DNA was extracted for each sample individually using a mechanical disruption and hot/cold phenol and phenol:chloroform extraction as previously described [16], with the modification of 25:24:1 phenol:chloroform:isoamyl alcohol in place of phenol:chloroform. DNA samples were resuspended in Tris Buffer and quantified with a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA). The V4 hypervariable region of the bacterial 16S rRNA gene was amplified by polymerase chain reaction (PCR) using barcoded, 1-step sequencing primers as previously described [17]. PCR reactions were performed using 25 ng template DNA, 0.5 μL forward and reverse primers at 10 mmol/L, 12.5 μL KAPA 2xHiFi Master Mix (Roche, Rotkreuz, Switzerland), and nuclease free water (IDT, Coralville, Iowa, USA) to a total volume of 25 μL. Grey water samples were processed identically to sand samples. Water was used in place of template DNA for PCR negative controls, which were processed identically to sand samples.
PCR reaction conditions were as follows: 95 °C for 3 min, 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, followed by a final extension at 72 °C for 5 min. PCR products were quantified on a 1% (wt/vol) low-melt agarose gel, desired PCR products (~ 380 bp) were excised and recovered via gel extraction using a 96-well Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA), and quantified using a Qubit High-Sensitivity assay kit (Thermo Fisher Scientific) and a Synergy 2 Multi-Mode plate reader (BioTek, Winooski, VT). DNA samples were equimolarly pooled to 4 nmol/L and sequenced on an Illumina Miseq (Illumina, Inc., San Diego, CA) using a 500-cycle MiSeq v2 sequencing kit with custom sequencing primers [17].
Sequence cleanup and classification
Paired-end reads were demultiplexed on the Illumina MiSeq and processed using mothur v.1.44.1 [18], as previously described [17]. Briefly, paired-end reads were assembled into contiguous sequences and those having ambiguous base calls, homopolymers longer than 8 bp, and lengths less than 200 bp or greater than 500 bp were removed. Sequences were aligned to the V4 region of the SILVA 16S rRNA gene reference database (v138) [19] and those not aligned were removed. Chimeric sequences were detected using Uchime [20] and removed. Taxonomic classification was performed using the SILVA taxonomic database (v138) with an 80% minimum confidence cut-off. Sequences classified as unknown, Archaea, Eukaryota, chloroplast, or mitochondria were removed, as were singleton sequences. Operational taxonomic units (OTUs) were clustered at 97% sequence similarity using the OptiClust method [21] and were taxonomically classified against the SILVA database with 80% minimum confidence. Representative sequences were determined using “get.oturep” with method = distance and were further taxonomically identified using blastn against the National Center for Biotechnology Information’s (NCBI) nucleotide database [22]. Samples with Good’s coverages, estimates of sample diversity captured by sequencing data, < 0.95 were removed [23]. Resultant samples were normalized to 16,000 sequences per sample. Alpha diversity metrics, Chao’s Richness and Shannon’s Diversity Index, and post-normalized Good’s coverage were calculated in mothur from the normalized OTU table. The sequencing dataset supporting the conclusions of this article is available in the NCBI’s Sequence Read Archive under BioProject number PRJNA724660 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA724660].
Sequence analysis in R
Statistical analyses were performed in R (v4.1.0) [24]. Bacterial communities were assessed between seasons (summer or winter), between sampling locations for each season individually (summer: cow pen, grey water, wet recycled sand, 4-d sand, and 14-d sand; winter: grey water, wet recycled sand, 2-d sand, 7-d sand), and between depths (surface or 7-in subsurface). When comparing depths, data were subset to only locations having both surface and 7-in samples (summer: 4-d and 14-d; winter: 2-d and 7-d) and were compared separately for summer and winter samples. For subsequent analyses, surface and subsurface samples for each location were grouped together as there was no significant difference in bacterial communities of these locations. When testing seasonal effects on alpha and beta diversity, summer cow pen samples were removed as they had no analogue in the winter sample set and the remaining locations were seasonally grouped as there were no direct analogs between summer and winter locations. Alpha diversity was assessed using Shannon’s Diversity Index, a measure of richness and evenness, and Chao’s Richness estimate. Normality was assessed using the Shapiro-Wilk test. Significance was determined using ANOVA or the Kruskal-Wallis (KW) test, depending on normality. Post-hoc pairwise comparisons were similarly performed using Tukey’s HSD or the Wilcoxon Rank Sum test in the case of significance (P < 0.05).
Beta-diversity was evaluated using Bray-Curtis dissimilarity, calculated using the R package vegan v2.5–7 [25], and both unweighted and weighted UniFrac metrics, calculated using a rooted neighbor-joining tree of OTUs in the R packages phangorn::import_mothur_dist (v2.7.1) [26], ape::bionj (v5.5) [27], and phyloseq::UniFrac (v1.36.0) [28, 29]. Beta-diversity was visualized in nonmetric multi-dimensional scaling (nMDS) plots with square root transformed data using vegan and ggplot2 v.3.3.5 [30]. Beta-dispersion was tested (vegan::betadisper) and, finding no violation of this assumption in our analyses, permutational multivariate ANOVA (vegan::adonis2) was used to assess significance. Pairwise comparisons were assessed using vegan::adonis2 and P-values were FDR-corrected for multiple comparisons. OTUs driving community differences between sampling groups were identified by the similarity percentages (SIMPER) function (vegan::simper) for seasonal and location analyses. OTUs explaining > 1% of the difference between groups were subjected to Kruskal-Wallis tests, with FDR-correction applied to confirm differential abundance with P < 0.05.
The core bacterial community was assessed by first identifying the OTUs present in all individual samples from each sampling location. These OTUs were then compared across all summer and winter sampling locations. Those found in all locations were considered the “core” microbiota. Heat trees describing the core microbiome were generated from a neighbor-joining tree of OTUs using the heat_tree function from the R package metacoder [31]. Differential abundance of core taxa between summer and winter was assessed using the Wilcoxon rank sum test with FDR correction. Log2 ratio of median relative abundance counts for taxa with adjusted P > 0.05 were adjusted to zero for plotting.
Bacterial plating and counts
Presence of viable bacteria was assessed for one sample collected from several sampled locations. Samples were thawed and 5 mL of each were placed into sterile 50-mL conical tubes. Immediately after, 45 mL of PBS was added before being vortexed. After mixing, 0.1 mL was removed and serially diluted in PBS. Each dilution was then plated on trypticase soy agar with 5% sheep’s blood (Becton Dickson, Franklin Lakes, NJ) and incubated at 37 °C for 24 h before counting colony forming units (CFUs) to determine total aerobic bacterial counts.
Results
Sampling and sequencing summary
A total of 1,833,757 raw sequences were generated from 26 summer samples (947,496 sequences), 18 winter samples (884,245 sequences), and 4 PCR negative controls (2,016 sequences). After cleanup, 1,390,389 sequences remained, including 736,707 from summer, 653,099 from winter, and 583 from the negative controls, resulting in 7,159 unique OTUs representing 34 phyla, 419 families, and 920 genera. The 2 never-before-used summer sand samples (SHP2, SHP16) and the 4 PCR negative controls were removed during normalization of sample sequence counts to 16,000 sequences per sample, with each having < 220 sequences per sample. This resulted in 24 summer and 18 winter samples that were used in all subsequent analysis. Pre-normalized Good’s coverages of the remaining samples ranged from 0.969 to 0.992 and post-normalized Good’s coverages ranged from 0.936 to 0.984. See Supplementary Table S1, Additional File 2 for additional details. The resulting summer samples averaged 16,072 ± 159 SD sequences per sample from 5,423 unique OTUs. Winter samples averaged 15,969 ± 197 SD sequences per sample from 4,488 unique OTUs.
Bacterial diversity and sand depth
Bacterial community diversity was first compared between samples collected at the surface and 7-in below the surface of drying recycled sand piles. Analyses were performed separately for summer and winter samples. No differences in alpha diversity were found across sampling depths for either summer (Chao: P = 0.15, KW, Shannon: P = 0.52, KW) or winter samples (Chao: P = 0.15, KW, Shannon: P = 0.09, ANOVA). Alpha diversity metrics are visualized in Supplementary Fig. S2, Additional File 1. Bray-Curtis Dissimilarity also did not differ by depth in either summer (P = 0.54, ANOVA) or winter samples (P = 0.14, ANOVA), as visualized in Supplementary Fig. S3, Additional File 1. Similar results were obtained using UniFrac, both weighted (summer: P = 0.39, winter: P = 0.14, ANOVA) and unweighted (summer: P = 0.65, winter: P = 0.17, ANOVA). As a result, surface and 7-in samples were combined for subsequent analyses.
Bacterial diversity across seasons
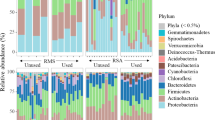
Comparison of bacterial community compositions between summer and winter samples reveal seasonal differences. Taxonomically, the 6 phyla with the highest relative abundances in all summer samples were the Proteobacteria (37% ± 15% SD), Bacteroidetes (30% ± 9%), Firmicutes (22% ± 13%), Actinobacteria (6% ± 4%), Spirochaetota (1% ± 2%), and Campilobacterota (1% ± 1%). The 5 genera with the largest relative abundances include the Acinetobacter (17% ± 12%), Psychrobacter (3% ± 2%), Pseudomonas (3% ± 2%), Proteiniphilum (3% ± 2%), and Flavobacterium (2% ± 1%). Similarly, winter samples were dominated by bacteria in the phyla Firmicutes (35% ± 10%), Proteobacteria (25% ± 8%), Bacteroidetes (24% ± 5%), Actinobacteria (12% ± 5%), Campilobacterota (2% ± 1%), and Spirochaetota (1% ± 1%). The 5 genera with the highest relative abundances in winter include Psychrobacter (7% ± 4%), Corynebacterium (6% ± 3%), Erysipelothrix (5% ± 1%), an unclassified Lactobacillales (5% ± 2%), and Facklamia (4% ± 2%). See Supplementary Figs. S4 and S5, Additional File 1, for more detail on bacterial community compositions.
Seasonal alpha and beta diversity analyses were performed without summer cow pen samples as there was no corresponding winter samples collected. For alpha diversity, which describes the diversity of individual samples, we found Chao’s Richness, an estimate of the number of unique species in a sample, to be greater in summer samples relative to winter samples (P = 0.025, KW). Shannon’s Diversity, which considers both the richness and evenness of a sample, did not differ by season (P = 0.239, ANOVA), suggesting summer samples contain additional rarer OTUs than winter samples (Supplementary Fig. S6, Additional File 1). Beta diversity, which describes the differences in community composition between samples, was determined using the Bray-Curtis Dissimilarity, a metric that considers both richness and evenness, and revealed the bacterial community composition of summer and winter samples to be distinct (P < 0.001, ANOVA) (Supplementary Fig. S7, Additional File 1). Similar results were also obtained using weighted and unweighted UniFrac distances (both P < 0.001, ANOVA), metrics that consider OTUs taxonomic relatedness in addition to richness (unweighted) and both richness and evenness (weighted). A total of 13 OTUs were predicted by SIMPER to drive the differences between summer and winter bacterial communities, of which 11 were differentially abundant by season. Of those 11, only 3 had higher mean relative abundances in summer than in winter: OTU1 and OTU2, which both classified to the genus Acinetobacter, and OTU15 classified to the genus Proteiniphilum. The remaining 8 OTUs were found in higher mean relative abundances in the winter samples, including OTU5, which classified as belonging to the genus Corynebacterium. See Supplementary Table S2, Additional File 2, for additional detail. Summer and winter samples were analyzed separately for subsequent analyses.
Bacterial diversity of summer sampling locations
The microbial diversity was compared across the 5 summer sampling locations: cow pens, grey water, wet recycled sand, 4-d sand, 14-d sand. Chao’s Richness did not differ across locations (P = 0.185, KW) (Fig. 1A). Shannon’s Diversity differed across sampling locations (P = 2.9e-3, KW) with the cow pen, grey water, and wet recycled sand samples being the most diverse. 4-d sand had the next highest diversity, being significantly lower diversity than the first 3 locations, and 14-d sand being the least diverse (Fig. 1A). The Bray-Curtis dissimilarity also differed between locations (P = 1e-4, ANOVA), with all 5 sampling locations being distinct except for the grey water and wet recycled sand samples (P = 0.1, ANOVA) (Fig. 2A and Supplementary Table S3, Additional File 2). Similar results were obtained using weighted and unweighted UniFrac and can be found in Supplementary Table S3, Additional File 2. A total of 34 OTUs were found to be differentially abundant between locations. Specifically, 6 OTUs were in lower abundance in 14-d sand compared to cow pen samples, including those in the genera Moheibacter, Ornithobacterium, and Aequorivita. We found 3 OTUs increased in abundance in the 14-d sand samples, relative to cow pen sand, including OTU1 and OTU2, which both classified to the genus Acinetobacter, and OTU3, which classified to the genus Pseudomonas. For additional detail, see Supplementary Table S4, Additional File 2.

Alpha diversity of summer and winter sampling locations. Boxplots of Chao’s Richness and Shannon’s Diversity Index values of samples from each of the summer (A) and winter (B) sampling locations. Differing letters indicate a significant difference between locations within each season (P < 0.05) using the Wilcoxon Rank Sum test for summer (A) and Tukey’s HSD for winter (B) locations. Pairwise comparisons of Chao’s Richness were not performed for summer locations since no overall differences were found (P > 0.05). Surface and 7-in samples are not differentiated as no differences were found between them across locations (P > 0.05)

Differences of bacterial community compositions across summer and winter sampling locations. Non-metric multidimensional scaling (nMDS) plot of the Bray-Curtis dissimilarity for microbial communities of recycled sand samples colleted in (A) summer and (B) winter. Each dot represents the bacterial community of a single sample and are colored by location. Ellipses represent standard error around centriod of each location and are colored by location. Surface and 7-in samples are not differentiated as no differences were found between them across locations (P > 0.05). Stress: A = 0.07, B = 0.052
Bacterial diversity of winter sampling locations
Bacterial community diversity was compared across the 4 winter sampling locations: grey water, wet recycled sand, 2-d sand, and 7-d sand. Community diversity differed across winter sampling locations, but to a lesser extent than summer samples. Both Chao’s Richness (P = 0.048, ANOVA) and Shannon’s Diversity (P = 0.015, ANOVA) differed slightly across the 4 winter locations (grey water, wet recycled sand, 2-day sand, and 7-day sand). Pairwise comparisons of individual locations revealed that Chao’s Richness of wet recycled sand was greater than 7-d sand (P = 0.03, Tukey HSD), Shannon’s Diversity of grey water was greater than 7-d sand (P = 0.026, Tukey HSD), and Shannon’s Diversity of wet recycled sand and 2-d sand trended towards being greater than 7-d sand (P = 0.055 and P = 0.059 resp., Tukey HSD) (Fig. 1B). Bray-Curtis dissimilarity and weighted UniFrac distances between winter locations trended towards a difference (P = 0.059 and P = 0.051 resp., ANOVA) while unweighted UniFrac distances did differ across locations (P = 0.005, ANOVA). Pairwise comparisons found 7-d sand to differ from both grey water and 2-day sand samples for all metrics, and from wet recycled sand when using unweighted UniFrac (Fig. 2B and Supplementary Table S3, Additional File 2). However, no OTUs predicted by SIMPER were differentially abundant between locations after FDR correction (Supplementary Table S5, Additional File 2].
A Core microbiota
The core microbiota was determined, which consisted of 141 OTUs belonging to 12 phyla and 97 genera and represents 68.45% ± 10.33% SD of the relative bacterial abundance of each sampling location. Across sampling locations, the relative abundance of the core microbiota ranged from 44.8% to 78.1% of summer locations and 67.9% to 80.2% of winter locations. The most abundant genera of the core microbiota include Acinetobacter (10% ± 12%), Psychrobacter (5% ± 4%), Corynebacterium (4% ± 3%), Pseudomonas (3% ± 3%), and an unclassified Lactobacillales (3% ± 2%). To better resolve the taxonomic identity of the core OTUs initially identified as “unclassified Lactobacillales” (OTU4 and OTU123), we conducted a BLASTN analysis of their consensus sequences against the NCBI’s non-redundant (nr) nucleotide database. The OTUs had a 98.42% and 99.21% sequence identity match, respectively, to several species in the genus Enterococcus, suggesting that these unclassified Lactobacillales likely belong to this genus. We also found these abundant core genera to be differentially abundant across seasons (Fig. 3). Of the most abundant genera, Acinetobacter was more abundant in summer than in winter samples, whereas Psychrobacter, Corynebacterium, and the unclassified Lactobacillales (Enterococcus) were more abundant in winter samples. Core OTUs classified to the genus Pseudomonas were not differentially abundant between seasons.

The core microbiota of a bedding sand recycling system. A heat-tree of the core microbiota of a bedding sand recycling system. Node and line widths increase with the mean relative abundance of the core taxonomic groups across all samples. Colors indicate the log2 ratio of the median counts of taxanomic groups between summer and winter samples. Increasing magenta indicates increased abundance in summer samples, cyan for increased abundance in winter samples, and grey represents no seasonal difference. Log2 ratio of median counts were set to zero for taxnomic groups having non-significant differential abundance by season (P < 0.05), per FDR-corrected pairwise Wilcoxon rank sum tests of median abundance counts
Viable bacterial counts
Viable aerobic bacterial counts were assessed for all summer sampling locations and two winter locations. Recovery from summer sand sampling locations ranged from 1.06 × 107 to 9.18 × 108 CFU/g of sand and 6.39 × 106 CFU/mL from summer grey water. Winter grey water resulted in 2.98 × 107 CFU/mL and winter 7-d surface sand contained 5.40 × 107 CFU/g. The remaining winter sand samples were not available for this analysis. See Supplementary Table S6, Additional File 2 for additional detail.
Discussion
In this study, we sought to characterize the bacterial community composition of a bedding sand recycling system on a Wisconsin dairy farm with respect to both location and season. Our compositional analysis revealed the 6 most abundant phyla for summer and winter samples to be identical, suggesting broad seasonal commonality. The most abundant phyla included several often found in the environment and in the bovine gastrointestinal tract including the Bacteroidetes, Firmicutes, Spirochaetota, Campilobacterota (formerly class Epsilonproteobacteria), and Proteobacteria, as well as the primarily soil-associated phylum Actinobacteria (Supplemental Figs. S3 and S4, Additional File 1) [32,33,34,35,36,37,38,39,40]. Both seasons were dominated by the Bacteroidetes, Firmicutes, and Proteobacteria, with the Proteobacteria being the most abundant phylum in summer samples and Firmicutes being the most abundant in winter samples. These results are largely consistent with those of a recent study focused on the microbiota of recycled bedding sand and its association with hock lesions and digital dermatitis in dairy cattle [41], although in their study, Actinobacteria was the most abundant phylum. This discrepancy may be due to their bedding system, which consisted of sand on top of compact soils, rather than concrete, thereby increasing the prevalence of soil Actinobacteria in their sand samples. This discrepancy may also result from several additional factors including variability in sand recycling system, season of collection, farming practices, or the surrounding environment.
Of the top 5 genera for each season, the relative abundances of Acinetobacter, Pseudomonas, and Corynebacterium are of particular note as these genera contain species that are known pathogens of dairy cattle and prior work has suggested that bacteria in the environment or bedding can be a leading cause of diseases on dairy farms [8, 42]. In our analysis, Acinetobacter was the most abundant genus across the sand recycling system. Acinetobacter are aerobic, rod-shaped Gram-negative bacteria, and notable for their ability to acquire resistance to multiple classes of antibiotics and survive for long periods of time on dry surfaces [43,44,45]. Though widely distributed in nature and not considered major mastitis pathogens in the US, studies of Chinese and Korean dairy herds have found Acinetobacter spp. to be increasingly isolated from mastitic milk samples [46,47,48,49]. Hoque et al. also found Acinetobacter to be the predominantly abundant genus in milk from Indian dairy cattle with clinical mastitis using shotgun metagenomics [50].
Similarly, Pseudomonas and Corynebacterium also contain species that are environmentally distributed and are known causative agents for bovine mastitis, specifically P. aeruginosa and C. bovis [51, 52]. Given the prevalence of Pseudomonas in our analysis, we note that the farm sampled in this study experienced cases of mastitis believed to be caused by Pseudomonas during our sampling period (D. Sockett, WI Veterinary Diagnostic Laboratory, personal communication, 2018). Although the methods used in our study are unable to conclusively differentiate taxa to the species-level, our findings suggest a potential route of transmission for Pseudomonas as a mastitis pathogen, and future work using culture-based approaches is warranted.
We next sought to understand how season impacted microbiota composition. Our data reveal that summer samples have increased richness, relative to winter samples, but we found no seasonal difference in Shannon’s Diversity metric, which considers both richness and abundance. This indicates that summer samples are primarily enriched in low abundance taxa and that the overall relative abundances of community members do not significantly shift between seasons (Supplementary Fig. S6, Additional File 1). Although the relative abundances did not strongly differ, the compositions of these communities differed between seasons (Supplementary Fig. S7, Additional File 1), which agrees with a previous study reporting that bacterial populations of bedding materials vary seasonally, likely due to changes in temperature and humidity [9, 53]. We note that the average temperature and humidity in May in southern Wisconsin is 20 °C and 66% humidity while in January the average temperature is − 3 °C and 75% humidity [54]. Given these findings, we posit that the microbial variation we observed is due to changes in overall temperature and humidity, and that this extends to all stages of the sand recycling process.
Our analyses further revealed enrichment of Acinetobacter OTUs in summer samples and the enrichment of several OTUs, including one Corynebacterium OTU, in winter samples (Supplemental Table S2, Additional File 2). These findings are also consistent with a previous report indicating that culturable mastitis pathogen counts increase in summer months [9]. Although less work has been done to understand the seasonal dynamics of Acinetobacter and Corynebacterium in dairy bedding, the incidences of human clinical infections of Acinetobacter baumannii are known to greatly increase in summer months, as are the relative abundances of freshwater Acinetobacter spp. [55, 56]. Similarly, a survey of milk isolates collected by the Wisconsin Veterinary Diagnostic Lab from 1,994 to 2,001 found the incidence of Corynebacterium bovis isolations to differ seasonally, though with higher incidences in summer months than in winter months [57].
Comparisons of bacterial community compositions between stages of the sand recycling system revealed high heterogeneity in summer samples but less heterogeneity in winter. For the summer samples, bacterial communities of the cow pen, grey water, and freshly recycled sand all had equally high diversity which then declined in the 4-d sand and further declined in the 14-d sand (Fig. 1A). Community compositions were similarly stratified with 4-d and 14-d communities being distinct from grey water and freshly recycled sand; however, communities of cow pen samples were also distinct from all other locations (Fig. 2A). These results suggest that the bacterial communities in cow pen sand are significantly modified by the sand recycling process. Identification of the bacterial taxa driving the differences between cow pen sand and 14-d sand (sand that is ready for re-bedding) revealed only 6 OTUs to have reduced abundance in 14-d sand relative to cow pen sand. Surprisingly, 3 OTUs were enriched in 14-d sand relative to sand from cow pens, 2 of which were classified as Acinetobacter and the third as Pseudomonas. Importantly, both Acinetobacter and Pseudomonas increased in relative abundance with time in the recycling room, with a 2–4-fold increase in Acinetobacter abundance and a 7-fold increase in Pseudomonas abundance (Supplementary Fig. S4, Additional File 1). These results may reflect that both organisms are known biofilm producers, which may allow them to resist removal from sand granules during washing, providing a competitive advantage [51, 58, 59]. Additionally, Acinetobacter spp. such as A. baumannii are well characterized as having high survivability on dry surfaces for long periods [44, 45]. However, the moisture content of the sand was not directly measured so additional studies are required to confirm the impact of moisture in this system. Additionally, with our current sampling design, we cannot discern whether these genera are surviving washing and then re-colonizing the sand or are being introduced from the surrounding farm environment as the sand sits in piles the sand recycling room.
Our study also found the bacterial communities in winter samples to be less heterogeneous across sampling locations, with only 7-d sand having decreased richness and diversity and a distinct community composition from several other sampled locations (Figs. 1B and Fig. 2B). No OTUs were found to drive the differences in diversity between 7-d sand compositions and 2-d sand, grey water, or wet recycled sand compositions, after correcting for multiple testing (Supplementary Table S5, Additional File 2). This is likely due to a combination of low sample size and reduced heterogeneity in the winter samples. The impact of drying on winter sand, especially relative to summer samples, is difficult to determine in our study as dryness was not measured and we performed 1-day convenience sampling and were subject to sand availability on that day. Similarly, cow pen samples were not readily available for winter sampling, preventing comparison of the community composition of 7-d sand with cow pen sand. We additionally found no difference in bacterial diversity or composition of sand collected at the top of sand piles and sand collected at lower depths for any of the samples despite the sand at the top being dry and the sand within the pile being visibly wet (Supplementary Figs. S2 and S3, Additional File 1).
A final goal of our study was to determine if a core microbiota was present across the sand recycling system. Members of the core microbiota we identified accounted for a surprisingly large portion of the bacterial community relative abundance for all locations (mean 68.45% ± 10.33% SD), which suggests a large portion of the recycled sand microbiota may be stable and consistent across seasons. The top 4 genera of the core microbiota included Acinetobacter, Corynebacterium, Psychrobacter, and Pseudomonas, with Acinetobacter enriched in summer samples, Corynebacterium and Psychrobacter enriched in winter, and Pseudomonas not differentially abundant by season, matching our earlier seasonal comparison of the total bacterial community (Fig. 3).
In addition, the most abundant genera identified in this study, Acinetobacter, Corynebacterium, Psychrobacter, Pseudomonas, and Enterococcus, are also common organisms associated with bulk tank raw milk [48, 60,61,62,63]. Although this study did not assess the microbial quality of the milk, the environment in which cattle are housed has been implicated as a source of milk contamination [64, 65]. While sand is viewed an ideal choice for bedding because it carries a decreased bacterial load in comparison to organic bedding materials, recycling sand may not necessarily eliminate the risk of bulk tank milk contamination.
We suggest that the presence and prevalence of several bacterial genera known to cause mastitis and bulk tank contamination warrant further investigation of the recycled sand microbiota, as it is possible that sand recycling does not eliminate sand bedding as a potential reservoir for these pathogens. As such, future work should seek to increase the number of farms to determine the variability of recycled sand communities across different farms and sand recycling systems. We note that the results of our study are constrained to the analysis of bacterial DNA relative abundances. While the enumeration of bacterial microflora in sand was found to be approximately 107–108 colony forming units per gram of sand, the incorporation of qPCR combined with a larger sample set from several farms would provide quantitative descriptions of the effects of sand recycling on total and specific bacterial abundances. Culturing and/or qPCR could thereby confirm if mastitis or enteric pathogens can be viably passaged through the sand recycling system across seasons. We also note that is study is unable to definitively identify the mechanisms driving seasonal and locational differences in bacterial community composition, thereby requiring more controlled future studies to discern the roles of dryness, climate, and environment in shaping this system. Finally, several of the most abundant genera we identified are characterized by their ability to accrue multi-drug resistance [51, 59, 66]. Focused work on the antibiotic resistance gene (ARG) carriage of both pathogenic and commensal bacteria could reveal the role of recycled bedding sand systems in ARG accumulation and dissemination.
Conclusions
In conclusion, we found that the bacterial community composition of a WI dairy farm bedding sand recycling system is both dynamic across seasons and stages of the sand recycling process, with a large “core” microbiota accounting for most of the bacterial community reads at each sampling location. The identification of an abundant core microbiota, and the presence and enrichment of several genera with known mastitis pathogen species suggests that bedding sand recycling systems may serve as bacterial reservoirs that warrant further investigation.
Availability of data and materials
The datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive repository under BioProject number PRJNA724660 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA724660].
References
Nebel RL, McGilliard ML. Interactions of high milk yield and reproductive performance in dairy cows. J Dairy Sci. 1993;76(10):3257–68. https://doi.org/10.3168/jds.S0022-0302(93)77662-6.
Stevenson JS. Reproductive management of dairy cows in high milk-producing herds. J Dairy Sci. 2001;84:E128–43. https://doi.org/10.3168/jds.S0022-0302(01)70207-X.
Windig JJ, Calus MPL, Veerkamp RF. Influence of herd environment on health and fertility and their relationship with milk production. J Dairy Sci. 2005;88(1):335–47. https://doi.org/10.3168/jds.S0022-0302(05)72693-X.
Lombard JE, Tucker CB, von Keyserlingk MAG, Kopral CA, Weary DM. Associations between cow hygiene, hock injuries, and free stall usage on US dairy farms. J Dairy Sci. 2010;93(10):4668–76. https://doi.org/10.3168/jds.2010-3225.
Gomez A, Cook NB. Time budgets of lactating dairy cattle in commercial freestall herds. J Dairy Sci. 2010;93(12):5772–81. https://doi.org/10.3168/jds.2010-3436.
van Gastelen S, Westerlaan B, Houwers DJ, van Eerdenburg FJCM. A study on cow comfort and risk for lameness and mastitis in relation to different types of bedding materials. J Dairy Sci. 2011;94(10):4878–88. https://doi.org/10.3168/jds.2010-4019.
Bak AS, Herskin MS, Jensen MB. Effect of sand and rubber surface on the lying behavior of lame dairy cows in hospital pens. J Dairy Sci. 2016;99(4):2875–83. https://doi.org/10.3168/jds.2015-9937.
Hogan JS, Smith KL, Hoblet KH, Todhunter DA, Schoenberger PS, Hueston WD, et al. Bacterial counts in bedding materials used on nine commercial dairies. J Dairy Sci. 1989;72(1):250–8. https://doi.org/10.3168/jds.S0022-0302(89)79103-7.
Rowbotham RF, Ruegg PL. Bacterial counts on teat skin and in new sand, recycled sand, and recycled manure solids used as bedding in freestalls. J Dairy Sci. 2016;99(8):6594–608. https://doi.org/10.3168/jds.2015-10674.
Bradley AJ, Leach KA, Green MJ, Gibbons J, Ohnstad IC, Black DH, et al. The impact of dairy cows’ bedding material and its microbial content on the quality and safety of milk – a cross sectional study of UK farms. Int J Food Microbiol. 2018;269:36–45. https://doi.org/10.1016/j.ijfoodmicro.2017.12.022.
Patel K, Godden SM, Royster E, Crooker BA, Timmerman J, Fox L. Relationships among bedding materials, bedding bacteria counts, udder hygiene, milk quality, and udder health in US dairy herds. J Dairy Sci. 2019;102(11):10213–34. https://doi.org/10.3168/jds.2019-16692.
Hogan J, Smith KL. Managing environmental mastitis. Vet Clin North Am Food Anim Pract. 2012;28(2):217–24. https://doi.org/10.1016/j.cvfa.2012.03.009.
Kristula MA, Rogers W, Hogan JS, Sabo M. Comparison of bacteria populations in clean and recycled sand used for bedding in dairy facilities. J Dairy Sci. 2005;88(12):4317–25. https://doi.org/10.3168/jds.S0022-0302(05)73118-0.
Kull JA, Ingle HD, Black RA, Eberhart NL, Krawczel PD. Effects of bedding with recycled sand on lying behaviors, udder hygiene, and preference of lactating Holstein dairy cows. J Dairy Sci. 2017;100(9):7379–89. https://doi.org/10.3168/jds.2016-12307.
Rowbotham RF, Ruegg PL. Associations of selected bedding types with incidence rates of subclinical and clinical mastitis in primiparous Holstein dairy cows. J Dairy Sci. 2016;99(6):4707–17. https://doi.org/10.3168/jds.2015-10675.
Stevenson DM, Weimer PJ. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl Microbiol Biotechnol. 2007;75(1):165–74. https://doi.org/10.1007/s00253-006-0802-y.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–20. https://doi.org/10.1128/AEM.01043-13.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41. https://doi.org/10.1128/AEM.01541-09.
Pruesse E, Quast C, Knittel K, Fuchs BM, Glo FO, Ludwig W. SILVA : a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–96. https://doi.org/10.1093/nar/gkm864.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200. https://doi.org/10.1093/bioinformatics/btr381.
Westcott SL, Schloss PD. OptiClust, an improved method for assigning amplicon-based sequence data to operational taxonomic units. mSphere. 2017;2(2):1–11.
Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1–2):203–14. https://doi.org/10.1089/10665270050081478.
Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40(3):237–64. https://doi.org/10.1093/biomet/40.3-4.237.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2021. https://www.R-project.org
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package. R package version 2.5–7. 2020. https://CRAN.R-project.org/package=vegan.
Schliep K. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27(4):592–3. https://doi.org/10.1093/bioinformatics/btq706.
Paradis E, Schliep K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35(3):526–8. https://doi.org/10.1093/bioinformatics/bty633.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217.
Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4(1):17–27. https://doi.org/10.1038/ismej.2009.97.
Wickham H. Ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016. https://ggplot2.tidyverse.org
Foster ZSL, Sharpton TJ, Grünwald NJ. Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput Biol. 2017;13(2):1–15. https://doi.org/10.1371/journal.pcbi.1005404.
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, et al. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev. 2007;71(3):495–548. https://doi.org/10.1128/MMBR.00005-07.
Jewell KA, McCormick CA, Odt CL, Weimer PJ, Suen G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl Environ Microbiol. 2015;81(14):4697–710. https://doi.org/10.1128/AEM.00720-15.
Mao S, Zhang M, Liu J, Zhu W. Characterizing the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep. 2015;5:16116. https://doi.org/10.1038/srep16116.
Wesley IV, Wells SJ, Harmon KM, Green A, Schroeder-Tucker L, Glover M, et al. Fecal shedding of Campylobacter and Arcobacter spp. in dairy cattle. Appl Environ Microbiol. 2000;66(5):1994–2000. https://doi.org/10.1128/AEM.66.5.1994-2000.2000.
Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G. Environmental and gut Bacteroidetes: the food connection. Front Microbiol. 2011;2(93):1–16. https://doi.org/10.3389/fmicb.2011.00093.
Filippidou S, Wunderlin T, Junier T, Jeanneret N, Dorador C, Molina V, et al. A combination of extreme environmental conditions favor the prevalence of endospore-forming Firmicutes. Front Microbiol. 2016;7(1707):1–11. https://doi.org/10.3389/fmicb.2016.01707.
Nakagawa S, Takaki Y. Nonpathogenic Epsilonproteobacteria. eLS. 2009. doi:https://doi.org/10.1002/9780470015902.a0021895.
Paster BJ, Dewhirst FE. Phylogenetic foundation of spirochetes. J Mol Microbiol Biotechnol. 2000;2(4):341–4.
Gupta RS. The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol Rev. 2000;24(4):367–402. https://doi.org/10.1111/j.1574-6976.2000.tb00547.x.
Li H, Wang X, Wu Y, Zhang D, Xu H, Xu H, et al. Relationships among bedding materials, bedding bacterial composition and lameness in dairy cows. Asian-Australas J Anim Sci. 2020;34(9):1559–68. https://doi.org/10.5713/ajas.20.0565.
Pankey JW. Premilking udder hygiene. J Dairy Sci. 1989;72(5):1308–12. https://doi.org/10.3168/jds.S0022-0302(89)79238-9.
Nocera FP, Attili AR, De Martino L. Acinetobacter baumannii: its clinical significance in human and veterinary medicine. Pathogens. 2021;10(2):1–13. https://doi.org/10.3390/pathogens10020127.
Wendt C, Dietze B, Dietz E, Rüden H. Survival of Acinetobacter baumannii on dry surfaces. J Clin Microbiol. 1997;35(6):1394–7. https://doi.org/10.1128/jcm.35.6.1394-1397.1997.
Wagenvoort JHT, Joosten EJAJ. An outbreak Acinetobacter baumannii that mimics MRSA in its environmental longevity. J Hosp Infect. 2002;52(3):226–7. https://doi.org/10.1053/jhin.2001.1294.
Al Atrouni A, Joly-Guillou ML, Hamze M, Kempf M. Reservoirs of non-baumannii Acinetobacter species. Front Microbiol. 2016;7:49. https://doi.org/10.3389/fmicb.2016.00049.
Song X, Huang X, Xu H, Zhang C, Chen S, Liu F, et al. The prevalence of pathogens causing bovine mastitis and their associated risk factors in 15 large dairy farms in China: an observational study. Vet Microbiol. 2020;247:108757. https://doi.org/10.1016/j.vetmic.2020.108757.
Gurung M, Nam HM, Tamang MD, Chae MH, Jang GC, Jung SC, et al. Prevalence and antimicrobial susceptibility of Acinetobacter from raw bulk tank milk in Korea. J Dairy Sci. 2013;96(4):1997–2002. https://doi.org/10.3168/jds.2012-5965.
Nam HM, Kim JM, Lim SK, Jang KC, Jung SC. Infectious aetiologies of mastitis on Korean dairy farms during 2008. Res Vet Sci. 2010;88(3):372–4. https://doi.org/10.1016/j.rvsc.2009.12.008.
Hoque MN, Istiaq A, Rahman MS, Islam MR, Anwar A, Siddiki AMAMZ, et al. Microbiome dynamics and genomic determinants of bovine mastitis. Genomics. 2020;112(6):5188–203. https://doi.org/10.1016/j.ygeno.2020.09.039.
Park H, Hong M, Hwang S, Park Y, Kwon K, Yoon J, et al. Characterisation of Pseudomonas aeruginosa related to bovine mastitis. Acta Vet Hung. 2014;62(1):1–12. https://doi.org/10.1556/avet.2013.054.
Gonçalves JL, Tomazi T, Barreiro JR, Beuron DC, Arcari MA, Lee SHI, et al. Effects of bovine subclinical mastitis caused by Corynebacterium spp. on somatic cell count, milk yield and composition by comparing contralateral quarters. Vet J. 2016;209:87–92. https://doi.org/10.1016/j.tvjl.2015.08.009.
Godden S, Bey R, Lorch K, Farnsworth R, Rapnicki P. Ability of organic and inorganic bedding materials to promote growth of environmental bacteria. J Dairy Sci. 2008;91(1):151–9. https://doi.org/10.3168/jds.2007-0415.
Current Results. Current Results Publishing Ltd., British Columbia, Canada. 2021. https://www.currentresults.com/Weather/Wisconsin/average-wisconsin-weather.php. Accessed 16 July 2021.
Kurihara MN, Sales RO, Silva KE, Maciel WG, Simionatto S. Multidrug-resistant Acinetobacter baumannii outbreaks: a global problem in healthcare settings. Rev Soc Bras Med Trop. 2020;53:e20200248. https://doi.org/10.1590/0037-8682-0248-2020.
Adewoyin MA, Okoh AI. Seasonal shift in physicochemical factors revealed the ecological variables that modulate the density of Acinetobacter species in freshwater resources. Int J Environ Res Public Health. 2020;17(10):3606. https://doi.org/10.3390/ijerph17103606.
Makovec JA, Ruegg DPL. Antimicrobial resistance of bacteria isolated from dairy cow milk samples submitted for bacterial culture: 8,905 samples (1994-2001). J Am Vet Med Assoc. 2006;222(11):1582–9.
Ude S, Arnold DL, Moon CD, Timms-Wilson T, Spiers AJ. Biofilm formation and cellulose expression among diverse environmental Pseudomonas isolates. Environ Microbiol. 2006;8(11):1997–2011. https://doi.org/10.1111/j.1462-2920.2006.01080.x.
Qi L, Li H, Zhang C, Liang B, Li J, Wang L, et al. Relationship between antibiotic resistance, biofilm formation, and biofilm-specific resistance in Acinetobacter baumannii. Front Microbiol. 2016;7:483. https://doi.org/10.3389/fmicb.2016.00483.
Addis MF, Tanca A, Uzzau S, Oikonomou G, Bicalho RC, Moroni P. The bovine milk microbiota: insights and perspectives from-omics studies. Mol BioSyst. 2016;12(8):2359–72. https://doi.org/10.1039/C6MB00217J.
Hahne J, Kloster T, Rathmann S, Weber M, Lipski A. Isolation and characterization of Corynebacterium spp. from bulk tank raw cow's milk of different dairy farms in Germany. PLoS One. 2018;13(4):e0194365.
Jayarao BM, Wang L. A study on the prevalence of gram-negative bacteria in bulk tank milk. J Dairy Sci. 1999;82(12):2620–4. https://doi.org/10.3168/jds.S0022-0302(99)75518-9.
McAuley CM, Britz ML, Gobius KS, Craven HM. Prevalence, seasonality, and growth of enterococci in raw and pasteurized milk in Victoria, Australia. J Dairy Sci. 2015;98(12):8348–58. https://doi.org/10.3168/jds.2015-9335.
Elmoslemany AM, Keefe GP, Dohoo IR, Jayarao BM. Risk factors for bacteriological quality of bulk tank milk in Prince Edward Island dairy herds. Part 2: Bacteria count-specific risk factors. J Dairy Sci. 2009;92(6):2644–52. https://doi.org/10.3168/jds.2008-1813.
Vissers MM, Driehuis F. On-farm hygienic milk production. In: Tamine AY, editor. Milk processing and quality management. Chichester: Wiley-Blackwell; 2009. p. 1–22.
Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis. 2008;197(8):1079–81. https://doi.org/10.1086/533452.
Acknowledgements
We would like to thank the Suen lab for their support, careful reading of this manuscript, and invaluable suggestions.
Funding
This study was funded by the Walter and Martha Renk Endowed Laboratory for Food Safety and the UW-Madison Food Research Institute. AJS and GS were supported by a United States Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA) Food Safety Challenge Grant #20017–68003-26500. GS was also supported by a USDA NIFA Agricultural and Food Research Initiative Foundational Grant Foundation grant.
#2020–67015-31576.
Author information
Authors and Affiliations
Contributions
HP was responsible for study design, sample collection, processing, and co-contributed to data analysis and writing. AS supported sample processing and co-contributed to data analyses and writing. NA contributed to study design and data analysis. DS contributed to study design and sample collection. GS reviewed data analyses and manuscript drafts. CC was corresponding author and was responsible for overseeing study design, sample collection, processing, analyses, and revisions of data analyses and manuscript drafts. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1: Supplementary Fig. S1
Additional File 1.docx; Diagram of the sand recycling process and its stages. Supplementary Fig. S2 Additional File 1.docx; Violin plots of Chao’s Richness (A) and Shannon’s Diversity (B) of samples collected from the surface (top) of the drying recycled sand piles or 7-in (7in) below the surface. Supplementary Fig. S3 Additional File 1.docx; Non-metric multidimensional scaling (nMDS) plot of the Bray-Curtis dissimilarity of microbial communities from the surface (top) and 7-in subserface (7in) samples of drying recycled sand piles. Supplementary Fig. S4 Additional File 1.docx; Stacked bar plots of the relative abundances of the 6 most abundant phyla (A) and 10 most abundant genera (B) in grey water and sand at all locations in the recycling process during summer sampling. Supplementary Fig. S5 Additional File 1.docx; Stacked bar plots of the relative abundances of the 6 most abundant phyla (A) and 10 most abundant genera (B) in grey water and sand at all locations of the recycling process during winter sampling. Supplementary Fig. S6 Additional File 1.docx; Violin plots of Chao’s Richness (A) and Shannon’s Diversity (B) of samples collected in summer and winter. Supplementary Fig. S7 Additional File 1.docx; Non-metric multidimensional scaling (nMDS) plot of the Bray-Curtis dissimilarity for microbial communities of recycled sand samples collected during summer and winter sampling.
Additional file 2: Supplementary Table S1
Additional File 2.xlsx; A table of samples, their coverage, read counts, alpha diversity metrics, and metadata. Supplementary Table S2 Additional File 2.xlsx; List of OTUs predicted by SIMPER to drive differences found between summer and winter samples. Supplementary Table S3 Additional File 2.xlsx; P-values of pair-wise comparisons of Bray-Curtis Dissimilarity and UniFrac distances between summer and winter locations. Supplementary Table S4 Additional File 2.xlsx; List of OTUs predicted by SIMPER to drive differences found between summer sampling locations. Supplementary Table S5 Additional File 2.xlsx; List of OTUs predicted by SIMPER to drive differences found between winter sampling locations. Supplementary Table S6 Additional File 2.xlsx; A table of aerobic viable plate counts for several summer and winter sampling locations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pilch, H.E., Steinberger, A.J., Sockett, D.C. et al. Assessing the microbiota of recycled bedding sand on a Wisconsin dairy farm. J Animal Sci Biotechnol 12, 114 (2021). https://doi.org/10.1186/s40104-021-00635-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-021-00635-6