
Abstract
Background
Mastitis in dairy cows caused by Staphylococcus aureus is a major problem hindering economic growth in dairy farms worldwide. It is difficult to prevent or eliminate due to its asymptomatic nature and long persistence of infection. Although transcriptomic responses of bovine mammary gland cells to pathogens that cause mastitis have been studied, the common responses of peripheral blood leukocytes to S. aureus infection across two consecutive generations of dairy cattle have not been investigated.
Methods
In the current study, RNA-Seq was used to profile the transcriptomes of peripheral blood leukocytes sampled from S. aureus-infected mothers and their S. aureus-infected daughters, and also healthy non-infected mothers and their healthy daughters. Differential gene expression was evaluated as follows: 1) S. aureus-infected cows versus healthy non-infected cows (S vs. H, which include all the mothers and daughters), 2) S. aureus-infected mothers versus healthy non-infected mothers (SM vs. HM), and 3) S. aureus-infected daughters versus healthy non-infected daughters (SMD vs. HMD).
Results
Analysis of all identified expressed genes in the four groups (SM, SMD, HM, and HMD) showed that EPOR, IL9, IFNL3, CCL26, IL26 were exclusively expressed in both the HM and HMD groups, and that they were significantly (P < 0.05) enriched for the cytokine-cytokine receptor interaction pathway. A total of 17, 13 and 10 differentially expressed genes (DEGs) (FDR Padj. < 0.1 and |FC| > 1.2) were detected in the three comparisons, respectively. DEGs with P < 0.05 and |FC| > 2 were used for functional enrichment analyses. For the S vs. H comparison, DEGs detected included CCL20, IL13 and MMP3, which are associated with the IL-17 signaling pathway. In the SM vs. HM and SMD vs. HMD comparisons, five (BLA-DQB, C1R, C2, FCGR1A, and KRT10) and six (BLA-DQB, C3AR1, CFI, FCAR, FCGR3A, and LOC10498484) genes, respectively, were involved in the S. aureus infection pathway.
Conclusions
Our study provides insights into the transcriptomic responses of bovine peripheral blood leukocytes across two generations of cattle naturally infected with S. aureus. The genes highlighted in this study could serve as expression biomarkers for mastitis and may also contain sequence variation that can be used for genetic improvement of dairy cattle for resilience to mastitis.
Similar content being viewed by others

Background
Bovine mastitis is an inflammation-driven disease of the mammary gland in cows. It normally occurs in response to infection by one of a number of pathogenic microorganisms including Escherichia coli and Staphylococcus aureus [1, 2]. It is widely recognized that the high incidence of bovine mastitis, coupled with associated animal welfare problems and the use of antimicrobials, means that this disease is one of the major challenges facing the dairy industry in the twenty-first century [3]. S. aureus is an important type of Gram-positive bacteria and is defined as a “contagious pathogen” [4]. S. aureus can adapt to the mammary gland environment and establish subclinical infections [1, 5]. The symptoms of S. aureus mastitis are usually less severe than mastitis caused by infection with E. coli, and may even be asymptomatic; however, the infection can persist for long periods and can also exhibit marked resistance to antibiotics [6]. S. aureus typically spreads among cattle within dairy herds during the process of milking [7], making prevention, control and elimination particularly difficult. Therefore, bovine mastitis caused by this bacterial pathogen represents a substantial economic burden to dairy production and also poses a significant public health risk through milk consumption.
Global functional genomics technologies, such as RNA sequencing (RNA-Seq), which is enabled by high-throughput sequencing (HTS), are ideally suited for investigating the complex host-pathogen interaction underlying mastitis disease caused by S. aureus infection and may also provide relevant data for elucidating the molecular mechanisms associated with resilience to mastitis disease. RNA-Seq enables expression quantification analysis of thousands of genes simultaneously [6, 8], thereby facilitating identification of individual genes that exhibit the largest expression changes in response to a biological perturbation such as infection by a bacterial pathogen. Several transcriptomics studies have investigated the bovine mammary gland tissue response to S. aureus [9] and E. coli [10], and also the hepatic tissue response to E. coli infection and lipopolysaccharide (LPS) challenge [11, 12]. However, to-date, no studies have been carried out on the peripheral blood transcriptome for S. aureus infection in cows and their offspring simultaneously.
In the current study, using RNA-Seq, we characterized the transcriptomes of peripheral blood leukocyte (PBL) samples collected from S. aureus-positive mother-daughter pairs and S. aureus-negative control mother-daughter pairs. The main objective of this work was to identify candidate blood-based transcriptional biomarkers for bovine mastitis caused by S. aureus and to provide new insights into host-pathogen interaction and the genetics of disease resistance.
Methods
Animal selection and sampling
All procedures for collection of animal blood and milk samples were approved by the Animal Welfare Committee of China Agricultural University, Beijing, China. All experiments were conducted according to the regulations and guidelines established by this committee (permit number: DK996).
In the present study, a lactating dairy cow and her lactating mother were defined as a mother-daughter pair. A total of 38 mother-daughter pairs were selected based on pedigree information and somatic cell counts (SCC) from a lactating herd of Holstein cows (n ≈ 1,200) at a dairy farm near Beijing, China. The SCC data for three consecutive months were measured and recorded. The Dairy herd improvement (DHI) records were provided by the Dairy Data Centre of China (www.holstein.org.cn). For the present study, milk SCC values of less than 100,000 cells/mL from both mothers and daughters were considered to be healthy [13]. Conversely, SCC values larger than 100,000 cells/mL were considered for diagnosis of subclinical mastitis. Detailed information on cow selection is provided in Fig. 1a and Table 1.

Identification of S. aureus isolated from milk samples from Holstein cows. a Workflow of sample selection. A total of 38 mother-daughter pairs were selected from 1,200 lactating dairy cattle according to pedigree and SCC records. After S. aureus identification from milk samples, four S. aureus-positive and four S. aureus-negative pairs were used for subsequent studies. b Specific PCR and electrophoresis map of the S. aureus thermonuclease (nuc) gene (279 bp). SA: abbreviation of S. aureus. +: S. aureus-positive control; −: S. aureus-negative control. SM: S. aureus mastitis mother; HM: healthy mother; SMD: S. aureus mastitis daughter; HMD: healthy daughter. c Sequence alignment of partial nuc gene. The upper lines were amplified from samples collected from the cows’ milk; the lower line is nuc sequence amplified from one of the S. aureus-positive samples. Identity = 95.68%
For S. aureus identification, a total of 30 mL of fresh milk was collected and mixed from all of the four lactating quarters of each cow. Subsequently, based on the bacteriological culture and PCR results, four S. aureus-positive mother-daughter pairs were selected and separated into the S. aureus mother group (SM, n = 4) and the S. aureus daughter group (SMD, n = 4). Similarly, four S. aureus-negative mother-daughter pairs were separated into the healthy mother group (HM, n = 4) and the healthy daughter group (HMD, n = 4) (Fig. 1a).
S. aureus isolation and identification
Bacteriological culture of milk samples was carried out according to National Mastitis Council standards [14]. A volume of 3 mL milk was mixed into trypticase soy broth containing 7.5% NaCl and cultured at 37 °C for 18–24 h. After that, a total of 10 mL culture was placed into Baird-Parker agar plates with tellurite and 5% egg yolk and cultured at 37 °C for 18–24 h. Two suspected colonies from each sample with surrounding clear zones were transferred to trypticase soy agar plated for DNA collection. Following this step, PCR amplification and sequencing of the S. aureus thermonuclease gene (nuc) [15] was performed (Fig. 1b and c). The PCR reaction was performed in 25 μL, containing 3 μL of genomic DNA (30–50 ng/μL), 1 μL of each primer (10 μmol), 12.5 μL of Taq™ Mix (1.25 units/25 μL reaction) and 7.5 μL of ddH2O. PCR was performed using the following thermocycler program: 94 °C for 10 min; 35 cycles of 94 °C for 30 s, 59 °C for 30 s and 72 °C for 30 s; 72 °C for 7 min.
Blood collection, RNA extraction and RNA-sequencing
A 20-mL blood sample was obtained from the caudal vein from each animal for buffy coat (leukocytes) collection using 15 min centrifugation at 3,000 r/min.
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to isolate total RNA from leukocytes according to the manufacturer’s protocol. The RNA quality was checked on a 1% agarose gel and quantified using a Qubit RNA Assay Kit and a Qubit 2.0 Fluorometer (ThermoFisher Scientific, Waltham, MA USA). RNA integrity was assessed with the BioAnalyzer 2100 System (Agilent Technologies, Santa Clara, CA, USA). All 16 RNA samples had an RNA integrity number (RIN) larger than 7.0 (Supplementary Table S1). The 28S:18S rRNA ratios of all samples were larger than 1.7 (Supplementary Table S1). An equivalent amount (4 μg) of total PBL RNA purified from each animal was used to construct RNA-Seq libraries with the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA). Finally, the libraries were sequenced using 150 bp paired-end reads with the Illumina HiSeq X Ten System (CapitalBio Technology, Beijing, China).
Quality control for raw data
Trimmomatic software version 0.38 [16] was used to filter out the adapter sequence and low-quality bases/reads with the default parameters (http://www.usadellab.org/cms/?page=trimmomatic). Further quality assessment of the sequence reads was then undertaken using FastQC version 0.11.8 [17]. After these QC procedures were completed, the sequence read data were used for the downstream analyses in the computational workflow.
Reads alignment and abundance estimation
The Bos taurus ARS-UCD1.2 reference assembly (FASTA format) and annotated gene model (GTF format) were downloaded from the Ensembl database (ftp.ensemble.org/pub/release-96/gtf/bos_taurus). The QC-assessed sequence read data for each sample were aligned to the reference genome using STAR with the basic options [18]. Transcript abundance was quantified using featureCounts [19] in the R subread package [20] under the default setting, and read counts were calculated to estimate the transcript expression levels. Genes with read counts more than ten in at least two samples were defined as expressed genes. The rlog-normalized read count was calculated with DESeq2 [21], which was then used to performed differential expression analysis.
The transcriptional responses to S. aureus infection were investigated by comparing differentially expressed genes between the infected and non-infected healthy control groups. We firstly analyzed gene expression levels in the S group (n = 8, 4 animals each in the SM and SMD groups) and H group (n = 8, 4 animals each in the HM and HMD groups), and compared the differentially expressed genes (DEGs) between the two groups. Detection of DEGs was also performed for the comparisons of the mother groups (SM vs. HM) and the daughter groups (SMD vs. HMD).
Functional enrichment and annotation
Gene set enrichment analysis was performed for all the expressed genes detected between the S and H groups, using the GSEA software package (v4.0.3) with the Human NCBI Gene ID Molecular Signature Database (MSigDB) version 7.0 and Hallmark database version 7.0 [22,23,24]. Gene sets were considered significant when P < 0.05 and FDR < 0.25 [25, 26].
The DEGs (P < 0.05 and |FC| > 2) in the comparisons of S vs. H, SM vs. HM and SMD vs. HMD were used to perform the following functional enrichment analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses of gene clusters were performed using clusterProfiler [27], with results exhibiting P < 0.05 considered significant. An interaction network analysis was also conducted using Ingenuity Pathway Analysis (IPA; Qiagen, Redwood City, CA, USA) [28].
Validation of RNA-Seq results through reverse transcription quantitative real-time PCR (RT-qPCR)
To quantitatively assess the reliability of our sequencing data, the expression levels of seven (SM vs. HM comparison) and ten (SMD vs. HMD comparison) randomly selected genes were estimated with RT-qPCR in the same samples used for RNA-Seq (Supplementary Table S1). The RNA samples were reverse transcribed into cDNA using the PrimeScript™ RT reagent kit according to the manufacturer’s instructions (Takara Bio, Dalian, China). Real-time detection of specific PCR products was performed with the Sybr Green I Master Mix Kit (Roche Diagnostics, Mannheim, Germany) on the LightCycler 480 II (Roche Diagnostics Ltd., Basel, Swetzerland), according to the manufacturer’s protocol. The oligonucleotide primers used for the RT-qPCR analysis are provided in Supplementary Table S2. The thermocycler program used was as follows: one cycle of preincubation at 95 °C for 10 min, 45 cycles of amplification (95 °C for 10 s, 60 °C for 10 s, and 72 °C for 10 s). Duplicate RT-qPCR assays were performed on each cDNA sample and relative expression values were calculated using the 2–△△Ct method with the bovine glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) as the internal reference [29, 30]. The log2|FC| values were calculated from RT-qPCR data to directly compare to the RNA-Seq results.
Statistical analysis
A linear regression analysis was conducted using the Prism software package (version 8; GraphPad Software, San Diego, CA, USA) to evaluate gene expression levels among experimental groups. For RT-qPCR data, the significant differences between groups were examined with the Student’s t-test.
Results
Transcriptomic data and detection of expressed genes in peripheral blood leukocyte samples
In the present study, an average of 22,435,225 (ranging from 18,801,274 to 25,620,906) paired-end reads were generated as raw data from the 16 RNA-Seq libraries. After filtering, an average of 20,955,429 reads (ranging from 16,864,277 to 23,829,973) were retained for each library. The average uniquely mapping rate was 94.11% (Supplementary Table S3). There were 10,287 detectable genes expressed in both the SM and SMD groups, and 10,248 gene expressed in the HM and HMD groups (Fig. 2a). A total of 10,086 detectable expressed genes were shared across the four groups. A total of 201 genes were identified that were exclusively expressed in the two consecutive generations of animals naturally infected with S. aureus (SM and SMD), which may be associated with susceptibility to S. aureus-induced mastitis. Conversely, 162 genes were exclusively expressed in the two consecutive generations of healthy control animals (HM and HMD), which may be associated with resilience to S. aureus-induced mastitis.

Comparison of gene expression profiles between mothers and daughters. a Venn diagram showing genes only expressed in S. aureus-positive mother-daughter pairs (purple circle), genes only expressed in S. aureus-negative mother-daughter pairs (yellow circle), and genes in intersection are common to both groups. b Linear regression analysis of expression levels for 201 genes exclusively expressed in the SM and SMD groups. c Linear regression analysis of expression levels for 162 genes exclusively expressed in the HM and HMD groups. d Significantly enriched (P < 0.05) KEGG pathways of genes exclusively expressed in the S. aureus-positive groups (SM and SMD). e Significantly enriched (P < 0.05) KEGG pathways of genes exclusively expressed in the S. aureus-negative groups (HM and HMD)
Furthermore, the correlation between the gene expression profiles (log2|FC| of 4000 randomly selected genes) in the two generations of SM vs. HM and SMD vs. HMD was significant (R2 = 0.3410, P < 0.0001, Supplementary Fig. S1). This result indicates that gene expression changes due to S. aureus infection challenge were moderately conserved between the two generations. The linear regression analyses were also performed using the normalized read counts of the 201 exclusively expressed genes (R2 = 0.9991, P < 0.0001, Fig. 2b) in the two generations of animals naturally infected with S. aureus (SM and SMD) and the 162 exclusively expressed genes (R2 = 0.9921, P < 0.0001, Fig. 2c) in the two generations of healthy controls (HM and HMD).
Transcriptome changes in peripheral blood leukocytes from S. aureus-positive cattle compared to S. aureus-negative control cattle
It is well established that S. aureus, as an intracellular pathogen of mammals, has evolved a wide range of mechanisms for immunosuppression and immunoreaction [31, 32]. To investigate host-pathogen interaction in bovine PBL infected with S. aureus, the transcriptomes of infected cattle and non-infected healthy control animals were compared. Although no significant DEGs were detected using an FDR Padj. threshold of 0.05, a total of 17, 13 and 10 DEGs were observed for the three comparisons (Table 2, 3 and 4), respectively, using the criteria of FDR Padj. < 0.1 and |FC| > 1.2 [33,34,35]. Furthermore, a total of 301, 283 and 260 DEGs (P < 0.05, log2|FC| > 2) (Supplementary Table S5, 6 and 7 and Fig. S2) were used for functional enrichment analysis. The expression heat maps of the 301, 283 and 260 genes of the three comparisons revealed different transcriptional profiles between S. aureus-positive and S. aureus-negative samples (Fig. 3). To validate the RNA-Seq results, seven and ten genes were selected for RT-qPCR validation in the SM vs. HM and SMD vs. HMD comparisons (Fig. 4), respectively. Results from this analysis showed that the gene expression patterns obtained using RNA-Seq were consistent with the results generated from RT-qPCR (Fig. 4a and c). The correlation coefficients between the RNA-Seq and RT-qPCR results for the genes in the SM vs. HM and SMD vs. HMD comparisons were 0.97 and 0.90 (P < 0.0001), respectively (Fig. 4b and d).

Transcriptomic changes of PBL in the three comparisons. Cluster analyses and heat maps of DEGs in (a) S vs. H, (b) SM vs. HM and (c) SMD vs. HMD. The different columns represent different samples and the different rows denote different DEGs. Red and blue show increased and decreased expression, respectively

RT-qPCR validation of RNA-Seq results. a and c RT-qPCR confirmation results for the randomly selected DEGs from the SM vs. HM comparison and the SMD vs. HMD comparison, respectively. b and d Regression analysis of the log2|FC| values between the RNA-Seq and RT-qPCR validation of the SM vs. HM and the SMD vs. HMD comparisons, respectively. The GAPDH gene was used as an internal reference control gene. * means P < 0.05. ** means P < 0.01
Functional enrichment and annotation
The GSEA analysis performed between S and H groups revealed that a total of 38 gene sets were upregulated in S group compared to the controls, of which six gene sets were significant at FDR < 0.25 and P < 0.05. Significantly enriched gene sets (FDR < 0.25 and P < 0.05) in the S group are shown in Supplementary Fig. S3 and the enrichment information is summarized in Supplementary Table S4.
KEGG pathway analysis of 201 genes exclusively expressed in S. aureus-positive animals revealed nine significantly enriched KEGG pathways (P < 0.05) (Fig. 2d). Whereas genes exclusively expressed in S. aureus-negative animals revealed four significantly enriched KEGG pathways (P < 0.05) (Fig. 2e). Five genes (EPOR, IL9, IFNL3, CCL26, and IL26) exclusively expressed in S. aureus-negative animals were enriched in the cytokine-cytokine receptor interaction KEGG pathway.
KEGG pathway analysis of the 301 DEGs in the S vs. H comparison revealed 15 significant enriched pathways (P < 0.05) (Fig. 5a). Most of these pathways were associated with the immune response or inflammation. For example, the IL-17 signaling pathway was enriched by three DEGs (CCL20, IL13, and MMP3). Furthermore, ten KEGG pathways were significantly enriched (P < 0.05) by DEGs in the SM vs. HM comparison and, among these, S. aureus infection was the most significantly enriched pathway (Fig. 5b). Moreover, DEGs in the comparison of SMD vs. HMD were significantly (P < 0.05) enriched in ten pathways, including S. aureus infection, phagosome. Importantly, six DEGs were significantly involved in the S. aureus infection pathway (Fig. 5c). The top ten biological processes identified using GO enrichment analysis are shown in Supplementary Fig. S4.

KEGG pathway enrichment of DEGs. a KEGG pathways of DEGs in the comparison of S vs. H and expression differences of the DEGs involved in the enrichment of the IL-17 signaling pathway. b KEGG pathways of DEGs in the comparison of SM vs. HM and expression differences of the DEGs involved in the enrichment of the S. aureus infection pathway. c KEGG pathways of DEGs in the comparison of SMD vs. HMD and expression differences of the DEGs involved in the enrichment of the S. aureus infection pathway
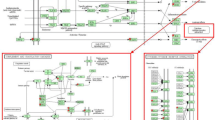
To construct interaction networks, the DEGs in each comparison were further analyzed using the IPA software tool. Interestingly, the PI3K family is shared across all networks generated for the three comparisons and interacted with 13, 6 and 11 molecules in comparisons of S vs. H, SM vs. HM and SMD vs. HMD, respectively (Figs. 6, 7 and 8). Moreover, the IL12 complex and kinase AKT both play key roles in the SM vs. HM and SMD vs. HMD comparison (Figs. 7 and 8). Collectively, these functional analysis results suggested that the immune responses were activated in PBL from S. aureus-infected dairy cattle.

IPA interaction network generated from the DEG list in the S vs. H comparison. Red denotes gene nodes that were increased in expression, and green denotes nodes that were decreased in expression in S samples compared to H samples. The red circle denotes a commonly shared node in the three comparisons

IPA interaction network generated from the DEG list in the SM vs. HM comparison. Red denotes gene nodes that were increased in expression, and green denotes nodes that were decreased in expression in SM samples compared to HM samples. The red circle denotes a commonly shared node in the three comparisons

IPA interaction network generated from the DEG list in the SMD vs. HMD comparison. Red denotes gene nodes that were increased in expression, and green denotes nodes that were decreased in expression in SMD samples than HMD samples. The red circle denotes a commonly shared node in the three comparisons
Finally, the linear regression results between normalized read counts and SCC showed that significant correlations were observed between the expression levels of DEGs (CXCL9, SOCS1, LOC508858, and CYP2E1) and SCC (Fig. 9), suggesting these genes might be served as potential molecular biomarkers of mastitis caused by infection with S. aureus.

Linear regression analysis between normalized read counts and SCC. The X-axis indicates normalized read counts of DEGs, and the Y-axis indicates SCC. a-d) represent CXCL9, SOCS1, LOC508858, and CYP2E1, respectively
Discussion
In recent years, health traits, including mastitis resistance, are increasingly being incorporated into the breeding goals of the modern dairy industry [36,37,38]. Mastitis is a low heritability trait [39, 40] and it is therefore difficult to achieve significant improvements using conventional breeding strategies. Therefore, it is important to investigate the complex host-pathogen interaction underlying mastitis disease caused by S. aureus infection from the perspective of the peripheral blood transcriptome. In previous genome-wide association studies (GWAS), a number of candidate genes (TRAPPC9 [41], mTORC1 [42], JAK2 and STAT5A [43]) were observed to be associated with mastitis-related traits such as SCC. However, transcriptome profiles of S. aureus infection in both cows and their offspring have not been studied in detail. In the current study, for the first time, transcriptome profiles of PBL in response to S. aureus infection were investigated in two generations of dairy cattle.
Previous studies have identified many genes as potential expression biomarkers for bovine mastitis caused by infection with S. aureus mastitis [6, 44]. However, the concordance among these results is not high, possibly as a consequence of environmental factors and varying genetic backgrounds. To address this issue, eight mother-daughter pairs (four S. aureus-infected pairs and four uninfected mother-daughter pairs) from the same farm were used as experimental subjects for the current study. Three well-defined animal selection criteria were used for comparative analyses of the transcriptomes of PBL sampled from two generations of dairy cattle infected with S. aureus-induced mastitis: 1) availability of mother-daughter pairs, 2) three consecutive months of SCC score recordings, and 3) unambiguous S. aureus detection and identification.
For dairy cattle, to produce 1 kg of milk, 400 to 500 kg of blood must pass through the udder [45]. Consequently, PBL represents a valuable sample resource to evaluate the condition of udder health. In our previous study [44], we infected udder quarters with S. aureus (1 × 106 CFU/mL), then conducted mammary biopsies, and collected udder tissues for RNA extraction and transcriptome analysis using RNA-Seq. Mammary biopsies cause trauma and stress; therefore, the use of an accessible and informative tissue such as peripheral blood from naturally infected animals in a production population substantially mitigates animal welfare issues. Thus, PBL samples from two generations of S. aureus-infected cows were used to perform transcriptome sequencing.
Our study is the first to describe transcriptomic profiles of PBL in S. aureus-infected and uninfected dairy cattle across two generations. More than 160 genes were observed solely in the healthy mother-daughter pairs compared to the S. aureus-infected mother-daughter pairs, an observation that may be relevant to identification of genes underpinning resilience to S. aureus mastitis (Fig. 2a). These genes were observed to be significantly enriched in four biological pathways with the cytokine-cytokine receptor interaction pathway directly related to the immune response. Five genes (EPOR, IL9, IFNL3, CCL26, and IL26) that were involved in this pathway might serve as potential molecular markers for breeding programs that enhance resistance to S. aureus infection and mastitis (Fig. 2e). Thus, these immune-related genes warrant further study in a larger group of animals across two generations.
EPOR encodes the erythropoietin receptor, which is a member of the cytokine receptor family. It can activate JAK2 tyrosine kinase that, in turn, activates a range of intracellular pathways. A previous study observed that the IL9 gene emerged as a central node in the network associated with Streptococcus agalactiae-induced subclinical mastitis [46]. In this regard, we have also identified IL9 as a key gene associated with S. aureus-induced subclinical mastitis. Interestingly, sequence variation at IFNL3 has been shown to be associated with hepatitis B virus (HBV) infection in a Chinese human population [47]. CCL26 encodes a secreted protein involved in immunoregulatory and inflammatory processes, which Anna et al. observed to be associated with IL4-mediated signaling pathways in bovine macrophages challenged in vitro with S. agalactiae [48]. The protein encoded by IL26 is a member of the IL10 family of cytokines [49], which has been shown to exhibit anti-bacterial activity against a wide range of bacteria including S. aureus [50], suggesting a broader role in host defenses against bacteria [51].
As expected, a small number of DEGs (Fig. 3) were detected using FDR Padj. < 0.1, a less stringent statistical threshold. Chinese dairy cattle represent a relatively outbred animal population with significant variation in genetic background [52]. S. aureus can readily adapt to its host through evasion of almost every facet of the immune system [32]; therefore, S. aureus-infected cows may exhibit less severe and milder immune responses, which may account for the relatively small number of DEGs. In this regard, Fang et al. also reported modest differential gene expression in bovine mammary gland induced by a low dose of S. aureus to simulate naturally infected mastitis [44]. It would therefore be instructive to perform a substantially larger in vitro challenge experiment using varying S. aureus challenge doses.
Indoleamine 2,3-dioxygenase 2 encoded by IDO2 is an immunomodulatory molecule with potential effects on various diseases including cancer and autoimmune conditions. A previous study demonstrated that IDO2 functions as a modifier in B cells to control pathogenic inflammation and autoimmunity [53]. NR4A1, which encodes the nuclear receptor subfamily 4 group A member 1 protein, was differentially expressed in the comparison of SM vs. HM for the current study. NR4A1 has been shown to play a key role as a regulator of the immune response to apoptotic cells [54].
In the present study, immune-related pathways were enriched by different but complementary functional enrichment and annotation methods (KEGG, IPA and GSEA). Three DEGs (CCL20, IL13, and MMP3) were found to be involved in the IL-17 signaling pathway in the S vs. H comparison (Fig. 5a). The interleukin 17 (IL-17) family are proinflammatory cytokines that play crucial roles in both acute and chronic inflammatory responses [55]. Moreover, there are five (BLA-DQB, C1R, C2, FCGR1A, and KRT10) (Fig. 5b) and six genes (BLA-DQB, C3AR1, CFI, FCAR, FCGR3A, and LOC10498484) (Fig. 5c) enriched in the S. aureus infection pathway in the SM and SMD groups, respectively. The BLA-DQB gene encodes the bovine major histocompatibility complex, class I, DQ beta protein, which has been hypothesized to play a key role in the synchronization of the immune response to mastitis-causing bacteria in bovine udder secretory tissues [56]. Interestingly, BLA-DQB exhibited contrary directions of expression in SM and SMD compared with the healthy controls, which might be due to the differences in the duration of S. aureus infection [57], differences in age [58], parity [59] and lactation period [35] of the cattle.
The IPA analysis revealed an interaction network involving the phosphoinositide 3-kinase (PI3K) family in all three comparisons (Fig. 6, 7 and 8). The PI3K family of enzymes are involved in myriad cellular functions, including cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking. In particular, PI3K proteins have important functions in immunobiology [60]. IL12 (Fig. 7 and 8) encodes an important cytokine that can regulate both innate and adaptive immune responses during infection [61]. Kinase AKT (Fig. 7 and 8) is involved in the regulation of the development and function of innate immune cells, including neutrophils, macrophages, and dendritic cells [62].
The cytokine-cytokine receptor signaling pathway genes exclusively expressed in uninfected mother-daughter pairs (EPOR, IL9, IFNL3, CCL26, and IL26) and the DEGs involved in immune-related pathways identified in the current study (including CCL20, IL13, MMP3, and BLA-DQB), may contain sequence polymorphisms that could serve as genetic markers for susceptibility to mastitis caused by S. aureus infection. Consequently, identification of SNPs within these genes and systematic evaluation through large-scale association studies using suitable animal cohorts can provide valuable information for future genome-enabled breeding programs to enhance mastitis resilience in dairy cattle.
Conclusions
In summary, the transcriptome profiles of PBL sampled from two consecutive generations of cows with naturally infected S. aureus mastitis and non-infected healthy control animals were investigated. Many of the genes highlighted in this study as being important in the bovine host response to infection with S. aureus may represent candidate PBL expression biomarkers for mastitis and may also contain sequence variation that can be leveraged for genomic selection of cattle less susceptible to mastitis disease.
Availability of data and materials
All genomic annotation data defining gene regions are available for download (ftp://ftp.ensemble.org/pub/release-96/gtf/bos_taurus). RNA-Seq data from China Agricultural University is available upon the agreement of China Agricultural University and should be requested directly from the authors.
Abbreviations
- S. aureus :
-
Staphylococcus aureus
- S:
-
S. aureus-infected cows
- H:
-
Healthy non-infected cows
- SM:
-
S. aureus-infected mastitis mothers
- HM:
-
Healthy non-infected mothers
- SMD:
-
S. aureus-infected daughters
- HMD:
-
Healthy non-infected daughters
- EPOR:
-
Erythropoietin receptor
- IL9:
-
Interleukin 9
- IFNL3:
-
Interferon lambda 3
- CCL26:
-
C-C motif chemokine ligand 26
- IL26:
-
Interleukin 26
- DEGs:
-
Differentially expressed genes
- FDR:
-
False discovery rate
- CCL20:
-
Chemokine (C-C motif) ligand 20
- IL13:
-
Interleukin 13
- MMP3:
-
Matrix metalloproteinase-3
- IL-17:
-
Interleukin 17
- BLA-DQB:
-
Bos taurus major histocompatibility complex, class II, QB beta
- C1R:
-
Complement C1r subcomponent
- C2:
-
Complement C2
- FCGR1A:
-
Fc fragment of IgG receptor Ia
- KRT10:
-
Keratin 10
- C3AR1:
-
Complement component 3a receptor 1
- CFI:
-
Complement factor I
- FCAR:
-
Fc fragment of IgA receptor
- FCGR3A:
-
Fc fragment of IgG, low affinity IIIa, receptor
- E. coli :
-
Escherichia coli
- RNA-Seq:
-
RNA sequencing
- HTS:
-
High-throughput sequencing
- LPS:
-
Lipopolysaccharide
- PBL:
-
Peripheral blood leukocyte
- SCC:
-
Somatic cell counts
- DHI:
-
Dairy herd improvement
- Nuc:
-
Thermonuclease
- PCR:
-
Polymerase chain reaction
- RIN:
-
RNA integrity number
- GSEA:
-
Gene set enrichment analysis
- MSigDB:
-
Molecular signature database
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- IPA:
-
Ingenuity Pathway Analysis
- RT-qPCR:
-
Reverse transcription quantitative real-time PCR
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- PI3K:
-
Phosphoinositide 3-kinases
- IL12:
-
Interleukin 12
- AKT:
-
Protein kinase B, also known as PKB
- CXCL9:
-
Chemokine (C-X-C motif) ligand 9
- SOCS1:
-
Suppressor of cytokine signaling 1
- CYP2E1:
-
Cytochrome P450 2E1
- GWAS:
-
Genome-wide association study
- TRAPPC9:
-
Trafficking protein particle complex 9
- mTORC1:
-
Cytosolic arginine sensor for mTORC1 subunit 1
- JAK2:
-
Janus kinase 2
- STAT5A:
-
Signal transducer and activator of transcription 5A
- IDO2:
-
Indoleamine 2,3-dioxygenase 2
- NR4A1:
-
Nuclear receptor subfamily 4 group A member 1
References
Schukken YH, Gunther J, Fitzpatrick J, Fontaine MC, Goetze L, Holst O, et al. Host-response patterns of intramammary infections in dairy cows. Vet Immunol Immunopathol. 2011;144(3–4):270–89.
Keane OM. Symposium review: Intramammary infections—major pathogens and strain-associated complexity*. J Dairy Sci. 2019;102(5):4713–26.
White DG, McDermott PF. Emergence and transfer of antibacterial resistance. J Dairy Sci. 2001;84:E151–E5.
Fox LK, Gay JM. Contagious mastitis. Vet clin North Am Food Anim pract. 1993;9(3):475–87.
Bradley A. Bovine mastitis: an evolving disease. Vet J. 2002;164(2):116–28.
Jensen K, Günther J, Talbot R, Petzl W, Zerbe H, Schuberth HJ, et al. Escherichia coli- and Staphylococcus aureus-induced mastitis differentially modulate transcriptional responses in neighbouring uninfected bovine mammary gland quarters. BMC Genomics. 2013;14:36.
Lingard D. Herd health: food animal production medicine, vol. 29. Place. Published; 1997.
Gilbert FB, Cunha P, Jensen K, Glass EJ, Foucras G, Robert-Granié C, et al. Differential response of bovine mammary epithelial cells to Staphylococcus aureus or Escherichia coli agonists of the innate immune system. Vet Res. 2013;44(1):40.
Lutzow YCS, Donaldson L, Gray CP, Vuocolo T, Pearson RD, Reverter A, et al. Identification of immune genes and proteins involved in the response of bovine mammary tissue to Staphylococcus aureus infection. BMC Vet Res. 2008;4(1):18.
Mitterhuemer S, Petzl W, Krebs S, Mehne D, Klanner A, Wolf E, et al. Escherichia coli infection induces distinct local and systemic transcriptome responses in the mammary gland. BMC Genomics. 2010;11(1):138.
Jørgensen HBH, Buitenhuis B, Røntved CM, Jiang L, Ingvartsen KL, Sørensen P. Transcriptional profiling of the bovine hepatic response to experimentally induced E. coli mastitis. Physiol Genomics. 2012;44(11):595–606.
Jiang L, Sørensen P, Røntved C, Vels L, Ingvartsen KL. Gene expression profiling of liver from dairy cows treated intra-mammary with lipopolysaccharide. BMC Genomics. 2008;9(1):443.
Sharma N, Singh NK, Bhadwal MS. Relationship of somatic cell count and mastitis: An overview. Asian-Australas J Anim Sci. 2011;24(3):429–38.
Hogan JS, National MC. Laboratory handbook on bovine mastitis. USA: National Mastitis Council; 1999.
Gao J, Ferreri M, Liu XQ, Chen LB, Su JL, Han B. Development of multiplex polymerase chain reaction assay for rapid detection of Staphylococcus aureus and selected antibiotic resistance genes in bovine mastitic milk samples. J Vet Diagn Investig. 2011;23(5):894–901.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
FastQC: A Quality Control Tool for High Throughout Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 4 Oct 2018.
Dobin A, Gingeras TR. Mapping RNA-seq reads with STAR. Curr Protoc Bioinformatics. 2015;51:11.4.1–9.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Liao Y, Smyth G, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47.
Varet H, Brillet-Gueguen L, Coppee JY, Dillies MA. SARTools: a DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One. 2016;11(6):e0157022.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25.
Anselmi G, Vaivode K, Dutertre CA, Bourdely P, Missolo-Koussou Y, Newell E, et al. Engineered niches support the development of human dendritic cells in humanized mice. Nat Commun. 2020;11(1):2054.
Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560(7718):325–30.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–7.
Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30(4):523–30.
Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques. 2004;37(1):112 -4, 6, 8-9.
Robinson TL, Sutherland IA, Sutherland J. Validation of candidate bovine reference genes for use with real-time PCR. Vet Immunol Immunopathol. 2007;115(1–2):160–5.
Kobayashi SD, DeLeo FR. Staphylococcus aureus protein A promotes immune suppression. mBio. 2013;4(5):e00764–13.
de Jong NWM, van Kessel KPM, van Strijp JAG. Immune Evasion by Staphylococcus aureus. Microbiol Spectr. 2019;7(2):1-27.
Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, et al. Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med. 2016;13(12):e1002201.
Liu L, Yang N, Xu G, Liu S, Wang D, Song J, et al. Transgenerational transmission of maternal stimulatory experience in domesticated birds. FASEB J. 2018;32(12):7002–17.
Seo M, Lee H-J, Kim K, Caetano-Anolles K, Jeong JY, Park S, et al. Characterizing milk production related genes in Holstein using RNA-seq. Asian-Australas J Anim Sci. 2016;29(3):343–51.
Miglior F, Fleming A, Malchiodi F, Brito LF, Martin P, Baes CF. A 100-year review: identification and genetic selection of economically important traits in dairy cattle. J Dairy Sci. 2017;100(12):10251–71.
Parker Gaddis KL, VanRaden PM, Cole JB, Norman HD, Nicolazzi E, Dürr JW. Symposium review: development, implementation, and perspectives of health evaluations in the United States. J Dairy Sci. 2020;103(6):5354–65.
Weigel KA, Shook GE. Genetic selection for mastitis resistance. Vet Clin North Am Food Anim Pract. 2018;34(3):457–72.
Govignon-Gion A, Dassonneville R, Baloche G, Ducrocq V. Multiple trait genetic evaluation of clinical mastitis in three dairy cattle breeds. Animal. 2016;10(4):558–65.
Urioste JI, Franzén J, Windig JJ, Strandberg E. Genetic relationships among mastitis and alternative somatic cell count traits in the first 3 lactations of Swedish Holsteins. J Dairy Sci. 2012;95(6):3428–34.
Wang X, Ma P, Liu J, Zhang Q, Zhang Y, Ding X, et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015;16(1):111.
Sahana G, Guldbrandtsen B, Thomsen B, Holm LE, Panitz F, Brøndum RF, et al. Genome-wide association study using high-density single nucleotide polymorphism arrays and whole-genome sequences for clinical mastitis traits in dairy cattle1. J Dairy Sci. 2014;97(11):7258–75.
Usman T, Yu Y, Liu C, Wang X, Zhang Q, Wang Y. Genetic effects of single nucleotide polymorphisms in JAK2 and STAT5A genes on susceptibility of Chinese Holsteins to mastitis. Mol Biol Rep. 2014;41(12):8293–301.
Fang L, Hou Y, An J, Li B, Song M, Wang X, et al. Genome-wide transcriptional and post-transcriptional regulation of innate immune and defense responses of bovine mammary gland to Staphylococcus aureus. Front Cell Infect Microbiol. 2016;6:193.
Moran J. Tropical dairy farming: feeding Management for Small Holder Dairy Farmers in the humid tropics. Australia: Land Links; 2005.
Lewandowska-Sabat AM, Kirsanova E, Klopp C, Solberg TR, Heringstad B, Østerås O, et al. Transcription profiling of monocyte-derived macrophages infected in vitro with two strains of Streptococcus agalactiae reveals candidate pathways affecting subclinical mastitis in cattle. Front Genet. 2019;10:689.
Chen J, Wang W, Li X, Xu J. A meta-analysis of the association between IL28B polymorphisms and infection susceptibility of hepatitis B virus in Asian population. BMC Gastroenterol. 2015;15:58.
Lewandowska-Sabat AM, Hansen SF, Solberg TR, Østerås O, Heringstad B, Boysen P, et al. MicroRNA expression profiles of bovine monocyte-derived macrophages infected in vitro with two strains of Streptococcus agalactiae. BMC Genomics. 2018;19(1):241.
Knappe A, Hör S, Wittmann S, Fickenscher H. Induction of a novel cellular homolog of Interleukin-10, AK155, by transformation of T lymphocytes with Herpesvirus Saimiri. J Virol. 2000;74(8):3881.
Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. 2015;16(9):970–9.
Stephen-Victor E, Fickenscher H, Bayry J. IL-26: An emerging proinflammatory member of the IL-10 cytokine family with multifaceted actions in antiviral, antimicrobial, and autoimmune responses. PLoS Pathog. 2016;12(6):e1005624.
Riquet J, Coppieters W, Cambisano N, Arranz J-J, Berzi P, Davis SK, et al. Fine-mapping of quantitative trait loci by identity by descent in outbred populations: application to milk production in dairy cattle. Proc Natl Acad Sci U S A. 1999;96(16):9252–7.
Merlo LMF, DuHadaway JB, Grabler S, Prendergast GC, Muller AJ, Mandik-Nayak L. IDO2 modulates T cell-dependent autoimmune responses through a B cell-intrinsic mechanism. J Immunol. 2016;196(11):4487–97.
Ipseiz N, Uderhardt S, Scholtysek C, Steffen M, Schabbauer G, Bozec A, et al. The nuclear receptor Nr4a1 mediates anti-inflammatory effects of apoptotic cells. J Immunol. 2014;192(10):4852–8.
Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the Yin and the Yang. Trends Immunol. 2017;38(5):310–22.
Kosciuczuk EM, Lisowski P, Jarczak J, Majewska A, Rzewuska M, Zwierzchowski L, et al. Transcriptome profiling of Staphylococci-infected cow mammary gland parenchyma. BMC Vet Res. 2017;13(1):161.
Garzoni C, Francois P, Huyghe A, Couzinet S, Tapparel C, Charbonnier Y, et al. A global view of Staphylococcus aureus whole genome expression upon internalization in human epithelial cells. BMC Genomics. 2007;8(1):171.
Steegenga WT, Boekschoten MV, Lute C, Hooiveld GJ, de Groot PJ, Morris TJ, et al. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Dordr). 2014;36(3):9648.
Salleh MS, Mazzoni G, Höglund JK, Olijhoek DW, Lund P, Løvendahl P, et al. RNA-Seq transcriptomics and pathway analyses reveal potential regulatory genes and molecular mechanisms in high- and low-residual feed intake in Nordic dairy cattle. BMC Genomics. 2017;18(1):258.
Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4(4):313–9.
Watford WT, Moriguchi M, Morinobu A, O'Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14(5):361–8.
Zhang Y, Wang X, Yang H, Liu H, Lu Y, Han L, et al. Kinase AKT controls innate immune cell development and function. Immunology. 2013;140(2):143–52.
Acknowledgments
The authors thank three reviewers for constructive comments and suggestions on the manuscript, Siyuan Mi, Liangyu Shi, Jiangang Qi and all the members of Molecular and Quantitative Genetics Laboratory (China Agricultural University) for sample collection; CapitalBio Technology (Beijing, China) for sequencing services; Gillian P. McHugo for helpful discussions. The support provided by the China Scholarship Council (201906350210) during a visit by Di Wang to University College Dublin is acknowledged.
Funding
This research was financially supported by the NSFC-PSF Joint Project (31961143009), Beijing Natural Science Foundation (6182021), Beijing Dairy Industry Innovation Team (BAIC06), Modern Agro-industry Technology Research System (CARS-36) and the Program for Changjiang Scholar and Innovation Research Team in University (IRT-15R62).
Author information
Authors and Affiliations
Contributions
Ying Yu and Di Wang conceived the study. Di Wang, Lei Liu, and Tao Duan collected the samples. Di Wang, Lei Liu, and Thomas J. Hall analyzed the data. Di Wang, Lei Liu, Serafino M.A. Augustino, Tao Duan, David E. MacHugh, Jinhuan Dou, Yi Zhang, Yachun Wang, and Ying Yu wrote and prepared the manuscript. All authors reviewed and contributed to the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal management and experimental procedures followed the animal care protocols approved by the China Agricultural University Animal Care and Use Ethics Committee.
Consent for publication
Not applicable.
Competing interests
The authors have declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary information
Additional file 1: Supplementary Figure S1.
Linear regression analysis of the log2|FC| of 4000 randomly selected gene expression values for the SM vs. HM and SMD vs. HMD comparisons.
Additional file 2: Supplementary Figur S2.
Volcano plots of DEGs for (A) the S vs. H comparison, (B) the SM vs. HM comparison, and (C) the SMD vs. HMD comparison.
Additional file 3: Supplementary Figure S3.
Gene set enrichment analysis (GSEA) plots depicting the enrichment of functional gene sets up-regulated in the S group compared to the control group (FDR < 0.25 and P < 0.05). (A-F): GSEA plot depicting the enrichment of functional gene sets up-regulated in oxidative phosphorylation, heme metabolism, fatty acid metabolism, Kras signaling up-regulation, inflammatory response and interferon-gamma response in the S vs. H comparison (FDR < 0.25 and P < 0.05).
Additional file 4: Supplementary Figure S4.
GO Biological Process enrichment of DEGs. (A) Top ten Biological Process GO categories of DEGs in the S vs. H comparison. (B) Top ten Biological Process GO categories of DEGs in the SM vs. HM comparison. (C) Top ten Biological Process GO categories of DEGs in the SMD vs. HMD comparison.
Additional file 5: Table S1.
Quality report of 16 RNA samples for RNA sequencing.
Additional file 6: Table S2.
Primer pairs of DEGs used for qRT-PCR validation.
Additional file 7: Table S3.
Summary of sequence reads aligenment
Additional file 8: Table S4.
Six significantly enriched upregulated gene sets.
Additional file 9: Table S5.
Differentially expressed genes detected in S vs. H
Additional file 10: Table S6.
Differentially expressed genes detected in SM vs. HM.
Additional file 11: Table S7.
Differentially expressed genes detected in SMD vs. HMD.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, D., Liu, L., Augustino, S.M.A. et al. Identification of novel molecular markers of mastitis caused by Staphylococcus aureus using gene expression profiling in two consecutive generations of Chinese Holstein dairy cattle. J Animal Sci Biotechnol 11, 98 (2020). https://doi.org/10.1186/s40104-020-00494-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-020-00494-7