
Abstract
Background
The aim of this study is to examine the inflammatory-cytokine expressions in the presence of non-cytotoxic dose of methylmercury (MeHg) in murine macrophages, which is suspected to play an important role in brain damage caused by MeHg exposure. We focused on murine macrophage inflammatory protein-2 (MIP-2), keratinocyte chemoattractant (KC), and monocyte chemoattractant protein-5 (MCP-5). MIP-2 and KC are murine functional homologues of human IL-8 and MCP-5 for human MCP-1. Furthermore, we examined the suppressive effect of N-acetyl-l-cysteine (NAC) on the MeHg-induced inflammatory cytokines.
Methods
In a murine RAW264.7 macrophage cell line, MeHg-induced cytokine expressions were measured using real-time PCR. The suppressive effect of NAC was examined by putting it into the culture medium together with MeHg (co-treatment). In addition, pre- and post-treatment experiments were conducted, in which the cells were treated with NAC before and after MeHg exposure, respectively.
Results
Exposure to a non-cytotoxic dose of MeHg up-regulated the mRNA expression of MIP-2 and MCP-5. On the other hand, KC expression was not induced in the presence of MeHg. Effect of MeHg on MIP-2 expressions was suppressed by pre-, co-, and post-treatment with NAC. However, the suppressive effect of pre-treatment was less than the post-treatment, which was as effective as co-treatment.
Conclusion
In functional homologues of human IL-8, only MIP-2 expression, not KC, was activated in the presence of non-cytotoxic dose of MeHg in murine RAW264.7 macrophage cell line. The more evident inhibitory effect of NAC observed in post-treatment experiments suggests a possible involvement of intracellular activities such as antioxidant effects.
Similar content being viewed by others


Background
Methylmercury (MeHg) is well known to have neurotoxicity, because MeHg passes through the blood–brain barrier and accumulates in the brain [1, 2]. MeHg shows toxicological effects even at low concentrations [3]. Cellular responses to low level of MeHg exposure involve inflammatory processes. The previous studies with non-lethal doses of MeHg, concentrations as low as 1–4 μM in human astrocytoma, and 1–10 μM in macrophage cell lines suggested a possible involvement of an early inflammatory signalling [4,5,6]. Inflammation and subsequent degeneration of neural tissues in the central nervous system involve glial cells and peripheral macrophages, which are main sources of various pro-inflammatory cytokines and chemokines [7,8,9]. In brain specimens of Minamata disease patients, strong macrophage infiltration can be found in the tissue surrounding their brain lesions. In a histochemical analysis of brain tissue obtained from those patients, mercury (Hg) was detected in macrophages throughout the brain [10].
In humans, IL-8 is reported to possess a crucial role in immune responses elicited in central nervous system [11], especially in the induction of chemotaxis against its target cells (e.g., monocytes and neutrophil). U937, a human macrophage cell line, showed the up-regulation of IL-8 in response to MeHg exposure [12]. Murine macrophage inflammatory protein-2 (MIP-2) and keratinocyte chemoattractant (KC) are functional homologues of IL-8 in human [13]. MIP-2 expression was reported to be activated in the liver of MeHg-exposed mice [14]. On the other hand, there are few studies that indicate the molecular responses of MIP-2 and KC against the direct exposure to MeHg in vitro.
Monocyte chemoattractant protein-1 (MCP-1) is also suspected to work as an alert system in response to MeHg exposure in the brain on the basis of experimental findings using mice whose murine MCP-1 homologue was knocked out [15]. In addition, at a non-cytotoxic concentration of 4 µM MeHg, Muniroh et al. [16] observed a significant induction of MCP-1 expression in U-87 MG cells, a model system cell line of human astrocytes. Murine MCP-5 was reported to be the most homologous chemokine to human MCP-1 [17, 18]. In fact, MCP-5 was structurally more similar to MCP-1 than JE, the putative murine homologue of MCP-1 [18]. However, there are no reports concerning the effect of MeHg on MCP-5 expression.
N-acetyl-l-cysteine (NAC), the acetylated precursor of l-cysteine, is a sulfhydryl-containing antioxidant, which has been used for the treatment of heavy metal toxicity and can act as an antiinflammatory agent [19]. It reduces reactive oxygen species levels by raising intracellular glutathione concentrations and/or playing directly as a free radical scavenger. NAC was used to modulate inflammatory pathways in peripheral and central nervous system and cytokine levels in neuropsychiatric disorders [19]. Furthermore, NAC was reported to act as a chelating agent for mercury and accelerates urinary excretion of MeHg in mice [20]. It should also be noted that a study has shown that NAC suppressed the MeHg-activated MCP-1 and IL-6 expressions in U-87 MG cell line [18], and IL-6 and IL-8 expressions in U937 macrophages [12].
Based on above background, we investigated the inflammatory responses to the non-cytotoxic concentration of MeHg, focusing on MIP-2, KC, and MCP-5 expressions as functional homologues of IL-8 and MCP-1 in mice. To expand the role of these genes for understanding the toxicity of MeHg in human, further in vivo mechanistic analysis using rodents will be necessary. In addition, we examined the suppressive effect of NAC on the MeHg-induced inflammatory cytokines.
Methods
Cell culture
A murine macrophage cell line, RAW264.7, was used in this study (Sumitomo Dainippon Pharma, Osaka, Japan). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, MO, USA) with 1% l-glutamine (Sigma-Aldrich, MO, USA), penicillin (100 U/mL), streptomycin (100 µg/mL) (Sigma-Aldrich, MO, USA), and 10% heat-inactivated fetal bovine serum (FBS) (Nichirei Biosciences, Tokyo, Japan) at 37 °C in a humidified incubator (5% CO2). An initial concentration of 2 × 104 cells/mL was used for each experiment.
Cytotoxicity test
Stock solution of MeHgCl (10 mM) (Tokyo Chemical Industry, Tokyo, Japan) was dissolved in Dulbecco’s PBS (Sigma-Aldrich, MO, USA) with l-cysteine (Hg:Cys = 1:1) and kept at − 80 °C until use. The stock was diluted with culture medium just before being added to the cells. NAC (Wako Pure Chemical Industries, Osaka, Japan) was dissolved in FBS-free DMEM, and the pH was adjusted to 7.4 with sodium hydroxide (NaOH).
For cytotoxicity assays, the cells were cultured for 24 h in 96-well plates and incubated with MeHg (0.1–100 µM) for 24 h. Cell toxicity was assayed using the WST-8 Cell Counting kit, according to the manufacturer’s protocol (Wako Pure Chemical Industries, Osaka, Japan). In brief, the cells were washed, and Hank’s balanced salt solution (100 µL) and WST-8 solution (10 µL) were added to each well and kept for incubation at 37 °C in 5% CO2. The colour was quantified within 1–2 h by a micro-plate spectrophotometer (TriStar LB941, Berthold Technologies, Germany) at 450 nm absorbance. Culture medium was used to adjust the absorbance values of the sample. Mean values and standard errors (SEs) were obtained from the results of four experiments.
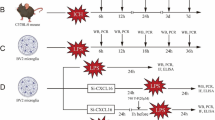
Treatments of MeHg and NAC
Non-cytotoxic concentrations of MeHg (0.5 or 2 µM) and NAC (1 or 20 mM) were used for further experiments. To examine the suppressive effects of NAC on MeHg-induced MIP-2, KC, and MCP-5 up-regulations, the following protocols were used:
Pre-treatment experiment
After 23 h of cell culture, cells were pre-incubated with NAC for 1 h, and the remaining NAC, if any, was washed out with a double volume of culture medium. Then, cells were incubated with the medium containing MeHg for 3 h. The incubation time of NAC was determined based on prior experiments.
Co-treatment experiment
After 24 h of cell culture, cells were treated with the medium containing both NAC and MeHg for 3 h, which was prepared and kept at the room temperature for 30 min in advance.
Post-treatment experiment
After 24 h of cell culture, cells were treated with the medium containing MeHg. Medium was washed out after 3 h of MeHg exposure and then NAC was added. Cells were harvested 30 h after the starting of cell culture.
Analysis of mRNA expression
Total RNA from the cells was extracted using an RNeasy Plus Mini kit (Qiagen), which eliminates genomic DNA contamination. Then, cDNA was synthesized from total RNA (600 ng), using QuantiTect Reverse Transcription (Qiagen) and stored at − 80 °C until use. The samples were prepared in three independent experiments. The mRNA expressions of cytokine genes were quantified with LightCycler instrument (Roche Diagnostics Japan, Tokyo, Japan), using the LightCycler FastStart DNA MasterPLUS SYBR Green I (Roche Diagnostics) following the manufacturer’s instructions. The primers for each gene were designed and synthesized on the basis of respective information in NCBI, using the software of Primer3 (http://frodo.wi.mit.edu/primer3/), and the target regions were 80–300 bp in length (Sigma-Aldrich Japan, Hokkaido).
PCR amplification was conducted as reported in the previous studies [21, 22] in a total volume of 20 µL containing cDNA and each primer (0.5 µM). Forward (F) and reverse (R) primers were:
β-actin, 5′-CGTGCGTGACATCAAAGAGAAG-3′ (F), 5′-ATGCCACAGGATTCCATACCC-3′(R);
KC, 5′-AGAACATCCAGAGCTTGAAGGTGTT-3′(F), 5′-GGACACCTTTTAGCATCTTTTGGACA-3′(R); MIP-2, 5′-AAGTTTGCCTTGACCCTGAA-3′(F), 5′-AGGCACATCAGGTACGATCC-3′(R) and MCP-5, 5′-TGGACCAGATGCGGTGAGC-3′ (F), 5′-GGCTGCTTGTGATTCTCCTGTAG-3′(R), respectively. The DNA polymerase was activated by initial denaturation at 95 °C for 10 min, and 45 amplification cycles were carried out. The cycle consists of denaturation at 95 °C for 10 s, annealing at 60 °C for 10 s, and extension 72 °C for 15 s. The fluorescent product was detected at the end of the extension period. All PCR assays were performed three times independently.
The data were analyzed using the Light Cycler analysis software version 4.1 (Roche Diagnostics Japan, Tokyo, Japan). Amplification specificity was confirmed by melting curve analysis of the PCR products. Threshold cycle (Ct) values of the target genes were normalized to the expression of the β-actin gene as an internal control gene. Unstimulated RAW264.7 cells harvested in 24 h were used as a standard, and unstimulated cells harvested after 30 h as a negative control. The relative expression in each sample to that of the control sample was calculated according to the 2−ΔΔCT method [23].
Statistical analysis
Mean values and corresponding SEs were calculated using the Stata 14.0 Software (StataCorp LLC, Texas, USA). The P value for the difference between two groups was calculated by the Mann–Whitney U test, and a non-parametric test for trend across ordered groups was performed. The level of significance was indicated by P < 0.05.
Results
Cytotoxicity of MeHg and NAC in RAW264.7 macrophage cell line
Cytotoxicity of MeHg on RAW264.7 cells was assayed after incubating cells for 24 h in the presence or absence of MeHg. As shown in Fig. 1, the maximum non-cytotoxic dose of MeHg was 2 µM. Therefore, for cytokine-induction experiments, the doses of 0.5 and 2 µM were used. NAC was not cytotoxic up to 50 mM (data not shown). On the basis of these results, 1 mM and 20 mM of NAC were selected for further experiments.

Cell viability after exposure to different doses of MeHg. MeHg (0.1–100 µM) exposure on RAW264.7 cell line and the cell viability by WST-8 cell counting Assay. Values represent the mean ± standard error of four experiments
Effect of MeHg on mRNA expressions of MIP-2, KC, and MCP-5
As shown in Fig. 2, MIP-2 mRNA was detected initially (in the absence of MeHg in the medium) in RAW264.7 cells. When calculating relative gene expressions, we used this initial value at time 0 as the reference. In response to 2 µM of MeHg, MIP-2 mRNA expression was significantly induced within 3 h (P = 0.0495). Activation of MIP-2 mRNA expression declined after 6 h of MeHg exposure. No significant expression was observed at 6 h or after. In the lower dose, no evident up-regulation was found.

MIP-2 mRNA expression at 3, 6, 12, and 24 h after MeHg exposure. RAW264.7 cells were treated with 0 µM (control), 0.5, 2 µM of MeHg, and mRNA expression levels were analyzed by real-time PCR. Values were normalized to the expression of β-actin and it represents three independent experiments. MIP-2 mRNA expression induced by MeHg treatment alone is compared with control and the significant value is labelled as "*"
KC mRNA expression was not significantly up-regulated by 2 µM of MeHg (Fig. 3). When we treated the cells with lipopolysaccharide (LPS) to confirm the KC expressions in RAW264.7 cells, we could detect the KC up-regulation at 3 h (data not shown).

KC mRNA expression at 3, 6, 12, and 24 h after MeHg exposure. RAW264.7 cells were treated with 0 µM (control), 0.5, and 2 µM of MeHg, and mRNA expression levels were analyzed by real-time PCR. Values were normalized to the expression of β-actin and it represents three independent experiments
MCP-5 expression was significantly induced within 3 h in response to 2 µM of MeHg (P = 0.0495). Activation of MCP-5 mRNA expression decreased after 6 h of MeHg treatment. In the lower dose, no obvious up-regulation was observed (Fig. 4).

MCP-5 mRNA expression at 3, 6, 12, and 24 h after MeHg exposure. RAW264.7 cells were treated for 3, 6, 12, and 24 h with 0 µM (control), 0.5, and 2 µM of MeHg and mRNA expression levels were analyzed by real-time PCR. Values were normalized to the expression of β-actin and it represents three independent experiments. MCP-5 mRNA induced by MeHg treatment alone is compared with control and the significant value is labelled as "*"
Effect of different timing of NAC treatment on mRNA expression of MIP-2
Figure 5 shows the effect of NAC pre-treatment, co-treatment, and post-treatment on MIP-2 mRNA expression induced by MeHg exposure. For this experiment, MIP-2 was selected, because its response to MeHg was remarkable. The NAC treatment decreased MeHg-induced MIP-2 expression in pre-treatment (P = 0.020), co-treatment (P = 0.020), and post-treatment (P = 0.020) when the NAC treatment groups and non-treatment groups were compared. In pre-treatment, MeHg-induced MIP-2 expressions were significantly inhibited by NAC regardless of dose (P = 0.0495 for both doses). In co-treatment, regardless of NAC dose, MIP-2 gene expressions were significantly suppressed (P = 0.0495 for both doses). In post-treatment, NAC also inhibited MIP-2 expressions significantly (P = 0.0495 for both doses). Significant dose-dependent reduction of MIP-2 was observed only in the post-treatment (P for trend 0.017).

Effect of NAC to suppress MeHg-induced MIP-2 mRNA expression. RAW264.7 cells were treated with 1 or 20 mM NAC at 1 h before (pre-treatment), with (co-treatment), or 1 h/3 h after 2 µM MeHg (post-treatment) for 3 h of incubation. mRNA expression levels of MIP-2 were analyzed by real-time PCR values which were normalized to the expression of β-actin and it represents three independent experiments. MIP-2 mRNA expression induced by MeHg treatment alone is compared with control, P = 0.0369 (labelled as #), MeHg + NAC treatment (1, 20 mM) effects are compared with the MeHg treatment alone and the significant values (P = 0.0495) were shown in the figure (labelled as *)
The post-treatment of NAC was more effective than co-treatment regardless of dose (P = 0.0495 for both doses) and post-treatment of NAC was more effective than pre-treatment only when NAC dose was 20 mM (P = 0.0495).
Discussion
In this study, exposure to non-cytotoxic doses of MeHg (2 µM) up-regulated MIP-2 and MCP-5 mRNA expressions in RAW264.7 macrophage cell line. To our knowledge, the present study is the first report regarding the activation of MIP-2 and MCP-5 expression by MeHg in vitro. Note that murine MIP-2 and KC are functional homologues of human IL-8 [15, 16]. The MIP-2 but not KC expression was significantly activated by MeHg, indicating that only the MIP-2, as a part of human IL-8 homologue, mainly plays a role in inflammatory response against MeHg exposure in murine macrophages. MIP-2 was reported to be more potent than KC in leukocyte recruitment, endothelial cell chemotaxis [24, 25], and cyclophilin-A-induced neutrophil migration [26]. In neutrophil emigration in cremaster muscle, MIP-2 and KC did not differ significantly [27, 28]. Some studies have proved that MIP-2 alone can recruit chemotactic cells, but when both MIP-2 and KC are present, the recruitment is maximal. On the other hand, Tanimoto et al. [26] reported that, on the basis of different studies, MIP-2 and KC also complemented each other in their roles and functions. The expression patterns of KC and MIP-2 are tissue-specific, and therefore, their roles in chemotactic cells recruitment are different from tissue to tissue [29, 30]. The functional regulation of IL-8 in the presence of MeHg was different between human and mice. These results will be useful for further in vivo functional analysis of these gene expressions using mice.
Muniroh et al. [4] and Yamamoto et al. [5] also showed suppressive effects of NAC on MeHg-induced cytokine expressions. The importance of the antioxidant mechanism of NAC and its antiinflammatory effect is emphasized in reference to a report in which NAC inhibits LPS-induced cytokine activation [30]. In the present study, the up-regulation of MIP-2 in response to exposure to MeHg was suppressed by NAC pre-, co-, and post-treatments. In the pre-treatment experiment, the medium containing NAC was washed out before the MeHg exposure, and hence, the NAC could only act intracellularly. In the co-treatment experiment, the medium was mixed previously with NAC and MeHg. If the co-treatment was most effective, it suggests a strong involvement of chelating effect of NAC in the medium. In the post-treatment, the medium containing MeHg was washed out before addition of NAC into the medium. Since the post-treatment was as effective as co-treatment, there is no evidence to indicate an extracellular effect such as a chelating effect. The inhibitory effects of NAC shown in pre- and post-treatments indicate the involvement of intracellular activities, including antioxidant effects.
Conclusion
In murine RAW264.7 macrophage cell line, the MIP-2, functional homologue of IL-8, was mainly involved in the inflammatory response induced by MeHg exposure. The MCP-5, a murine homologue of human MCP-1, was also involved in such an inflammatory response. The remarkable suppressive effect of NAC on MeHg-induced inflammatory-cytokine expressions observed in post-treatment experiments would suggest a possible involvement of intracellular activities such as the antioxidant effects.
Abbreviations
- MeHg:
-
methylmercury
- MIP-2:
-
macrophage inflammatory protein-2
- KC:
-
keratinocyte chemoattractant
- MCP-5:
-
monocyte chemoattractant protein-5
- NAC:
-
N-acetyl-l-cysteine
- MCP-1:
-
monocyte chemoattractant protein-1
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- LPS:
-
lipopolysaccharide
References
Aschner M, Aschner JL. Mercury neurotoxicity: mechanisms of blood-brain barrier transport. Neurosci Bio Behav Rev. 1990;14:169–76.
World health organization. Exposure to mercury: a major public health concern WHO, Public Health and Environment, Geneva. 2007. http://www.who.int/phe/news/Mercury-flyer.pdf. Accessed 30 Mar 2017.
Karagas MR, Choi AL, Oken E, Horvat M, Schoeny R, Kamai E, Cowell W, Grandjean P, Korrick S. Evidence on the human health effects of low-level methylmercury exposure. Environ Health Perspect. 2012;120:799–806.
Muniroh M, Khan N, Koriyama C, Akiba S, Vogel CFA, Yamamoto M. Suppression of methylmercury-induced IL-6 and MCP-1 expressions by N-acetylcysteine in U-87MG human astrocytoma cells. Life Sci. 2015;134:16–21.
Yamamoto M, Khan N, Muniroh M, Motomura E, Yanagisawa R, Matsuyama T, Vogel CFA. Activation of interleukin-6 and -8 expressions by methylmercury in human U937 macrophages involves RelA and p50. J Appl Toxicol. 2017;37:611–20.
Crespo-Lopez ME, Costa-Malaquias A, Oliveira EHC, Miranda MS, Arrifano GPF, Souza-Monteiro JR, Sagica FE, Fontes-Junior EA, Maia CSF, Macchi BM. Nascimento JLM is low non-lethal concentration of methylmercury really safe? A report on genotoxicity with delayed cell proliferation. PLoS ONE. 2016. https://doi.org/10.1371/journal.pone.0162822.
Das S, Basu A. Inflammation: a new candidate in modulating adult neurogenesis. J Neurosci Res. 2008;86:1199–208.
Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–59.
Widera D, Holtkamp W, Entschladen F, Niggemann B, Zanker K. MCP-1 induces migration of adult neural stem cells. Eur J Cell Biol. 2004;83:381–7.
Takeuchi T, Eto K, Tokunaga H. Mercury level and histochemical distribution in a human brain with Minamata disease following a long-term clinical course of twenty-six years. Neurotoxicology. 1989;10:651–7.
Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. 2010;30:459–73.
Filippo KD, Robert BH, Laschinger M, Hogg N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signalling pathways. J Immunol. 2008;180:4308–15.
Wu Q, Li WK, Zhou ZP, et al. The Tibetan medicine Zuotai differs from HgCl2 and MeHg in producing liver injury in mice. Regul Toxicol Pharmacol. 2016;78:1–7.
Godefroy D, Gosselin RD, Yasutake A, Fujimura M, Combadiere C, Maury-Brachet R, Laclau M, Rakwal R, Melik-Parsadaniantz S, Bourdineaud JP, Rostene W. The chemokine CCL2 protects against methylmercury neurotoxicity. Toxicol Sci. 2012;125:209–18.
Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K. Properties of the novel proinflammatory supergene ‘‘intercrine’’ cytokine family. Annu Rev Immunol. 1991;9:617–48.
Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med. 1997;185:99–109.
Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830:4117–29.
Aremu DA, Madejczyk MS, Ballatori N. N-acetylcysteine as a potential antidote and biomonitoring agent of methylmercury exposure. Environ Health Perspect. 2008;116:26–31.
Yamamoto M, Hirano S, Vogel CF, Cui X, Matsumura F. Selective activation of NF-kappaB and E2F by low concentration of arsenite in U937 human monocytic leukemia cells. J Biochem Mol Toxicol. 2008;22:136–46.
Yamamoto M, Takeya M, Ikeshima-Kataoka H, Yasui M, Kawasaki Y, Shiraishi M, Majima E, Shiraishi S, Uezono Y, Sasaki M, Eto K. Increased expression of aquaporin-4 with methylmercury exposure in the brain of the common marmoset. J Toxicol Sci. 2012;37:749–63.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2−∆∆CT method. Methods. 2001;25:402–8.
Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155:2158–64.
Zwijnenburg PJ, Polfliet MM, Florquin S, Van den Berg TK, Dijkstra CD, van Deventer SJ, Roord JJ, van der Poll T, van Furth AM. CXC-chemokines KC and macrophage inflammatory protein-2 (MIP-2) synergistically induce leukocyte recruitment to the central nervous system in rats. Immunol Lett. 2003;85:1–4.
Heine SJ, Olive D, Gao JL, Murphy PM, Bukrinsky MI, Constant SL. Cyclophilin A cooperates with MIP-2 to augment neutrophil migration. J Inflamm Res. 2011;4:93–104.
Liu L, Puri KD, Penninger JM, Kubes P. Leukocyte PI3Kgamma and PI3K delta have temporally distinct roles for leukocyte recruitment in vivo. Blood. 2007;110:1191–8.
Zhang XW, Liu Q, Wang Y, Thorlacius H. CXC chemokines, MIP-2 and KC, induce P-selectin-dependent neutrophil rolling and extravascular migration in vivo. Br J Pharmacol. 2001;133:413–21.
Tanimoto N, Terasawa M, Nakamura M, Kegai D, Aoshima N, Kobayashi Y, Nagata K. Involvement of KC, MIP-2, and MCP-1 in leukocyte infiltration following injection of necrotic cells into the peritoneal cavity. Biochem Biophys Res Commun. 2007;361:533–6.
Endlich B, Armstrong D, Brodsky J, Novotny M, Hamilton TA. Distinct temporal patterns of macrophage-inflammatory protein-2 and KC chemokine gene expression in surgical injury. J Immunol. 2002;168:3586–94.
Rovai LE, Herschman HR, Smith JB. The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J Leukoc Biol. 1998;64:494–502.
Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–9.
Authors’ contributions
Conceived and designed the experiments: SA, CK, MY, and NA. Performed the experiments: JD, NA, and MM. Analyzed the data: NA, MY, CK, and SA. Wrote the manuscript: DJ, NA, MM, SA, MY, and CK. Software analysis: NA and SA. All the authors read and approved the final manuscript.
Acknowledgements
We wish to thank Joint Research Laboratory of Kagoshima University Graduate School of Medicine and Dental Sciences for providing their facilities.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The study was financed by institutional funds.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in the published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
David, J., Nandakumar, A., Muniroh, M. et al. Suppression of methylmercury-induced MIP-2 expression by N-acetyl-l-cysteine in murine RAW264.7 macrophage cell line. Eur J Med Res 22, 45 (2017). https://doi.org/10.1186/s40001-017-0287-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-017-0287-4