
Abstract
Cnidium officinalis rhizomes were immersed in 80% MeOH. The extract was fractionated to water, n-butanol, and ethyl acetate fractions (Fr). Open column chromatography was repeatedly carried out on n-butanol and ethyl acetate Fr using silica gel, octadecyl silica gel, and Sephadex LH-20 as the stationary phase affording five phenyl alkanoids 1–5 including two new ones. The molecular structures including stereochemistry were decided based on spectroscopic interpretation of nuclear magnetic resonance, mass spectrometry, and infrared spectroscopy as well as chemical reaction. Three known compounds, coniferyl alcohol methyl ether (1), vanillin (2), and coniferyl aldehyde (3), were reported in the beginning for this plant by authors. Two new phenyl alkanoids were named, 7-methoxyeugenol and cnidiumoside.
Similar content being viewed by others
Introduction
Cnidium officinalis Makino (Umbelliferae) is a perennially grown herb either semi-shaded or not shaded in moist soil. It originates from China but is now extensively cultivated in Korea, Japan, and China [1]. The ground parts have been used as a medicinal, conspicuous aroma, and as condiment materials in beverages, baking, cosmetics, and the pharmaceutical industry [1]. The dried rhizomes (Cnidii Rhizoma) have been especially utilized in East-Asia countries for the treatment of female menstrual disorders and headaches through a decrease in inflammation and an improvement in blood circulation [2, 3]. Many studies have also reported the rhizomes as having anti-cancer [4], analgesic [5], antibacterial [6], anticonvulsive [7], anti-inflammatory [4, 5], febrifuge, hypotensive [8], sedative [9], and vasodilator [10] effects. The major constituents of the rhizomes were revealed to be phthalides, alkaloids, ceramides, polyphenols, and flavonoids [1]. Phthalides in particular are the most important constituents copiously contained in the essential oil [6]. Many reported biological activities of C. officinalis rhizomes are due to the presence of phthalides [6]. Phenols are also major constituents of rhizomes and are reported to have various pharmacological activities [11]. However, phenyl alkanoids of this plant are rarely studied. The authors isolated five phenyl alkanoids from C. officinalis rhizomes in this study. Among them, two compounds were revealed to be new and three others have never been reported for C. officinalis. This paper describes the isolation procedure for phenyl alkanoids and the structure determination including stereostructures.
Materials and methods
Plant materials
Cnidium officinalis rhizomes were provided and identified by Dr. J. T. Jeong, Department of Herbal Crop Research, RDA, Korea. A standard sample (NPCL-20200023) was put up at NPCL Laboratory, KyungHee University, Yongin, Korea.
General experimental procedures
The instruments and materials used for the isolation and identification of the phenyl alkanoids were the same as those in literatures [12]. The silica gel and the octadecyl silica gel (ODS) resins used for column chromatography (CC) were Kiesel gel 60 (Merck, Darmstadt, Germany) and the Lichroprep RP-18 (40–60 m\(\mu\), Merck) respectively. Sephadex LH-20 was purchased from Amersham Biosciences (Uppsala, Sweden). Thin layer chromatography (TLC) was carried out using Kiesel gel 60 F254 and RP-18 F254S (Merck) TLC plates, and the spots were detected using a UV lamp Spectroline Model ENF-240 C/F (Spectronics Corporation, Westbury, NY, USA) and a 10% H2SO4 solution. Deuterium solvents were purchased from Merck Co. Ltd and Sigma Aldrich Co. Ltd (St. Louis, MO, USA). Nuclear magnetic resonance (NMR) spectra were recorded on a 600 MHz FT-NMR spectrometer (Bruker AVANCE 600, Billerica, MA, USA). Infrared (IR) spectra were obtained using a Perkin Elmer Spectrum One FT-IR spectrometer (Buckinghamshire, England). The specific rotation value was measured with JASCO P-1010 digital polarimeter (Tokyo, Japan). ESIMS spectra were recorded on a AB SCIEX Q-TOF 5600 (Framingham, MA, USA). Solvents were supplied by Burdick & Jackson (Muskegon, MI, USA).
Isolation of phenyl alkanoids from Cnidium officinalis rhizomes
C. officinalis rhizomes were dried at room temperature and 10 kg of dried materials were powdered and soaked overnight in ethanol (70%, 54 L × 2) at room temperature. The obtained solution was evaporated using rotary vacuum evaporator at 40℃ affording a brownish extract (2.1 kg). The residue was divided using systemic solvent fractionation using polarity as ethyl acetate (COE, 280 g), n-butanol (COB, 125 g), and water (COW, 1.695 kg) Fr. The column chromatography (CC) for COE (270 g) and COB (120 g) was performed as seen in Figs. 1, 2. Finally, five phenyl alkanoids (1, COE-11–5; 2, COE-13–2-3; 3, COE-13–2-5; 4, COE-13–11-12–2; 5, COB-13–9-13–2) were isolated.

Isolation of phenyl alkanoids from the rhizomes of Cnidium officinalis

Chemical structure of phenyl alkanoids from the rhizome of Cnidium officinalis Makino. 1, coniferyl alcohol methyl ether; 2, vanillin; 3, coniferyl aldehyde; 4, 7S-7-methoxyeugenol; 5, cnidiumoside
Coniferyl alcohol methyl ether (1): Rf value on silica gel TLC, 0.72, n-hexane–ethyl acetate (1:1); Rf value on octadecyl silica gel TLC, 0.85, acetone–H2O (3:1); IR (LiF plates) νmax 3393, 2933, 1676, 1599, 1512, 1463 cm−1; 13C and 1H NMR (CMR and PMR): Tables 1 and 2. EI-MS: m/z 180 [M]+.
Vanillin (2): Rf value on silica gel TLC, 0.29, n-hexane– ethyl acetate (2:1); Rf value on octadecyl silica gel TLC, Rf 0.70, acetone–H2O (2:1); IR (LiF plates) νmax 3234, 2923, 1675, 1585, 1509 cm−1; CMR and PMR: Tables 1 and 2. EI-MS: m/z 152 [M]+.
Coniferyl aldehyde (3): Rf value on silica gel TLC, 0.20, n-hexane– ethyl acetate (2:1); Rf value on octadecyl silica gel TLC, Rf 0.72, acetone–H2O (2:1); IR (LiF plates) νmax 3183, 2919, 1748, 1660, 1584, 1512 cm−1; CMR and PMR: Tables 1 and 2. EI-MS: m/z 178 [M]+
7-Methoxyeugenol (4): Rf value on silica gel TLC, 0.76, n-hexane– ethyl acetate (1:1); Rf value on octadecyl silica gel TLC, 0.39, acetone–H2O (2:1); [α]D + 23.2 (c 0.12, CH3OH); IR (LiF plates) νmax 3153, 2916, 1673,1594, 1510, 1450 cm−1; CMR and PMR: Tables 1 and 2. EI-MS: m/z 194.0940 [M]+ (Calcd for C11H14O3 194.0937).
Cnidiumoside (5): Rf value on silica gel TLC, 0.54, chloroforem–methanol–H2O (6:4:1); Rf value on octadecyl silica gel TLC, 0.19, methanol–H2O (1:2); [α]D + 15.7 (c 0.10, CH3OH); IR (LiF plates) νmax 3425, 2996, 1675, 1465 cm−1; CMR and PMR: Tables 1 and 2. High resolution positive ESI–MS: m/z 845.3072 [M + H]+ (Calcd for C38H53O21 845.3074).
Acid hydrolysis of cnidiumoside (5)
The solution of 10 mg of 5 in 5 mL of 1N HCl was refluxed for 2 hrs. 15 mL of H2O was added and treated with ethyl acetate (20 mL × 2). The ethyl acetate phase was concentrated in vacuo and purified via open silica gel CC (2 × 8 cm) using n-hexane-ethyl acetate (2:1) as the eluting solution to give a dehydrodiconiferyl alcohol (5a, 4 mg).
Dehydrodiconiferyl alcohol (5a): [α]D -49 (c 0.18, CHCl3); PMR (600 MHz, CDCl3, δH, coupling pattern, J in Hz) 6.84 (1H, d, 1.9, H-2), 6.82 (1H, s, H-2′ or H-6′), 6.79 (1H, s, H-2′ or H-6′), 6.50 (1H, dd, 1.9, 8.3, H-6), 6.46 (1H, d, 8.3, H-5), 6.18 (1H, br. d, 16.0, H-7′), 6.14 (1H, dt, 16.0, 5.2, H-8′), 5.50 (1H, d, 6.8, H-7), 4.20 (2H, dd, 5.2, 1.3, H-9′), 3.89 (1H, dd, 11.9, 5.5, H-9a), 3.88 (3H, s, 3-OMe or 3′-OMe), 3.81 (1H, dd, 11.9, 6.3, H-9b), 3.78 (3H, s, 3-OMe or 3′-OMe), 3.56 (1H, ddd, 6.8, 6.3, 6.5, H-8).
Results and discussion
Five phenyl alkanoids 1–5 were isolated from the C. officinalis rhizome through repeated open column chromatography using octadecyl silica gel, silica gel, and Sephadex LH-20 as the stationary phase. 1–3 were identified as coniferyl alcohol methyl ether (1) [13], vaniline (2), and coniferyl aldehyde (3) [14], respectively, from the spectroscopic data and confirmed by previously reported data.
Compound 4 showed a molecular ion peak at m/z 194.0940 [M]+ in the high resolution EI-MS, establishing the molecular formula as C11H14O3 (Calcd. for C11H14O3 194.0937). The IR data exhibited the absorbing bands due to hydroxyl (3153 cm−1) and olefin (1673, 1594 cm−1) groups. CMR spectrum (150 MHz, CDCl3) showed 11 signals including two methoxy signals (δC 56.06, OMe-3; 56.40, OMe-7) suggesting 4 to be a phenylpropanoid. Two oxygenated olefin quaternaries (δC 145.36, C-4; 146.84, C-3), one olefin quaternary (δC 132.99, C-1), four olefin methines (δC 109.14, C-2; 114.24, C-5; 120.32, C-6; 139.00, C-8), one exomethylene (δC 116.21, C-9), and one oxygenated methine (δC 84.64, C-7) were detected. PMR spectrum (600 MHz, CDCl3) included the olefin signals due to a 1,2,4-trisubstituted benzene moiety [δH 6.81, dd, J = 1.8, 7.8 Hz, H-6 (δH, coupling pattern, J in Hz), H-5; 6.86, d, 1.8, H-2; 6.88, d, 7.8), the signals derived from a vinyl moiety (5.20, br. d, 10.2, H-9a; 5.26, br. d, 17.4, H-9b; 5.93, ddd, 6.6, 10.2, 17.4, H-8), an oxygenated methine (4.55, d, 6.6, H-7), and two methoxies (3.31, s, OMe-7; 3.90, s, OMe-3) signals. The NMR data described above were almost identical to eugenol [15] excluding an oxygenated methine and a methoxy moieties instead of a methylene moiety, suggesting that an additional methoxy moiety was linked to C-7. The HMBC spectrum affirmed it; a methoxy proton (δH 3.31, OMe-7) correlated with an oxygenated methine carbon (δC 84.64, C-7), which also correlated with three olefin methine protons (δH 5.93, H-8; 6.81, H-6; 6.86, H-2) and an exomethylene proton (δH 5.20, 5.26, H-9). As a result, the planar structure of compound 4 was decided as a 2-methoxy-4-(1-methoxy-2-propen-1-yl) phenol, 7-methoxyeugenol. The stereostructure of chiral center, C-7, could be determined as S by comparison of a specific rotation value, [α]D + 23.2, with a previously reported value for 7-hydroxyeugenol [16, 17]. Compound 4 was identified to be 7S-7-methoxyeugenol.
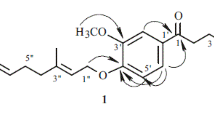
Compound 5 exhibited a molecular ion peak at m/z 845.3072 [M + H]+ in the high resolution ESI–MS, established the molecular formula as C38H52O21 (Calcd. for C38H53O21 845.3072). The IR data showed the absorption bands due to hydroxyl (3153 cm−1) and phenyl (1673, 1594 cm−1) moieties. Though CMR spectrum (150 MHz, CD3OD) showed about 50 carbon signals, some of them were revealed from those of impurities included in compound 5. Among them, 38 carbon signals were defined as those of the compound. The 20 signals including two methoxy signals as those of the aglycone moiety and 18 carbon signals due to three hexoses were detected, suggesting that 5 was a lignan triglyceride. CMR spectrum showed two hemiacetals (δC 102.89, C-1′′; 104.71, C-1′′′) and one hemiketal (δC 99.34, C-2′′′′) carbon signals as well as PMR spectrum showed only two hemiacetal proton signals (4.37, d, 7.8, H-1′′′; 4.89, d, 7.8, H-1′′), indicating that three sugars in compound 5 were composed of two aldohexoses and one ketohexose. CMR chemical shifts of three sugars confirmed the presence of one terminal β- d -galactopyranosyl (δC 62.95, C-6′′; 69.54, C-4′′; 71.99, C-2′′; 75.33, C-3′′; 77.99, C-5′′; 102.89, C-1′′) and one terminal β-d-allulofuranosyl (δC 62.65, C-6′′′′; 64.69, C-1′′′′; 71.42, C-3′′′′; 71.79, C-4′′′′; 83.46, C-5′′′′; 99.34, C-2′′′′) moieties as well as one β- d -glucopyranosyl (δC 71.49, C-4′′′; 75.05, C-2′′′; 78.23, C-5′′′; 78.35, C-3′′′; 104.71, C-1′′′) with a down-shifted oxygenated methylene signal (δC 66.06, C-6′′′) due to a glycosidation shift [18] indicating a terminal sugar to be connected to the hydroxyl group at C-6′′′ of a glucopyranosyl moiety. The coupling constants of two hemiacetal protons were 7.8 and 7.8 Hz confirming the anomer hydroxyl groups of both aldohexose moieties to have a β-configuration. The carbon signals of the aglycone moiety included four oxygenated olefin quaternaries (δC 145.71, C-3′; 147.74, C-4; 149.29, C-4′; 151.04, C-3), three olefin quaternaries (δC 129.89, C-5′; 132.91, C-1′; 138.22, C-1), seven olefin methines (δC 111.50, C-2; 112.37, C-2′; 116.92, C-6′; 118.13, C-5; 119.62, C-6; 127.86, C-8′; 132.05, C-7′), one oxygenated methine (δC 89.01, C-7), two oxygenated methylenes (δC 63.99, C-9′; 72.57, C-9), two methoxies (δC 56.92 × 2), and one methine (δC 53.39, C-8) signals. The oxygenated methylene carbon was shifted down-field compared to the usual one, confirming it to be linked to a sugar. PMR spectrum exhibited proton signals due to one 1,2,4-trisubstituted benzene ring (6.98, br. d, 8.4, H-6; 7.08, br. s, H-2; 7.14, d, 8.4, H-5), one 1,2,3,5-tetrasubstituted benzene ring (6.85, br. s, H-2′; 7.01, br. s, H-6′), a double bond with a trans configuration (6.23, dt, 15.6, 5.0, H-8′; 6.53, br. d, 15.6, H-7′), one oxygenated methine (5.70, d, 8.0, H-7), two oxygenated methylenes (4.20, d, 5.0, H-9′; 3.77, overlapped, H-9a; 4.24, dd, 4.0, 10.2, H-9b), two methoxies (3.84, s, OMe-3; 3.89, s, OMe-3′), and one methine (δH 3.64, overlapped, H-8) moieties. The aforementioned NMR data suggested the aglycone to be a neolignan with a coniferyl alcohol and a dehydroconiferyl alcohol moieties. In the HMBC spectrum, the oxygenated methine proton signal (δH 5.70, H-7) showed cross peaks with one oxygenated olefin quaternary (δC 149.29, C-4′), two olefin quaternaries (δC 129.89, C-5′; 138.22, C-1), two olefin methines (δC 111.50, C-2; 119.62, C-6), one oxygenated methylene (δC 72.57, C-9), and one methine (δC 53.39, C-8) carbon signals. The methine proton signal (δH 3.64, H-8) correlated with the carbon signals due to one oxygenated olefin quaternary (C-4′), two olefin quaternaries (C-1 and C-5′), one olefin methine (δC 116.92, C-6′), and one oxygenated methylene (C-9) moieties. Therefore, the two phenylpropanoid moieties of neolignan were linked at C-7 and C-4′ through an ether bond and at C-8 and C-5′. The anomer proton signals of one β-d-galactopyranosyl and one β-d-glucopyranosyl moieteies (δH 4.37, H-1′′′ and 4.89, H-1′′) showed correlations with the oxygenated olefin quaternary (δC 147.74, C-4) and the oxygenated methylene (C-9) carbon signals, respectively, as well as the anomer carbon signal of a β-d-glucopyranosyl moiety (δC 104.71, C-1′′′) correlated with the proton signals of an oxygenated methylene moiety (δH 3.77 and 4.24, H-9). Long-range coupling between the anomer carbon signal of the β-d-allulopyranosyl moiety (δC 99.34, C-2′′′′) and the oxygenated methylene proton signals (δH 3.47 and 3.65, H-6′′′) of the β-d-glucopyranosyl moiety in the HMBC spectrum were detected. The HMBC correlations described above concluded the linkage position of three sugars as seen in Fig. 1. The relative stereostructure between H-7 and H-8 was determined to be a trans configuration because the cross peak was not observed between H-7 (δH 5.70) and H-8 (δH 3.64) but was observed between H-7 and H-9 (δH 3.77 and 4.24) in the NOESY spectrum. The glycoside, compound 5, was treated with 1 N HCl and refluxed at 90℃ for two hours to finally yield the aglycone, dehydrodiconiferyl alcohol (5a). The dehydroconiferyl alcohol moiety with two chiral carbons, C-7 and C-8, has four stereoisomers, that showed typical specific rotation value [19]. 5a had an erythro-configuration between C-7 and C-8 and the specific rotation value was measured to be -49, confirming the absolute stereostructue of C-7 and C-8 to be R and S, respectively. Finally, the chemical structure of compound 5 was determined to be (2R,3S)-4-O-β-d-galactopyranosyl-9-[(6-O-β-d-allulofuranosyl-β-d-gucopyranosyl)oxy] dehydrodiconiferyl alcohol as seen in Fig. 1. It was revealed to be a new compound, named cnidiumoside.
Availability of data and materials
The data and materials used in this study are available under permission from the corresponding author on reasonable request.
Change history
07 January 2022
A Correction to this paper has been published: https://doi.org/10.1186/s13765-021-00658-7
References
Li W, Tang Y, Chen Y, Duan JA (2012) Advances in the chemical analysis and biological activities of chuanxiong. Molecules 17:10614–10651
Zhou J, Qu F (2009) Treating gynaecological disorders with traditional Chinese nedicine: A review. Afr J Tradit Complement Altern Med 6:494–517
Shin HY, Lee SH, Kim HR, Kim JH, Yang SB, Cho SY, Park JM, Ko CN, Park SU (2020) A review of clinical research trends in the treatment of primary headache disorders with pharmacopuncture. J Int Korean Med 39:1191–1205
Lee KY, Kim JH, Kim EY, Yeom M, Jung HS, Sohn Y (2019) Water extract of Cnidii Rhizoma suppresses RANKL-induced osteoclastogenesis in RAW 264.7 cell by inhibiting NFATc1/c-Fos signaling and prevents ovariectomized bone loss in SD-rat. BMC Complement Altern Med 19:207–219
Cho SK, Kwon OI, Kim CJ (1996) Anti-inflammatory and analgesic activities of the extracts and fractions of Cnidii Rhizoma. Kor J Pharmacogn 27:282–287
Sim Y, Shin S (2014) Antibacterial activities of the essential oil from the leaves and rhizomes of Cnidium officinale Makino. J Essent Oil Res 26:452–457
Jeong JE, Lee YJ, Choi YA, Park JM, Lee SM, Jo NY, Lee EY, Lee CK, Roh JD (2021) Seizure after subdural hematoma treated with combination western-Korean medicine. J Acupunct Res 38:72–78
Jeong JB, Ju SY, Park JH, Lee JR, Yun KW, Kwon ST, Lim JH, Chung GY, Jeong HJ (2009) Antioxidant activity in essential oils of Cnidium officinale Makino and Ligusticum chuanxiong hort and their inhibitory effects on DNA damage and apoptosis induced by ultraviolet B in mammalian cell. Cancer Epidemiol 33:41–46
Lee JT, Park JH, Lee KH (2005) Effect of methanol extract of Cnidii rhizoma on the function of receptors for GABA and glycine. J Korean Acad Pediatr Dent 32:55–66
Kim SD, Kim GW, Shin HM (2006) Effects of Cnidium officinale, Petasites japonicas, Coptis chinonsis extract mixture on vasodilation. Korean J Orient Physiol & Pathol 20:1620–1624
Adil M, Ren X, Jeong BR (2019) Light elicited growth, antioxidant enzymes activities and production of medicinal compounds in callus culture of Cnidium officinale Makino. J Photochem Photobiol B 196:111509
Kim HG, Jung YS, Oh SM, Oh HJ, Ko JH, Kim DO, Kang SC, Lee YG, Lee DY, Baek NI (2020) Coreolanceolins A-E, new flavanones from the flowers of Coreopsis lanceolata, and their antioxidant and anti-inflammatory effects. Antioxidants 9:539
Naito T, Niitsu K, Ikeya Y, Okada M, Mitsuhashi H (1992) A phthalide and 2-farnesyl-6-methyl benzoquinone from Ligusticum chuangxiong. Phytochem 31:1787–1789
Zhao YX, Luo JR, Li CS, Yi TM, Zhou J (2008) Palaeophytochemical constituents from the miocene-fossil wood of Picea likiangensis in Xun-dian of Yunnan, China. Bull Korean Chem Soc 29:1613–1616
Elgendy EM, Khayyat SA (2008) Oxidation reactions of some natural volatile aromatic compounds: Anethole and eugenol. Russ J Org Chem 44:823–829
Stambasky J, Malkov VA, Kocovsky P (2008) Synthesis of enantiopure 1-arylprop-2-en-1-ols and their tert-butyl carbonates. J Org Chem 73:9148–9150
Zhang SY, Fan CL, Huang XJ, Huang WH, Liu XH, Wang L, Ye WC (2015) A new phenylpropanoid and a new isoflavone glycoside from Shenqi Fuzheng Injection. J Asian Nat Prod Res 17:274–279
Kim HG, Oh HJ, Ko JH, Song HS, Lee YG, Kang SC, Lee DY, Baek NI (2019) Lanceoleins A-G, hydroxychalcones, from the flowers of Coreopsis lanceolata and their chemopreventive effects against human colon cancer cells. Bioorg Chem 85:274–281
Hirai N, Okamoto M, Udagawa H, Yamamuro M, Kato M, Koshimizu K (1994) Absolute configuration of dehydrodiconiferyl alcohol. Biosel Biotech Biochem 58:1679–1684
Acknowledgements
This work was supported by the “Cooperative Research Program for Agriculture Science & Technology Development” (Project no. PJ01420403), Rural Development Administration, Republic of Korea.
Funding
Rural Development Administration (RDA), Republic of Korea (Project no. PJ01420403) funded this study.
Author information
Authors and Affiliations
Contributions
N-IB planned the study and wrote the paper. H-GK, TNN, and Y-GL isolated phenyl alkanoids. MHL, DYL, Y-HL, and N-IB determined the chemical structures of phenyl alkanoids. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
1H and 13C NMR spectra of coniferyl alcohol methyl ether (1). Fig. S2. 1H and 13C NMR spectra of vanilline (2). Fig. S3. 1H and 13C NMR spectra of coniferyl aldehyde (3). Fig. S4. 1H and 13C NMR spectra of 7S-7-methoxyeugenol (4). Fig. S5. 1H-1H COSY and HSQC spectra of 7S-7-methoxyeugenol (4). Fig. S6. HMBC spectrum of 7S-7-methoxyeugenol (4). Fig. S7. 1H and 13C NMR spectra of cnidiumoside (5). Fig. S8. 1H-1H COSY and HMQC spectra of cnidiumoside (5). Fig. S9. HMBC spectra of cnidiumoside (5). Fig. S10. NOESY spectrum of cnidiumoside (5).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, HG., Nguyen, T.N., Lee, YG. et al. New phenylalkanoids from the rhizome of Cnidium officinalis Makino. Appl Biol Chem 64, 79 (2021). https://doi.org/10.1186/s13765-021-00650-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-021-00650-1