
Abstract
Melanoma is the leading cause of skin cancer deaths, and the poor prognosis of metastatic melanoma has made needs for a novel pharmacological treatment or efficient intervention. Carnosol, a major polyphenolic compound from Rosmarinus officinalis, has a wide range of biological activities including anti-cancer effect. However, the underlying molecular mechanisms of its anti-cancer effect remain poorly understood in malignant human melanoma cells. In the present study, we investigate the apoptotic effect and the underlying anti-cancer mechanisms of carnosol. Our results revealed that carnosol strongly induced apoptosis against human melanoma G361 cells in a dose- and time-dependent manner, and caused dramatical elevation in cellular reactive oxygen species (ROS) level during apoptosis. In mechanistic studies, carnosol treatment decreased protein level of anti-apoptotic B‑cell lymphoma 2 (Bcl-2) and B cell lymphoma-extra large (Bcl-xL), however, increased level of pro-apoptotic Bcl-2-associated X protein (Bax) protein. Moreover, carnosol escalated cellular level of p53, which was accompanied by a decline of mouse double minute 2 homolog (MDM2) level. Also, carnosol inhibited activation of Src and signal transducer and activator of transcription 3 (STAT3), therefore down-regulated STAT3-dependent gene expression, such as D-series cyclin and survivin. These changes by carnosol were attenuated by pre-treatment of N-acetyl cysteine, and abolished progression of carnosol-induced apoptosis. In conclusion, carnosol induced apoptosis in human melanoma G361 cells through ROS generation and inhibition of STAT3-mediated pathway. Our results provide molecular bases of carnosol-induced apoptosis, and suggest a novel candidate for human melanoma treatment.
Similar content being viewed by others


Introduction
Melanoma, a malignant tumor derived from melanocytes, causes the majority of deaths attributed to skin cancer. The incidence of melanoma, the most fatal form of skin cancer, has increased significantly over the past 20 years [1]. Melanoma has been treated by various ways. Surgical excision is a representative treatment in early stages of the disease [2], in addition, RAF and MEK kinase-targeted chemotherapy or immunotherapy using immune checkpoint inhibitors are approved for patients with advanced stage [3]. However, melanomas have diverse genetic alterations in key molecules on growth/proliferation signaling pathways and cell cycle control, such as BRAF, NRAS, KIT, PTEN and CDKN2A [4,5,6], and the poor prognosis-associated with metastatic melanoma has made it necessary for a novel pharmacological treatment options or more efficient chemotherapeutic intervention.
Apoptosis, known as programmed cell death, is triggered by extrinsic and intrinsic ligands. These two different apoptotic pathways are regulated by diverse intracellular signaling pathways and coordinated by a network of genes [7]. During the apoptotic process, activation of caspase cascade leads to the cleavage of essential proteins that is involved in anti-apoptotic pathways, such as Bcl-2 family proteins [8]. Apoptosis is a critical process for cellular homeostasis, therefore, apoptosis became a key target for the development of new anti-cancer therapies [9].
Reactive oxygen species (ROS) are generated by the mitochondria in most mammalian cells [10]. ROS have been related to diverse physiological and pathologic condition. Elevated ROS also act as signaling molecules in the maintenance of physiological functions [11], however, excessive ROS above the physiological concentration can lead to carcinogenesis or cell death [12].
Oxidative stress is due to the imbalance between scavenging activities of intracellular anti-oxidant and production of ROS [13], and also elevates intracellular levels of ROS that cause damage to various biological molecules, such as DNA, proteins and lipids [14]. In addition, oxidative stress has been reported to induce apoptosis via diverse pathways, such as endoplasmic reticulum (ER) stress and mitochondrial cascade [15]. Therefore, manipulating intracellular ROS levels is a way to selectively kill various cancer cells, which has been involved in the anti-cancer effects of several therapeutic agents, including paclitaxel, cisplatin, and TNF-related apoptosis-inducing ligand (TRAIL/Apo-2L) [16,17,18].
It has been demonstrated that signal transducer and activator of transcription 3 (STAT3), a member of the STAT family, plays critical roles in signaling of various cytokines, hormones, and growth factors on diverse human malignancies [19, 20]. STAT3 is constitutively activated in most human solid tumors, and constitutive activation of STAT3 signalling pathway has been demonstrated to mediate critical processes in tumor cells, such as survival, proliferation, angiogenesis, and metastasis [21,22,23]. In previous studies, many researchers indicated that STAT3 might be a novel target for anti-cancer therapy [24,25,26].
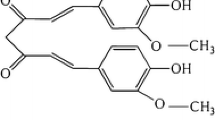
Rosmarinus officinalis, generally known as rosemary, is an aromatic evergreen herb which is in use of traditional medicine, fragrance, and spice. Rosmarinus officinalis contains rosmarinic acid, ursolic acid, betulinic acid, carnosic acid and carnosol as major bioactive polyphenolic components [27]. Among these components, carnosol (Fig. 1a) has a wide range of biological activities, including anti-cancer, anti-oxidant, and anti-inflammatory effects [28,29,30].
In this study, we aimed to investigate the underlying molecular mechanisms of anti-cancer effects of carnosol in malignant human melanoma G361 cells. Our results suggest that carnosol treatment induced apoptosis in G361 cells through oxidative stress and the interference of STAT3-mediated signaling pathway.
Materials and methods
Chemicals and reagents
Carnosol (purity 99%), N-acetyl cysteine (NAC), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and anti-β-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against cleaved caspase-3, -7, -9, poly(ADP-ribose) polymerase (PARP), Bcl-2, Bcl-xl, Bax, cytochrome c, STAT3, p-STAT3 (Y705), Src, p-Src, cyclin D1, D2, D3 and survivin were bought from Cell Signaling Technology Inc. (Beverly, MA, USA). Antibodies against p53, murine double minute-2 (Mdm2), and horse-radish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Paso Robles, CA, USA). The 2′,7′-dichlorofluorescein diacetate (DCF-DA) was procured from Invitrogen (Carlsbad, CA, USA). Hank’s balanced salt solution (HBSS) was purchased from the Meditech (Herndon, VA, USA).
Cell lines and cell culture
G361 cells were obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics (100 U/ml penicillin G and 100 mg/ml streptomycin) at 37 °C in a humidified incubator with 5% CO2-containing atmosphere. In all experiments, cells were seeded at 50–60% confluency. All chemicals were dissolved in ethanol and the final concentration of ethanol was less than 0.1% in culture media.
Cell viability assay
The effect of carnosol on cell viability was measured by MTT assay. Cells were plated at 2 × 103 cells/well onto a 96-well plate and incubated with 100 µl of carnosol-containing media at 37 °C for indicated time. Thereafter, 10 μl of MTT stock solution (5 mg/ml) was added to each well and incubated for 4 h. Medium was removed, insoluble formazan was dissolved in DMSO and absorbance at 550 nm was measured by using microplate reader (Tecan Trading AG, Switzerland). Cell viability was described as the relative percentage of control, and calculated based on technical triplicate repeats.
Annexin V-propidium iodide (PI) staining
Annexin V-PI staining was performed using fluorescein isothiocyanate (FITC)-Annexin V staining kit (BD Biosciences, San Jose, CA, USA) following the manufacturer’s instructions. Briefly, carnosol-treated cells were washed with PBS and resuspended in binding buffer containing Annexin V and PI. The fluorescence intensity of cells was analyzed by FACSVerse™ (BD Biosciences).
Immunoblot analysis
Cells were harvested and lysed with radioimmunoprecipitation assay (RIPA) buffer, and then cell lysates were quantified by using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA). Equal amount of cell lysates was denatured by boiling at 100 °C for 5 min in SDA-PAGE sample buffer. The cell lysates were separated on 8–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membrane. The blotto was blocked with 5% skim milk in Tris-buffered saline containing 0.1% of Tween 20 (TBS-T) for 1 h at room temperature. The blot membrane was washed for 3 times with TBS-T after blocking and hybridized with corresponding antibodies at 4 °C for overnight. After then, the blotto was washed for 3 times and incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies at room temperature. The membranes were washed 3 times for 10 min each with TBS-T buffer. Immunoblot s were developed using enhanced chemiluminescence (ECL) reagent (GE Healthcare, NJ, USA) and visualized with Imagequant™ LAS 4000 (Fujifilm Life Science, Japan).
Electrophoretic mobility gel shift assay (EMSA)
The EMSA for STAT3 DNA binding was performed using a DNA–protein binding detection kit according to the manufacturer’s protocol (GIBCO BRL, Grand Island, NY). Nuclear extracts were prepared from cells cultured with or without TQ. The STAT3 oligonucleotide probe 5′-AGC TTC ATT TCC CGT AAA TCC CTA-3′ (Bionics, South Korea) was labeled with [γ-32P] ATP and EMSA was performed according to the protocol previously described [31].
Measurement of ROS accumulation
Cells were treated with carnosol in the presence or absence of NAC for 12 h and then loaded with 25 μM of DCF-DA. After incubation for 30 min at 37 °C in 5% CO2-containing atmosphere, cells were washed twice with HBSS solution, and examined under a fluorescence microscope to detect the intracellular ROS. The positive control cells were treated with 500 μM of hydrogen peroxide (H2O2). For flow cytometry, cells were trypsinized and re-suspended in growth media, then cells were applied to FACSVerse™ for further analysis.
Statistical analysis
The data were expressed as mean ± SD based on at least triplicate repeats, and statistical analysis between each single experimental group was performed using the Student’s t-test, and p value less than 0.05 was considered statistically significant.
Results
Effect of carnosol on cell viability in human melanoma G361 cells
To examine the effect of carnosol on the viability of human melanoma G361 cells, we performed MTT assay after treatment of carnosol. As shown in Fig. 1b, carnosol significantly reduced cell viability with a time- and concentration-dependent manner in comparison to the untreated cells. Carnosol treatment also induced the apoptosis-like morphological changes as the dose increases in G361 cells (Fig. 1c). We further analyzed these morphological changes after carnosol treatment by Annexin V-PI staining and calculated the number of Annexin V(+)/PI(−) cells. At 100 μM of carnosol-treated group, the cells were revealed approximately 80% of the apoptotic cell population as compared to untreated cells after 24 h of treatment (Fig. 1d, e). These results showed that the treatment of carnosol to G361 cells dramatically increased apoptotic cell death as compared with untreated control cells.

Concentration- and time-dependent effect of carnosol on cell viability of G361 cells. a Chemical structure of carnosol. b Concentration- and time-dependent decrease of viability of human melanoma G361 cells after carnosol treatment. c The morphological changes of G361 cells after dose-dependent carnosol treatment after 24 h. d The FACS analysis for carnosol-treated G361 cells (20, 50, 100 μM) using Annexin V-PI staining. The data were presented by dot plots. Annexin V-positive cells were calculated and plotted as the mean ± S.D e *p < 0.05, **p < 0.01 compared to control group
Effect of carnosol on activation of caspases, levels of anti-apoptotic Bcl-2 family protein, and p53
The caspase cascade is one of the key components in induction of apoptosis, and is closely linked to mitochondrial proteins that control the mitochondrial membrane permeability [32]. To elucidate the involvement of caspases and Bcl-2 family proteins in carnosol-induced apoptosis, we assessed the cellular levels of cleaved caspases, Bcl-2, Bcl-xL, and Bax by immunoblot. After 24 h of carnosol treatment with indicated concentration, caspase-9, -7, and -3, which play a critical role in the execution-phase of apoptosis, were sequentially activated by proteolytic cleavage. In addition, cleaved PARP, a well-known substrate of active caspase-3, was detected in a dose-dependent manner (Fig. 2a). In addition, immunoblot analysis against anti-apoptotic Bcl-2 family proteins revealed that the expression levels of Bcl-2 and Bcl-xL were concentration-dependently decreased, while pro-apoptotic Bax protein was elevated its expression by carnosol treatment (Fig. 2b). As shown in Fig. 2c, carnosol increased expression level of p53, however, diminished protein expression level of Mdm2, a regulator of p53 through proteosomal degradation, in a concentration-dependent manner. These data suggest that carnosol induces apoptosis through the decrease of Bcl-2 and Bcl-xL and accumulation of p53 by elimination of Mdm2 in G361 cells.

Effect of carnosol on proteolytic cleavage of caspases and protein levels of anti- and pro-apoptotic Bcl-2 family and p53 in apoptosis of G361 cells. a, b The cellular levels of apoptosis marker proteins (cleaved caspase-9, -7, -3, and PARP) and anti- and pro-apoptotic proteins (Bax, Bcl-2, and Bcl-xL) in G361 cells after carnosol treatment (0, 20, 50, 100 μM) for 24 h. β-actin was as a loading control. c The expression levels of p53 and mdm2 in G361 cells with concentration-dependent carnosol treatment
Inhibition of STAT3 activation and STAT3-mediated gene expression by carnosol
STAT3 plays a key role in cell growth and apoptosis through transcriptional activation of diverse genes in response to cell stimuli [33]. To further analyze the underlying mechanism in carnosol-induced apoptosis, the cellular levels of p-STAT3 and the activity of STAT3-stimulating protein kinases, Janus kinases (JAK) and Src, were examined by immunoblot analysis. Treatment of carnosol in G361 cells inhibited phosphorylation of STAT3 on tyrosine 705 residue (Fig. 3a), while p-JAK and JAK were not detected under the same condition (data not shown). In addition, we observed that carnosol inhibited DNA-binding activity of STAT3, which is assessed by EMSA (Fig. 3b). Then, we assessed the expression levels of survivin and D-series cyclins, the representative STAT3-regulated cell proliferative genes. As shown in Fig. 3c, the expression levels of Cyclin D1, D2, and D3 were gradually decreased by carnosol treatment in a concentration-dependent manner, and the cellular level of survivin was also decreased from 20 μM of carnosol treatment. These results suggest that carnosol treatment suppresses activity of Src kinase to activate STAT3 and this inhibition down-regulates STAT3-mediated gene expression in G361 cells.

Carnosol-induced inhibition of STAT3-mediated signaling and gene expression in G361 cells. a G361 cells were treated with indicated concentrations of carnosol. The cellular levels of p-STAT3 (Y705), STAT3, p-Src, and Src were determined by immunoblot. b The DNA-bound STAT3 was analyzed after carnosol treatment. c Immunoblot analysis was assessed to elucidate the expression levels of cyclin D1, D2, D3, and survivin after 24 h treatment with indicated concentrations of carnosol
Involvement of ROS in carnosol-induced apoptosis
Since it is well known that the accumulation of intracellular ROS could induce apoptosis in various cancer cells, we examined the involvement of ROS in carnosol-induced apoptosis. The cells were treated with carnosol (20, 50, and 100 μM) for 12 h, and then we stained the cells with DCF-DA to measure intracellular total ROS (Fig. 4a, b). The carnosol-induced ROS generation was analyzed by fluorescence microscopy and flow cytometry. The treatment of carnosol in G361 cells led to the production of ROS in a concentration-dependent manner, especially, 100 μM of carnosol-treated G361 cells generated more ROS than H2O2-treated cells. However, carnosol-induced ROS generation was attenuated by pre-treatment of NAC (Fig. 5).

Increase in total ROS level after carnosol treatment in G361 cells. a Cells were treated with carnosol (20, 50 or 100 μM) for 12 h and cells were stained with 25 μM of DCF-DA. Then, intracellular accumulation of ROS was examined by fluorescence microscopy (×200) or analyzed by flow cytometry (b, left panel). H2O2 (500 μM)-treated cells were used as positive control. The increase in the intensity of DCF-DA fluorescence was compared to blank group, and plotted as a graph using geomean value (c, right panel). **p < 0.01 compared to control group

NAC-dependent elimination of carnosol-induced ROS generation. Cells were treated with NAC (5 mM) for 12 h with or without carnosol treatment (100 μM), then ROS levels were measured by fluorescence microscopy (a) or by flow cytometry (b) after DCF-DA staining. c The percent intensity of ROS generation compared to non-stained blank was calculated using geomean value and plotted as a graph. The statistical significance was determined by Student’s T-test against non-treated control. *p < 0.01 compared to control group
Prevention of carnosol-induced apoptosis by scavenging of ROS
To explore whether carnosol-induced apoptosis regulated by ROS, we assessed the amelioration of carnosol-induced apoptosis including cell viability, STAT3-mediated signal pathway, and proteolytic activation of caspases in NAC-treated G361 cells. Pre-treatment of NAC mitigated carnosol-induced changes on the phosphorylation of STAT3 and Src activity (Fig. 6a), DNA-binding activity of STAT3 (Fig. 6b), and level of cleaved caspases (Fig. 6c). The pre-treatment of NAC reduced expression level of p53 and cleaved PARP. These findings suggest that ROS play critical roles in carnosol-induced apoptosis in G361 cells.

The amelioration of carnosol-induced apoptosis by elimination of ROS. a The NAC-induced restoration in phosphorylation levels of STAT3 and Src (a), in DNA-bound STAT3 (b). c The diminution of cleaved apoptosis marker proteins and decrease of p53 level after NAC treatment in carnosol-treated G361 cells. d, e The decrease of apoptotic cell population by NAC treatment. The percent of apoptotic cell of each group was calculated and plotted. **p < 0.01 compared to control group
Discussion
Carnosol, a phenolic diterpene in rosemary, has been reported to exert anti-cancer activities on various cancer cells [34, 35]. However, the biochemical basis of carnosol-induced anti-cancer mechanism remains elusive. In this study, we discovered that carnosol could induce apoptosis in human melanoma G361 cells in a concentration- and time-dependent manner. This apoptosis-inducing effect corresponds to the apoptosis by carnosol in other cancer cells [34, 36, 37]. In this study, we determined the apoptosis-inducing effect of carnosol on G361 cells and demonstrated that carnosol treatment would induce apoptosis through ROS-dependent inhibition of STAT3 signaling pathway.
The proteolytic activation of caspase-9 following the release of cytochrome c from mitochondria and activation of apaf-1 by intrinsic apoptotic signals activates downstream executioner caspases, such as caspase-3 and -7, then, initiates apoptosis [38]. During mitochondria-mediated apoptosis, the transition of mitochondrial membrane potential is an irreversible step and this process is regulated by anti- or pro-apoptotic BCL-2 family proteins [39]. According to current reports, the inhibition of Bcl-2 and Bcl-xL, anti-apoptotic BCL-2 family proteins, could facilitate apoptosis and be an overcoming strategy for drug resistance in human melanoma [40, 41]. From our results, the carnosol treatment revealed the proteolytic activation of caspase-3 and the cleavage of PARP. In addition, carnosol showed inhibition of Bcl-2 and Bcl-xL expression, while the expression level of Bax was increased. These results suggest that carnosol might cause apoptosis through the mitochondria-mediated intrinsic pathway.
The cellular level of tumor suppressor p53 is regulated by its cytosolic repressor, Mdm2, through proteasomal degradation [42]. Negative regulation of Mdm2 by chemicals stabilizes p53, and induces growth arrest and apoptosis in human melanoma cells [43, 44]. In the present study, the level of p53 was increased following the decrease of Mdm2 after carnosol treatment. These results suggest that accumulation of p53 through negative regulation of Mdm2 might contribute to carnosol-induced apoptosis.
The mitochondrion is a major subcellular organelle producing ROS as a by-product of the oxidative phosphorylation process. The numerous researchers have been examined chemicals from natural products for their anti-cancer activity [45, 46], and their large part of anti-cancer activity relies on positive- or negative-regulation of ROS level [47]. In our study, carnosol treatment generated ROS from the lowest dose, 20 μM, and this increase in cellular ROS level was analogous to 500 μM of H2O2-treated positive control group. Moreover, carnosol treatment-induced ROS generation in G361 cells was decreased by pre-treatment of anti-oxidant, NAC, and this pre-treatment of the cells with NAC attenuated carnosol-induced cleavage of caspase-3 and PARP. These evidences showed that ROS plays pivotal roles in carnosol-induced apoptosis.
Separately, several reports support that the expression of Bcl-2 and Bcl-xL is mediated by STAT3 in human melanoma models [48], and abnormal activation of STAT3 exerts effects on proliferation, invasion and migration of melanoma cells [49]. Moreover, the inhibiting STAT3 signal exhibits cytotoxicity and down-regulated expression of Bcl-xL and metalloprotease-2 and -9 in melanoma cells [50]. We assessed the carnosol-induced changes on STAT3 signal pathway in G361 cells. STAT3 is activated by phosphorylation on tyrosine 705 residue by upstream protein kinases, such as JAK2 [51], and Src kinase [52], followed by homodimer formation. Our results revealed that carnosol treatment decreased phosphorylation on STAT3 by Src kinase, and this inhibition on STAT3 phosphorylation reduced STAT3-regulated genes including Cyclin D1, D2, D3, and survivin. However, we could not detect phosphorylation of JAK2 in this model (data not shown). These findings suggest that decreased expression of Cyclin D1, D2, D3, and survivin by carnosol treatment is connected to carnosol-induced apoptosis.
Taken together, we demonstrated that carnosol treatment-induced accumulation of p53, inactivation of STAT3-mediated signal, generation of ROS, and caspase cascade cause apoptosis in human melanoma G361 cells. Moreover, we described our proposed action mechanism of carnosol in Fig. 7. Our findings would provide a molecular basis of carnosol-induced apoptosis in human melanoma cells, and might contribute to development of anti-melanoma drugs using carnosol.

Schematic diagram of possible carnosol-induced apoptosis pathway in human melanoma G361 cells
Availability of data and materials
Not applicable.
References
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64:9–29
Ross MI, Gershenwald JE (2011) Evidence-based treatment of early-stage melanoma. J Surg Oncol 104:341–353
Luke JJ, Flaherty KT, Ribas A, Long GV (2017) Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 14:463–482
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, Baboud J (2015) Genomic classification of cutaneous melanoma. Cell 161:1681–1696
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L (2012) A landscape of driver mutations in melanoma. Cell 150:251–263
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Fischer U, Janicke RU, Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 10:76–100
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344
Nakamura H, Nakamura K, Yodoi J (1997) Redox regulation of cellular activation. Annu Rev Immunol 15:351–369
Fan L, Yin S, Zhang E, Hu H (2018) Role of p62 in the regulation of cell death induction. Apoptosis 23:187–193
Jung WW (2014) Protective effect of apigenin against oxidative stress-induced damage in osteoblastic cells. Int J Mol Med 33:1327–1334
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12:931–947
Fathallah-Shaykh HM (2005) Genomic discovery reveals a molecular system for resistance to oxidative and endoplasmic reticulum stress in cultured glioma. Arch Neurol 62:233–236
Jang HJ, Hwang S, Cho KY, Kim DK, Chay KO, Kim JK (2008) Taxol induces oxidative neuronal cell death by enhancing the activity of NADPH oxidase in mouse cortical cultures. Neurosci Lett 443:17–22
Previati M, Lanzoni I, Corbacella E, Magosso S, Guaran V, Martini A, Capitani S (2006) Cisplatin-induced apoptosis in human promyelocytic leukemia cells. Int J Mol Med 18:511–516
Perez-Cruz I, Carcamo JM, Golde DW (2007) Caspase-8 dependent TRAIL-induced apoptosis in cancer cell lines is inhibited by vitamin C and catalase. Apoptosis 12:225–234
Darnell JE Jr (1997) STATs and gene regulation. Science 277:1630–1635
Yu H, Lee H, Herrmann A, Buettner R, Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14:736–746
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19:2474–2488
Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 118:3367–3377
Devarajan E, Huang S (2009) STAT3 as a central regulator of tumor metastases. Curr Mol Med 9:626–633
Bai E, Yang L, Xiang Y, Hu W, Li C, Lin J, Dai X, Liang G, Jin R, Zhao C (2018) L61H46 shows potent efficacy against human pancreatic cancer through inhibiting STAT3 pathway. Cancer Manag Res 10:565–581
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, Liu W, Zhang Q, Yang Y (2018) Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis 9:422
Chen H, Pan J, Zhang L, Chen L, Qi H, Zhong M, Shi X, Du J, Li Q (2018) Downregulation of estrogen-related receptor alpha inhibits human cutaneous squamous cell carcinoma cell proliferation and migration by regulating EMT via fibronectin and STAT3 signaling pathways. Eur J Pharmacol 825:133–142
Andrade JM, Faustino C, Garcia C, Ladeiras D, Reis CP, Rijo P (2018) Rosmarinus officinalis L.: an update review of its phytochemistry and biological activity. Future Sci OA 4:FSO283
Dorrie J, Sapala K, Zunino SJ (2001) Carnosol-induced apoptosis and downregulation of Bcl-2 in B-lineage leukemia cells. Cancer Lett 170:33–39
Aruoma OI, Spencer JP, Rossi R, Aeschbach R, Khan A, Mahmood N, Munoz A, Murcia A, Butler J, Halliwell B (1996) An evaluation of the antioxidant and antiviral action of extracts of rosemary and Provencal herbs. Food Chem Toxicol 34:449–456
Bauer J, Kuehnl S, Rollinger JM, Scherer O, Northoff H, Stuppner H, Werz O, Koeberle A (2012) Carnosol and carnosic acids from Salvia officinalis inhibit microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther 342:169–176
Chae IG, Kim DH, Kundu J, Jeong CH, Kundu JK, Chun KS (2014) Generation of ROS by CAY10598 leads to inactivation of STAT3 signaling and induction of apoptosis in human colon cancer HCT116 cells. Free Radic Res 48:1311–1321
Ola MS, Nawaz M, Ahsan H (2011) Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 351:41–58
Yuan ZL, Guan YJ, Wang L, Wei W, Kane AB, Chin YE (2004) Central role of the threonine residue within the p + 1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol Cell Biol 24:9390–9400
Park KW, Kundu J, Chae IG, Kim DH, Yu MH, Kundu JK, Chun KS (2014) Carnosol induces apoptosis through generation of ROS and inactivation of STAT3 signaling in human colon cancer HCT116 cells. Int J Oncol 44:1309–1315
Giacomelli C, Natali L, Trincavelli ML, Daniele S, Bertoli A, Flamini G, Braca A, Martini C (2016) New insights into the anticancer activity of carnosol: p53 reactivation in the U87MG human glioblastoma cell line. Int J Biochem Cell Biol 74:95–108
Al Dhaheri Y, Attoub S, Ramadan G, Arafat K, Bajbouj K, Karuvantevida N, AbuQamar S, Eid A, Iratni R (2014) Carnosol induces ROS-mediated beclin1-independent autophagy and apoptosis in triple negative breast cancer. PLoS ONE 9:e109630
Ishida Y, Yamasaki M, Yukizaki C, Nishiyama K, Tsubouchi H, Okayama A, Kataoka H (2014) Carnosol, rosemary ingredient, induces apoptosis in adult T-cell leukemia/lymphoma cells via glutathione depletion: proteomic approach using fluorescent two-dimensional differential gel electrophoresis. Hum Cell 27:68–77
McIlwain DR, Berger T, Mak TW (2013) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5:a008656
Lopez J, Tait SW (2015) Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer 112:957–962
Karpel-Massler G, Ishida CT, Bianchetti E, Shu C, Perez-Lorenzo R, Horst B, Banu M, Roth KA, Bruce JN, Canoll P, Altieri DC, Siegelin MD (2017) Inhibition of mitochondrial matrix chaperones and antiapoptotic Bcl-2 family proteins empower antitumor therapeutic responses. Cancer Res 77:3513–3526
Mukherjee N, Almeida A, Partyka KA, Lu Y, Schwan JV, Lambert K, Rogers M, Robinson WA, Robinson SE, Applegate AJ, Amato CM, Luo Y, Fujita M, Norris DA, Shellman YG (2016) Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 7:84594–84607
Kim DH, Kundu JK, Surh YJ (2011) Redox modulation of p53: mechanisms and functional significance. Mol Carcinog 50:222–234
Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, Turner DC, Essaka DC, Stewart CF, Sosman JA, Kelley MC, Ecsedy JA, Johnston JN, Richmond A (2015) Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res 75:181–193
Wu CE, Esfandiari A, Ho YH, Wang N, Mahdi AK, Aptullahoglu E, Lovat P, Lunec J (2018) Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br J Cancer 118:495–508
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477
Newman DJ, Cragg GM (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75:311–335
Sullivan LB, Chandel NS (2014) Mitochondrial reactive oxygen species and cancer. Cancer Metab 2:17
Hatiboglu MA, Kocyigit A, Guler EM, Akdur K, Nalli A, Karatas E, Tuzgen S (2018) Thymoquinone induces apoptosis in B16-F10 melanoma cell through inhibition of p-STAT3 and inhibits tumor growth in a murine intracerebral melanoma model. World Neurosurg 114:e182–e190
Boudny V, Adamkova L, Souckova K, Lauerova L, Krejci E, Fait V (2008) Defective IFNalpha/gamma-induced STAT3 protein activation in human malignant melanoma cells. Mol Med Rep 1:909–915
Li T, Fu X, Tse AK, Guo H, Lee KW, Liu B, Su T, Wang X, Yu Z (2017) Inhibiting STAT3 signaling is involved in the anti-melanoma effects of a herbal formula comprising Sophorae Flos and Lonicerae Japonicae Flos. Sci Rep 7:3097
Slattery ML, Lundgreen A, Kadlubar SA, Bondurant KL, Wolff RK (2013) JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol Carcinog 52:155–166
Laird AD, Li G, Moss KG, Blake RA, Broome MA, Cherrington JM, Mendel DB (2003) Src family kinase activity is required for signal tranducer and activator of transcription 3 and focal adhesion kinase phosphorylation and vascular endothelial growth factor signaling in vivo and for anchorage-dependent and -independent growth of human tumor cells. Mol Cancer Ther 2:461–469
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1A02050495, J.-S. Choi) and by the Ministry of Science, ICT and Future Planning (2017R1A2B4009831, K.- S. Chun).
Author information
Authors and Affiliations
Contributions
SMC and DHK performed experiments and wrote the paper. KSC and JSC edited, revised the manuscript and supervised the work. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Choi, S.M., Kim, DH., Chun, KS. et al. Carnosol induces apoptotic cell death through ROS-dependent inactivation of STAT3 in human melanoma G361 cells. Appl Biol Chem 62, 55 (2019). https://doi.org/10.1186/s13765-019-0463-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-019-0463-z