
Abstract
Background
The association between epigenetic modification of the genes involved in the vitamin D metabolic pathway and vitamin D metabolites’ status has been elucidated incompletely. This study aims to review the studies on the mentioned association and create a brighter view of this topic.
Methods
A systematic literature search was conducted in Medline database (PubMed), Scopus, and Web of Science up to the end of November 2020. Original articles which reported the effect of epigenetic alteration—methylation level or its changes—of genes involved in vitamin D regulation on the vitamin D metabolites serum level or its changes were included. The National Institutes of Health (NIH) checklist was used to assess the quality of included articles.
Results
Among 2566 records, nine reports were included in the systematic review according to the inclusion and exclusion criteria. Studies discussed the contribution of methylation status of members of the cytochrome P450 family (CYP2R1, CYP27B1, CYP24A1), and Vitamin D Receptor (VDR) genes to vitamin D level variance. CYP2R1 methylation status could regulate the contributing factors affecting the vitamin D serum level and predict response to vitamin D supplementation. Studies revealed that impaired methylation of CYP24A1 occurs in response to an increase in serum level of 25-hydroxyvitamin D (25(OH)D). It is reported that the association between methylation levels of CYP2R1, CYP24A1, and VDR genes and 25(OH)D level is not affected by the methyl-donors bioavailability.
Conclusions
The epigenetic modification of the vitamin D-related genes could explain the vitamin D levels variation among populations. Large-scale clinical trials in various ethnicities are suggested to find the effect of epigenetics on vitamin D response variation.
Registration
The systematic review protocol was registered on PROSPERO (registration number: CRD42022306327).
Similar content being viewed by others

Background
Vitamin D is a steroid hormone with crucial roles in calcium hemostasis and different extra-skeletal pathways. In humans, vitamin D3 production initiates from 7-dehydrocholesterol (7-DHC) in the skin. The concentration of 7DHC depends on the activity of the 7 Dehydrocholesterol Reductase (DHCR7) enzyme, which converts 7DHC into cholesterol [1, 2]. Afterward, vitamin D requires two hydroxylation stages for turning to the active hormonal format (1,25(OH)2D). 25-hydroxyvitamin D (25(OH)D) is the most detectable vitamin D metabolite in humans and is used for determining the status of vitamin D in individuals. Several enzymes are known to be involved in 25-hydroxylation, including CYP2R1, CYP27A1, and CYP3A4—members of the cytochrome P450 family [3, 4]. Despite the variation of enzymes in 25-hydroxylation, CYP27B1 is recognized as the only 1α-hydroxylase in humans [5]. CYP24A1 is the catabolic enzyme that regulates the metabolic pathway and prevents toxic amounts of vitamin D metabolites [6].
More than 11,000 genes were known as the target of 1,25(OH)2D [7]. The complex of vitamin D receptor (VDR), 1,25(OH)2D, and retinoid X receptor (RXR) interact with gene response elements, and after recruitment of co-regulatory factors [8], modulation of gene expression would be achieved [9]. Enrolled co-regulatory factors and epigenetic modifiers (methyltransferases, histone acetyltransferases, and factors with histone acetylase activity) lead to varied responses of a specific gene to the 1,25(OH)2D in different cells and tissues.
Epigenomic studies evaluated the effects of chromatin-modifying and remodeling enzymes which act through interpretation, addition, or removal of post-translational DNA methylation or histone modification in the absence of genomic alterations [10,11,12]. It is shown that methylation of cytosine residues (the cytosine that is 5’ to guanine) of CpG islands (clusters of CpGs) located at the promoter region results in gene silencing. Regulation of chromatin accessibility to transcription factors via modification of histone protein tails (including methylation and acetylation) is another part of epigenetic alterations [13].
Epigenetics and vitamin D status is a developing field of research. Although the epigenetic effect of vitamin D on the transcription of target genes has been discussed, the effect of epigenetic modification on vitamin D level and its bioavailability has still been investigated incompletely [14,15,16,17,18,19,20]. This systematic review was conducted to summarize the literature that has assessed the association between epigenetic modification of genes involved in the vitamin D metabolic pathway and the status of vitamin D metabolites.
Methods
Search strategy
The current systematic review study was conducted according to the PRISMA 2020 (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement [21]. The protocol was registered on PROSPERO (ID: CRD42022306327). A systematic literature search was conducted in the Medline database (PubMed), Scopus, and Web of Science until the end of November 2020. The following search terms were used: ((“epigenetic” OR “epigenomic” OR “epigenomics” OR “methylation” OR “DNA methylation” OR “acetylation” OR “DNA acetylation”) AND ((“vitamin D” OR “25(OH)D” OR “25-hydroxyvitamin D” OR “hydroxycholecalciferols” OR “hypovitaminosis D” OR “vitamin D deficiency” OR “1,25(OH)2D” OR “cholecalc*” OR “serum vitamin D” OR “25-hydroxyvitamin D3” OR “Calcitriol”)). The reference lists of the included studies were checked to find undetected relevant studies.
Inclusion and exclusion criteria
Studies that assessed the effect of epigenetic modifications—methylation level or its changes—of genes involved in the vitamin D metabolic pathway on vitamin D metabolites status—serum level or its changes—and were reported in the English language were selected. Original research studies—with any design—that reported the mentioned association in humans, without any restriction of ethnicity, gender, race, and year of publication, were included. Duplicate publications and studies without enough data and information were excluded. Two expert reviewers independently assessed the title and abstract of studies to evaluate their inclusion eligibility. In case of disagreement between reviewers, the third reviewer (principal investigator) made the decision. Afterward, full-text of the articles were screened based on the inclusion and exclusion criteria, and any discrepancy between the reviewers was resolved by the principal investigator.
Data extraction and quality assessment
Following the full-text assessment of reports, reviewers extracted the following data from the included studies; first author name, year of publication, type of study, location of the studied population and their ethnicity, any specific characteristics of the studied population, the association that is studied, total sample size, evaluated gene, CpG ID, chromosome number and the CpG position on the chromosome, location type of the CpG site, association statistics (R2, r, and β), false discovery rate (FDR), and p-value. A third reviewer assessed all the extracted information. Quality of the included studies was assessed by the National Institutes of Health (NIH) study quality assessment tool [22]. Two separate reviewers scored each study based on the NIH tool. In cases of disagreement, the third reviewer’s opinion was sought.
Results
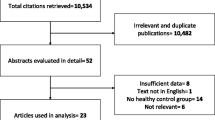
The flow diagram of the study selection process is shown in Fig. 1. The initial search identified 2566 records, and 1865 of them remained after excluding duplicates. After screening the title and abstracts, 128 reports remained for further assessment. The full texts of the reports were reviewed carefully, and finally, nine research studies were included in the systematic review. Characteristics of included studies are shown in Table 1. Quality assessment of the included studies is reported in Tables 2, 3 and 4.

Flow diagram of the study selection process (PRISMA 2020)
A cohort study on 80 elderly patients assessed the association between 25(OH)D status and methylation level of vitamin D metabolic genes, including CYP24A1, CYP27B1, CYP2R1, and VDR (peripheral blood cells were the source for DNA methylation analysis). The average methylation level among examined CpG sites was low. The methylation level of CYP2R1 and CYP24A1 were positively related together (R2: 0.17, p-value < 0.001). Bivariate analysis showed a weak negative correlation of 25(OH)D levels with methylation of CYP2R1 (R2: 0.05, p-value = 0.04) and CYP24A1 (R2: 0.06, p-value = 0.02), and a positive correlation with VDR (R2: 0.12, p-value = 0.001). There was no significant association between the 25(OH)D level and the methylation status of CYP27B1. Also, they evaluated the influence of methyl donor availability on the methylation status of mentioned genes. While the relationship of 25(OH)D levels and methylation of the genes in the presence of additional variables—i.e., serum folate, B12, and plasma homocysteine—were not altered, the potential role of serum folate and B12 in methylation regulation of these genes was discovered. Serum B12 level showed a positive correlation with the methylation status of CYP27B1, and serum folate level was related to VDR methylation. The adjusted model with vitamin D and calcium intake, age, sex, body mass index (BMI), cumulative irradiance, alcohol intake, and cigarette smoking history showed a better predictive value for 25(OH)D level (R2; 0.54, p-value < 0.001) in comparison to modeling without the inclusion of metabolic vitamin D genes methylation status (R2: 0.46, p-value < 0.001). In the mentioned adjusted model, CYP2R1 gene methylation status was a significant independent negative (β: -0.2, p-value = 0.03) predictor of 25(OH)D level, and VDR gene methylation was an independent positive predictive factor (β: 0.26, p-value = 0.005). Although there was no significant predictive value for CYP24A1 methylation individually in the described model, a significant predictive value of the interaction of CYP24A1 gene methylation and vitamin D intake was found (p interaction = 0.04). This suggests that increased methylation of CYP24A1 is not the direct cause of low serum vitamin D levels but is the response to increased vitamin D availability (increased intake). The significant interaction between CYP2R1 methylation status and the major determinants of serum vitamin D levels, including vitamin D intake (p interaction < 0.001), calcium intake (p interaction = 0.003), and cumulative irradiance (p interaction = 0.009), suggests that CYP2R1 methylation status regulates the effect of those contributing factors on the vitamin D level modulation. VDR gene methylation is positively associated with 25(OH)D levels in the mentioned model. This association remained when interaction with vitamin D intake was considered (p interaction; 0.04) or when corrected in the methyl donor model (R2; 0.21, p-value = 0.006). These findings suggest a negative feedback loop for maintaining vitamin D homeostasis [23].
One randomized clinical trial was conducted among 64 overweight/obese African Americans. The role of baseline DNA methylation (extracted from whole blood buffy coat) in the serum 25(OH)D level response to vitamin D supplementation was investigated. Due to the expected serum vitamin D level and actual post-test level, participants were categorized into high-response and low-response groups. Expected levels were estimated by the intervention dose, gender, age, body mass index, baseline serum vitamin D level, and seasonal variation. Twenty CpG sites located in the CYP family genes and VDR showed statistically significant associations with serum 25(OH)D response (Table 1). Also, the methylation level of cg07873128 located in the body region of the oxysterol binding protein like 5 (OSBPL5) gene was negatively correlated with response to vitamin D supplementation. They suggested that hypermethylation of the mentioned CpG site could cause cholesterol and calcium regulation impairment, resulting in decreased response to vitamin D supplementation. However, changes in the methylation level of cg07873128 (OSBPL5) were not associated with changes in serum 25(OH)D level (p-value = 0.6). Also, it is mentioned that only methylation changes of cg06368932 (CYP24A1) were positively associated with the changes in 25(OH)D levels [24].
One case-control study among the Chinese population (healthy participants and patients with pulmonary tuberculosis) sequenced 310 CpG sites in the promoter region of 5 candidate genes (CYP24A1, CYP27B1, CYP27A1, CYP2R1, and VDR). CYP27A1_3 was the only region that was significantly associated with 1,25(OH)2D level (r = 0.13, p-value = 0.045). They evaluated the correlation of methylation levels of these genes and 25(OH)D serum levels by four different models (Model 1: Cumulative methylation level was calculated by adding the frequency of all CpG sites in each region/ Model 2: Cumulative methylation level was calculated by adding the frequency of statistically significant CpG sites in each region/ Model 3: Inclusion of only hypermethylated CpG sites in the cases and exclusion of CpG sites with an inverse relationship between cases and controls/ Model 4: Inclusion of only statistically significant CpG sites after Bonferroni correction). Cumulative methylation levels of CYP24A1, CYP27B1, CYP27A1, or VDR genes were significantly associated with serum 25(OH)D levels in all four models. Although, CYP2R1 was significantly positively associated only in the third model [25].
One clinical trial among non-Hispanic white postmenopausal women evaluated whether methylation statuses of CYP genes were associated with 25(OH)D serum levels in response to vitamin D supplementation. Calcium and vitamin D (1100 IU/day) intervention on 446 subjects for at least 12 months was conducted. Of them, 18 responders (the highest 12-month increase in serum 25(OH)D) and 18 non-responders (the lowest 12-month increase in serum 25(OH)D) were selected. Methylation levels of the promoter regions of both CYP2R1 and CYP24A1 genes at baseline were significantly higher in non-responders compared to responders. However, no significant differences were found in methylation levels of CYP27B1 and CYP27A1 between responders and non-responders at baseline. It was found that only the CYP24A1 gene experienced methylation reduction in both responders and non-responders after a 12-month vitamin D supplementation. It should be added that time by treatment was also significant for only CYP24A1. A validation study on 145 participants was also conducted to confirm these results. The methylation level of eight CpG sites (among the 14 examined sites in 145 subjects) in the CYP2R1 gene was significantly negatively associated with the serum 25(OH)D increment in a 12-month period. The methylation average of 14 evaluated CpG sites of CYP2R1 was also negatively associated with the increment in 25(OH)D level after 12-month supplementation. Baseline DNA methylation of two CpG sites of CYP24A1 (among the 16 examined sites in 117 subjects) was also negatively associated with the vitamin D supplementation response. (Table 1) The contribution of CYP2R1 and CYP24A1 (10 statistically significant CpG sites together) to the vitamin D response variation was reported to be moderate (R2 = 6.4%) [26].
In the validation study, methylation reduction in each 14 CpG site of CYP2R1 at the 12-month visit was statistically significant compared to the baseline. It was also mentioned that the decrease in average methylation of the CYP2R1 CpG sites was significant (p-value = 0.001). Analysis of the CYP24A1 CpG sites revealed different reactions of the CpG sites to the vitamin D supplementation [26].
DNA extracted from peripheral blood lymphocytes of 384 individuals showed a weak association between CYP27B1 site 15–17 and 25(OH)D levels. Also, there was a tendency for a higher CYP27B1 methylation ratio with a lower 25(OH)D serum level [27].
A sub-cohort study on 1270 non-Hispanic white women examined the association of methylation status of 198 CpGs in or near vitamin D-related genes—i.e., VDR, RXRA (retinoid-x receptor-alpha), GC, CYP24A1, CYP27B1, CYP2R1, and DHCR7/NADSYN1—with vitamin D levels (DNA was extracted from whole blood samples). Among them, 23 CpG sites showed statistically significant associations with 25(OH)D levels. Unexpectedly, it was noted that most of the significant associated CpG sites were located within gene bodies. CpG sites at different parts of the genes showed dissimilar associations with 25(OH)D levels [28].
A 1416 mother/newborn pairs study investigated the association between mid-pregnancy maternal 25(OH)D levels and cord blood DNA methylation. Findings showed a weak association between the methylation status of CpG sites among CYP24A1, CYP27A1, CYP27B1, and CYP2R1 genes and vitamin D levels [29]. (Table 1)
A study on 86 twin pairs and their mothers investigated the relationship between maternal 25(OH)D serum level (at 28-week gestation), placental methylation of CYP24A1, and neonatal cord blood 25(OH)D concentration. Findings showed no correlation between placental CYP24A1 gene methylation level and maternal or neonatal 25(OH)D serum level. Also, it was mentioned that there was no association between CYP24A1 methylation changes and changes in maternal or neonatal 25(OH)D concentration[30].
One study investigated the relationship between maternal 25(OH)D status (measured at 34 weeks gestation) and the methylation status of the RXRA gene retrieved from the umbilical cord. Findings showed that maternal free vitamin D index (the ratio of serum 25(OH)D to vitamin D binding protein concentration) had a statistically significant negative association with RXRA CpG4/5 methylation percentage (β = -3.29 SD/unit, p-value = 0.03). However, the results showed that 25(OH)D or vitamin D binding protein serum level was not a predictive factor for the methylation status of any site at RXRA [31].
Discussion
Vitamin D affects the epigenome on multiple levels and has a substantial role in the epigenetic regulation of genes [19, 32]. On the other hand, epigenetic mechanisms could regulate vitamin D metabolites [32, 33]. Therefore, it could be suggested that pathologies that demonstrated any association with epigenetic effects of vitamin D metabolites could be attributed to complex processes involving epigenetic modulation of genes engaged in vitamin D metabolism [34]. The present study illustrated that the epigenetic modulation of vitamin D-related genes could be the reason for vitamin D level variance among the population. In addition to its role in normal variation, it can be a reason for vitamin D deficiency. Several studies described the association between the methylation status of those genes—CpG islands located at the promoters and within the gene locus [32]—and vitamin D levels. According to the studies, the methylation status of CYP2R1, CYP27B1, CYP24A1, and VDR genes is responsible for nearly 18% of the vitamin D level variance [23].
CYP2R1 methylation status can regulate the effect of the contributing factors (e.g., calcium and vitamin D intake, cumulative radiance) on vitamin D serum levels [23]. A study on African-American adolescents showed that the CpG site at the CYP2R1 gene showed lower methylation in participants with sufficient levels of vitamin D in comparison with the participants with vitamin D deficiency [35]. Also, a significant reduction in the mean methylation level of CYP2R1 CpG sites following a period of vitamin D supplementation was reported [26]. Most of the studies confirmed that the methylation status of CYP24A1 is regulated by the vitamin D level. Studies suggested that impaired methylation of CYP24A1 occurred in response to increased 25(OH)D serum levels [23, 26]; however, another study suggested that impaired methylation of CYP24A1 was a direct cause of 25(OH)D deficiency [35]. A clinical study showed that the increase in the 1,25(OH)2D level could increase the activity of CYP24A1 over a few hours [14]. Another study showed that in response to 1,25(OH)2D, a 6-8-fold increase in VDR and RXR at the promoter of CYP24A1 and a 3-fold increase in H4 acetylation of coding and promoter regions were observed in the mice [18]. Therefore, these all together result in the initiation of CYP24A1 transcription in response to increased vitamin D availability.
A weak association between higher methylation of the CYP27B1 gene and lower 25(OH)D serum level is shown [27]. This could mean that in case of 25(OH)D availability, CYP27B1 would be more translated. Contrarily, a study on individuals aged 65 years or more found no direct correlation between plasma 25(OH)D and the methylation status of CYP27B1 [23]. In one case-control study on 122 patients with pulmonary tuberculosis and 118 healthy controls, the methylation of a fragment (cumulative methylation of CpG sites at the specific region of the gene) at the CYP27A1 showed a positive correlation with 1,25-dihydroxyvitamin D level [25] which could suggest the inhibitory role of 1,25(OH)2D on 25-hydroxylation.
Vitamin D binding protein (DBP) (encoded by the GC gene) binds to approximately 85% of circulating vitamin D metabolites [8]. In one of the reviewed studies, the methylation status of 3 CpG sites at the GC gene showed statistically significant positive beta coefficients for serum 25(OH)D level [28]. Although variation in DBP level can result in variation of total 25(OH)D level, free 25(OH)D serum level does not change. Therefore, lower DBP cannot cause vitamin D deficiency symptoms even in undetectable amounts of 25(OH)D (supporting the free hormone hypothesis) [8].
Studies showed the correlation between RXRA and VDR genes’ methylation status and serum vitamin D levels. The epigenome-wide association study revealed 8 CpG sites at the RXRA gene that were significantly associated with serum 25(OH)D level [28]. A cross-sectional study described the positive correlation between VDR methylation status and plasma 25(OH)D level independently and when corrected for the effect of vitamin D intake or methyl donor serum bioavailability. Results showed the negative feedback mechanism between 25(OH)D level as a ligand and VDR as a receptor [23].
Despite the fact that some genes do not directly influence the vitamin D metabolic pathway, they can affect the serum level of vitamin D metabolites and therefore lead to vitamin D deficiency symptoms. A study on overweight/obese African Americans revealed a negative correlation between the baseline methylation level of a CpG site located in the body of the OSBPL5 gene and response to vitamin D supplementation [24]. It is considered that hypermethylation of the CpG site located in the body of the gene would downregulate the transcription of the OSBPL5 gene [24, 36]. However, there are inconsistent findings related to the association between methylation of CpG sites located in the body of the genes and transcription [28, 37].
NADSYN1 (nicotinamide adenine dinucleotide synthetase 1) gene, which is located near DHCR7, has no known direct biological effect on vitamin D metabolism [38]. However, a genome-wide association study (GWAS) found a strong association between SNPs in the DHCR7/NADSYN locus and serum level of vitamin D metabolites [39]. One reviewed study revealed 7 CpG sites at the NADSYN1 gene that showed statistically significant associations with 25(OH)D levels. Also, they discovered that methylation levels of 4 CpG sites at the DHCR7 gene showed statistically significant associations with 25(OH)D levels, and three of them showed positive beta coefficients [28]. (Table 1) Another genome-wide methylation study showed two CpG sites at the DHCR7 gene with significantly different methylation levels between vitamin D deficient participants and participants with desirable vitamin D levels. However, CpG sites showed different methylation patterns [35].
The change in 25(OH)D serum level after vitamin D supplementation varies among individuals. Some factors, including body weight, age, sex, type of vitamin D supplementation (D2 or D3), calcium intake, baseline serum 25(OH)D, and physical activity, might result in this variation. These factors can lead to up to 50% of the changes [40, 41]. A recent review article discussed the use of genetic risk scores (GRS)—which describes combined variants of vitamin D lowering alleles—in assessing the responsiveness to vitamin D supplementation and also recommending the optimal dosage according to the genetic score [42]. Besides genetic variation [43], epigenetics could be another reason for the mentioned differences. One of the reviewed studies suggested that subjects with high methylation rates of the CYP2R1 and CYP24A1 genes may need higher dosages of vitamin D supplementation to achieve optimal serum levels [26]. It was reported that the contribution of the methylation status of CYP24A1 and CYP2R1 could explain 6.4% of vitamin D response variation [26]. They discussed that the absence of an association between CYP27A1 and vitamin D response could be due to its lesser activity in 25-hydroxylation compared to CYP2R1[26]. Another study reported twenty CpG sites located in the VDR and CYP family genes that showed statistically significant association with serum 25(OH)D response [24].
Generally, DNA methylation can be sensitive to cigarette smoking, alcohol usage, BMI, and the availability of methyl donors like folate and B12 [28]. It is reported that the inclusion of serum B12, folate, and plasma homocysteine in the multivariable regression analysis would not alter the correlation of CYP2R1, CYP24A1, and VDR methylation status with plasma 25(OH)D level. However, a potential role for serum B12 and folate in the methylation regulation of CYP27B1 and VDR was reported, respectively [23]. Another study showed that high folate intake could effectively lead to reduced expression of CYP24A1 in the ascending colon [44].
Recently, a new concept called personalized response to vitamin D supplementation has been suggested [45]. The results of clinical trials like VitDbol and VitDmet about the effects of vitamin D supplementation on biochemical vitamin D-sensitive parameters, accessibility change of chromatin regions, and the response of transcriptome-wide vitamin D target genes have been published lately [46,47,48,49,50,51,52]. These trials revealed the difference in humans’ molecular response to vitamin D supplementation [45]. According to the fold change of 25(OH)D serum level and fold change of parameters or gene expression, three groups were proposed; low responder, mid responder, and high responder. Individuals as low responders are more susceptible to vitamin D deficiency disorder and should take higher daily doses of vitamin D than high responders to obtain the optimal hormonal activity of vitamin D and maximal disease protective effect [11, 45]. So, personalized vitamin D supplementation based on the personalized optimum vitamin D level can be considered instead of a general recommendation. It is mentioned that this index is not related to the geographic location of individuals and is independent of the serum 25(OH)D levels [45]. Furthermore, a review study on Mendelian Randomization studies suggested that the benefits of increased vitamin D levels could be attributed to the correction of clinical deficiency [42]. Therefore, it can be concluded that the evaluation of vitamin D serum levels based on general threshold metrics could not be a good indicator of the optimal vitamin D level for the desired effects.
It is suggested that genetic variation can only predict 20% of the variation in vitamin D response indices, while the remaining could be due to epigenetic variations [45]. We suggest that this variation can be due to the differences in the functional status of intracellular 25(OH)D transformers or regulation of VDR, RXRA, and the co-regulators. It can be hypothesized that variations in the amount or function of the megalin (which transfers DBP-bound format to cells) and HSP70 (the potential intracellular transporter) could be possible reasons for vitamin D response variation [8].
The use of PBCs as the source of DNA methylation analysis is a major limitation of the reviewed articles. Although the expression of the mentioned enzymes in immune cells is demonstrated, the regulation of vitamin D status majorly occurs in liver and kidney tissues. Furthermore, the presence of CYP27B1 regulation via 1,25(OH)2D/VDR in renal cells and the absence of it in macrophages—due to genetic and epigenetic mechanisms that result in tissue-specific actions of 1,25(OH)2D—points to this error in the generalizability of the results [8].
According to the discrepancy in the method of studies, different populations, and methods of vitamin D level and methylation status assessment, reviewed studies showed diversity in the correlated CpG sites of each gene and their association with vitamin D level. We suggest large-scale studies in different ethnicities to find the effect of epigenetic modulation of vitamin D-related genes on vitamin D response variation and serum level. It can determine the usability of the epigenetic profile for the recommendation of the appropriate vitamin D supplementation dosage. Afterward, we could analyze each person’s metabolic response to the vitamin D serum level to determine the goal serum level.
Conclusions
Epigenetic modification plays a crucial role in regulating vitamin D levels and vitamin D response variation. Epigenetic changes could be considered to recommend the appropriate dosage of vitamin D supplementation. Large-scale research studies among various ethnicities are recommended to report more precisely.
Data Availability
Not applicable.
Abbreviations
- NIH:
-
National Institutes of Health
- 25(OH)D:
-
25-Hydroxyvitamin D
- 7-DHC:
-
7-Dehydrocholesterol
- NADSYN1:
-
Nicotinamide adenine dinucleotide synthetase 1
- DHCR7:
-
7 Dehydrocholesterol Reductase
- VDR:
-
Vitamin D receptor
- 1,25(OH)2D:
-
1,25-Dihydroxyvitamin D
- RXR:
-
Retinoid X receptor
- FDR:
-
False discovery rate
- OSBPL5:
-
Oxysterol binding protein like 5
- DBP:
-
Vitamin D binding protein
- BMI:
-
Body mass index
- CYP2R1:
-
Cytochrome P450 Family 2 Subfamily R Member 1
- CYP27A1:
-
Cytochrome P450 Family 27 Subfamily A Member 1
- CYP3A4:
-
Cytochrome P450 Family 3 Subfamily A member 4
- CYP27B1:
-
Cytochrome P450 Family 27 Subfamily B Member 1
- CYP24A1:
-
Cytochrome P450 Family 24 Subfamily A Member 1
References
Prabhu AV, Luu W, Li D, Sharpe LJ, Brown AJ. DHCR7: a vital enzyme switch between cholesterol and vitamin D production. Prog Lipid Res. 2016;64:138–51.
Mitsche MA, McDonald JG, Hobbs HH, Cohen JC. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife. 2015;4:e07999.
Gupta RP, Hollis BW, Patel SB, Patrick KS, Bell NH. CYP3A4 is a human microsomal vitamin D 25-hydroxylase. J Bone Miner Res. 2004;19(4):680–8.
Roizen JD, Li D, O’Lear L, Javaid MK, Shaw NJ, Ebeling PR, et al. CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest. 2018;128(5):1913–8.
Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, et al. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11(13):1961–70.
Bikle Daniel D, Vitamin D, Metabolism. Mechanism of Action, and clinical applications. Chem Biol. 2014;21(3):319–29.
Saccone D, Asani F, Bornman L. Regulation of the vitamin D receptor gene by environment, genetics and epigenetics. Gene. 2015;561(2):171–80.
Bikle D, Christakos S. New aspects of vitamin D metabolism and action - addressing the skin as source and target. Nat Rev Endocrinol. 2020;16(4):234–52.
Carlberg C, Bendik I, Wyss A, Meier E, Sturzenbecker LJ, Grippo JF, et al. Two nuclear signalling pathways for vitamin D. Nature. 1993;361(6413):657–60.
Henikoff S, Greally JM. Epigenetics, cellular memory and gene regulation. Curr Biol. 2016;26(14):R644–8.
Carlberg C. Nutrigenomics of vitamin D. Nutrients. 2019;11(3):676.
Bahrami A, Sadeghnia HR, Tabatabaeizadeh SA, Bahrami-Taghanaki H, Behboodi N, Esmaeili H, et al. Genetic and epigenetic factors influencing vitamin D status. J Cell Physiol. 2018;233(5):4033–43.
Cross HS. Extrarenal vitamin D hydroxylase expression and activity in normal and malignant cells: modification of expression by epigenetic mechanisms and dietary substances. Nutr Rev. 2007;65(8 Pt 2):108–12.
Abboud M, Rybchyn MS, Rizk R, Fraser DR, Mason RS. Sunlight exposure is just one of the factors which influence vitamin D status. Photochem Photobiol Sci. 2017;16(3):302–13.
Saffery R, Ellis J, Morley R. A convergent model for placental dysfunction encompassing combined sub-optimal one-carbon donor and vitamin D bioavailability. Med Hypotheses. 2009;73(6):1023–8.
Novakovic B, Sibson M, Ng HK, Manuelpillai U, Rakyan V, Down T, et al. Placenta-specific methylation of the vitamin D 24-hydroxylase gene: implications for feedback autoregulation of active vitamin D levels at the fetomaternal interface. J Biol Chem. 2009;284(22):14838–48.
Wimalawansa SJ, Vitamin D, Deficiency. Effects on oxidative stress, Epigenetics, Gene Regulation, and aging. Biology. 2019;8(2).
Meyer MB, Zella LA, Nerenz RD, Pike JW. Characterizing early events associated with the activation of target genes by 1,25-dihydroxyvitamin D3 in mouse kidney and intestine in vivo. J Biol Chem. 2007;282(31):22344–52.
Carlberg C, Velleuer E. Nutrition and epigenetic programming. Curr Opin Clin Nutr Metab Care. 2023;26(3):259–65.
Carlberg C. Vitamin D and its target genes. Nutrients. 2022;14(7):1354.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ (Clinical research ed). 2021;372:n71.
Study Quality Assessment Tools | NHLBI., NIH [Internet]. [cited 2021 Sep 24]. Available from: https://www.nhlbi.nih.gov/health-topics/study-quality-assessment-tools.
Beckett EL, Duesing K, Martin C, Jones P, Furst J, King K, et al. Relationship between methylation status of vitamin D-related genes, vitamin D levels, and methyl-donor biochemistry. J Nutr Intermediary Metabolism. 2016;6:8–15.
Chen L, Dong Y, Chen J, Huang Y, Zhu H. Epigenetics predicts serum 25-Hydroxyvitamin D response to vitamin D3 supplementation in African Americans. Mol Nutr Food Res. 2020;64(1):1900738.
Wang M, Kong W, He B, Li Z, Song H, Shi P, et al. Vitamin D and the promoter methylation of its metabolic pathway genes in association with the risk and prognosis of tuberculosis. Clin Epigenetics. 2018;10(1):118.
Zhou Y, Zhao L-J, Xu X, Ye A, Travers-Gustafson D, Zhou B, et al. DNA methylation levels of CYP2R1 and CYP24A1 predict vitamin D response variation. J Steroid Biochem Mol Biol. 2014;144:207–14.
Wjst M, Heimbeck I, Kutschke D, Pukelsheim K. Epigenetic regulation of vitamin D converting enzymes. J Steroid Biochem Mol Biol. 2010;121(1):80–3.
O’Brien KM, Sandler DP, Xu Z, Kinyamu HK, Taylor JA, Weinberg CR, Vitamin D. DNA methylation, and breast cancer. Breast Cancer Res. 2018;20(1):70.
Suderman M, Stene LC, Bohlin J, Page CM, Holvik K, Parr CL, et al. 25-Hydroxyvitamin D in pregnancy and genome wide cord blood DNA methylation in two pregnancy cohorts (MoBa and ALSPAC). J Steroid Biochem Mol Biol. 2016;159:102–9.
Novakovic B, Galati JC, Chen A, Morley R, Craig JM, Saffery R. Maternal vitamin D predominates over genetic factors in determining neonatal circulating vitamin D concentrations. Am J Clin Nutr. 2012;96(1):188–95.
Harvey NC, Sheppard A, Godfrey KM, McLean C, Garratt E, Ntani G, et al. Childhood bone mineral content is associated with methylation status of the RXRA promoter at birth. J Bone Miner Res. 2014;29(3):600–7.
Fetahu IS, Höbaus J, Kállay E. Vitamin D and the epigenome. Front Physiol. 2014;5(164).
Snegarova V, Naydenova D. Vitamin D: a review of its Effects on Epigenetics and Gene Regulation. Folia Med (Plovdiv). 2020;62(4):662–7.
Karlic H, Varga F. Impact of vitamin D metabolism on clinical epigenetics. Clin Epigenetics. 2011;2(1):55–61.
Zhu H, Wang X, Shi H, Su S, Harshfield GA, Gutin B, et al. A Genome-Wide Methylation Study of Severe Vitamin D Deficiency in African American Adolescents. The Journal of Pediatrics. 2013;162(5):1004-9.e1.
Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–90.
Lee K, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4.
Kuan V, Martineau AR, Griffiths CJ, Hyppönen E, Walton R. DHCR7 mutations linked to higher vitamin D status allowed early human migration to Northern latitudes. BMC Evol Biol. 2013;13(1):144.
Ahn J, Yu K, Stolzenberg-Solomon R, Simon KC, McCullough ML, Gallicchio L, et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet. 2010;19(13):2739–45.
Mazahery H, von Hurst PR. Factors affecting 25-Hydroxyvitamin D concentration in response to vitamin D supplementation. Nutrients. 2015;7(7):5111–42.
Zittermann A, Ernst JB, Gummert JF, Börgermann J. Vitamin D supplementation, body weight and human serum 25-hydroxyvitamin D response: a systematic review. Eur J Nutr. 2014;53(2):367–74.
Hyppönen E, Vimaleswaran KS, Zhou A. Genetic determinants of 25-Hydroxyvitamin D concentrations and their relevance to Public Health. Nutrients. 2022;14(20):4408.
Bahrami A, Mehramiz M, Ghayour-Mobarhan M, Bahrami-Taghanaki H, Sadeghi Ardekani K, Tayefi M, et al. A genetic variant in the cytochrome P450 family 2 subfamily R member 1 determines response to vitamin D supplementation. Clin Nutr. 2019;38(2):676–81.
Cross HS, Lipkin M, Kállay E. Nutrients regulate the Colonic vitamin D system in mice: relevance for human Colon malignancy. J Nutr. 2006;136(3):561–4.
Carlberg C, Haq A. The concept of the personal vitamin D response index. J Steroid Biochem Mol Biol. 2018;175:12–7.
Neme A, Seuter S, Malinen M, Nurmi T, Tuomainen T-P, Virtanen JK, et al. In vivo transcriptome changes of human white blood cells in response to vitamin D. J Steroid Biochem Mol Biol. 2019;188:71–6.
Carlberg C, Seuter S, de Mello VDF, Schwab U, Voutilainen S, Pulkki K, et al. Primary vitamin D target genes allow a categorization of possible benefits of vitamin D3 supplementation. PLoS ONE. 2013;8(7):e71042.
Wilfinger J, Seuter S, Tuomainen T-P, Virtanen JK, Voutilainen S, Nurmi T, et al. Primary vitamin D receptor target genes as biomarkers for the vitamin D3 status in the hematopoietic system. J Nutr Biochem. 2014;25(8):875–84.
Ryynänen J, Neme A, Tuomainen T-P, Virtanen JK, Voutilainen S, Nurmi T, et al. Changes in vitamin D target gene expression in adipose tissue monitor the vitamin D response of human individuals. Mol Nutr Food Res. 2014;58(10):2036–45.
Saksa N, Neme A, Ryynänen J, Uusitupa M, de Mello VDF, Voutilainen S, et al. Dissecting high from low responders in a vitamin D3 intervention study. J Steroid Biochem Mol Biol. 2015;148:275–82.
Vukić M, Neme A, Seuter S, Saksa N, de Mello VDF, Nurmi T, et al. Relevance of vitamin D receptor target genes for monitoring the vitamin D responsiveness of primary human cells. PLoS ONE. 2015;10(4):e0124339.
Seuter S, Virtanen JK, Nurmi T, Pihlajamäki J, Mursu J, Voutilainen S, et al. Molecular evaluation of vitamin D responsiveness of healthy young adults. J Steroid Biochem Mol Biol. 2017;174:314–21.
Acknowledgements
Not applicable.
Funding
This study was conducted as project number 2400258, supported by Isfahan University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
RK: Contribution to the conception and design of the study, interpretation of data, and revision of the manuscript. AF: Contribution to the design of the study, acquisition and interpretation of data, drafting and revision of the manuscript. SV: Contribution to acquisition and interpretation of data, drafting of the manuscript. MH: Contribution to the conception and design of the study, interpretation of data, and revision of the manuscript. PP: Contribution to acquisition and interpretation of data, drafting of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Research ethics code: IR.MUI.RESEARCH.REC.1400.489.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Forouhari, A., Heidari-Beni, M., Veisi, S. et al. Effect of epigenetics on vitamin D levels: a systematic review until December 2020. Arch Public Health 81, 106 (2023). https://doi.org/10.1186/s13690-023-01122-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13690-023-01122-2