
Abstract
Background
Hearing loss in newborns and children is a public health concern, due to high prevalence and negative effects on their development. Early detection and intervention of childhood hearing loss may mitigate these negative effects. Population-based newborn hearing screening programs have been established worldwide to identify children at risk for congenital hearing loss and to follow children at risk for late onset or progressive hearing loss. This article presents the protocol for a systematic review that aims to review the risk factors associated with permanent hearing loss in children, including congenital, early, or late onset. Risk factors associated with progressive hearing loss will be investigated as a secondary aim.
Methods
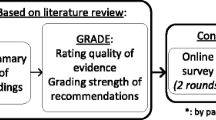
Scientific literature from the following databases will be investigated: MEDLINE, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R), Embase, and CINAHL. The primary outcome is a permanent bilateral or unilateral hearing loss with congenital onset or onset during childhood (birth to 18 years). The secondary outcome is progressive hearing loss. Studies must report data on risk factors associated with permanent hearing loss; risk factors may be present at birth or later and result in immediate or delayed hearing loss. Randomized controlled trials, quasi-experimental studies, nonrandomized comparative and non-comparative studies, and case series will be included. The risk of bias will be assessed using the Qualitative Assessment Tool for Quantitative Studies (McMaster University). If aggregation of data is possible for a subsection of studies, we will pool data using meta-analysis techniques. If aggregation of data is not possible, a qualitative synthesis will be presented. We will assess the quality and strength of the overall body of evidence using the Grading of Recommendations Assessment, Development and Evaluation (GRADE). The systematic review follows the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) recommendations.
Discussion
The resulting information will inform the update of a provincial audiological surveillance protocol for the Ontario Infant Hearing Program and will be applicable to early hearing detection and intervention (EHDI) programs worldwide.
Systematic review registration
We have registered the protocol in the International Prospective Register of Systematic Reviews (PROSPERO), registration number CRD42018104121.
Similar content being viewed by others


Background
Permanent hearing loss affects at least 1 to 2 per 1000 children at birth and has severe consequences for their development [1,2,3]. The negative consequences for language, cognitive, and social-emotional skills are particularly important when diagnosis is delayed for children with hearing loss, which delays access to early intervention programs [4,5,6,7]. There is good evidence that universal newborn hearing screening (UNHS) programs achieve early identification [8, 9]. When UNHS programs are implemented as part of a comprehensive early hearing detection and intervention program (EHDI), children with congenital and early onset hearing loss are diagnosed early and supported to develop language in their first months of life [3, 10].
In addition to screening newborns for hearing loss, there is a need to monitor children who are at risk of developing hearing loss after the neonatal period [11,12,13]. Prior to the population-based screening, it was difficult to identify whether hearing loss occurred congenitally, neonatally, or during early childhood beyond the neonatal period. Studies suggest that the prevalence of permanent hearing loss rises during childhood and that hearing loss occurs sometime after infancy in up to 25–50% of affected children [14,15,16]. Almost half of the children with hearing loss will experience deterioration in hearing during childhood [17, 18].
Children at risk for late onset hearing loss need to be closely monitored, even after passing a screening test, to ensure identification as early as possible and to avoid delays in treatment, unnecessary costs to the healthcare system, and additional parental stress [19, 20]. It is also important to monitor children with early onset hearing loss who are at risk for further deterioration in hearing (progressive hearing loss), so that appropriate intervention may be initiated in a timely manner. Surveillance programs are thus recommended as part of a comprehensive EHDI program but not universally applied [21, 22]. They aim to identify children at risk for late onset or progressive hearing loss and have been implemented in some UNHS or EHDI programs [11, 23, 24]. However, the effectiveness of these programs is not well established [25].
In 2007, the Joint Committee on Infant Hearing (JCIH) updated the list of risk factors associated with permanent congenital, delayed onset, or progressive hearing loss in childhood [21]. Due to the overlap between congenital/neonatal and delayed onset risk factors, the JCIH presented a single list. The JCIH recommends that all infants with these risk factors be closely monitored for hearing loss. The committee proposed a comprehensive diagnostic assessment in the neonatal period following a positive screening result and appropriate surveillance after a negative screening result. The JCIH list is widely adopted in a large number of UNHS programs, sometimes with adaptations. However, this list was published more than 10 years ago. Based on the evolution of care and new scientific knowledge during recent years, updating the risk factors to reflect current clinical practice is warranted [16]. Our systematic review aims to identify the most up-to-date risk factors for hearing loss in children and will include an examination of factors by the onset of hearing loss.
Methods
Objectives and research questions
The overall aim of this systematic review is to synthesize evidence on risk factors related to neonatal, early, and late onset hearing loss as well as progressive hearing loss in children. We will investigate all risk factors that have an immediate or delayed effect on hearing. The resulting information will inform surveillance protocols and will be applicable to EHDI programs worldwide.
The review aims to answer the following primary research question: what risk factors are associated with permanent hearing loss in children? Risk factors related to congenital, early, and late onset hearing loss will be examined.
A secondary objective of this review is to investigate the risk factors associated with progressive hearing loss in children with congenital, early, or late onset hearing loss.
Design
This systematic review protocol has been developed according to the Preferred Reporting Items for Systematic review and Meta-Analysis Protocols (PRISMA-P) (see Additional file 1). We have registered the protocol in the International Prospective Register of Systematic Reviews (PROSPERO), registration number CRD42018104121) [26].
Eligibility criteria
Primary research that meets the following criteria will be included in the systematic review.
Study designs
It is anticipated that literature in this field will consist largely of observational and descriptive studies. However, some risk factors are related to medical conditions or treatments and may be investigated through intervention studies. In an effort to collect the most comprehensive data, the following study designs will be included: randomized controlled trials, quasi-experimental studies, nonrandomized comparative studies (prospective, or retrospective cohort, case-control), nonrandomized studies without comparison group (e.g., prospective or retrospective cohort, cross-sectional), and case series. Consequently, we will exclude from this review case reports/case studies (less than five cases). Since we anticipate a very large number of articles, we have essentially extended the “case study” to include < 5 cases as we judged that these small cohorts would provide little additional information. We will also exclude editorials, viewpoints/opinion, literature reviews, conference abstracts, guidelines, recommendations, consensus papers, qualitative research, and gray literature, such as reports, thesis, and notes.
Studies not directly related to the review questions but that present pertinent information about the relationships between risk factors and hearing loss will also be included, as long as they meet the review inclusion criteria (e.g., population, exposure, outcomes).
Population
All children with permanent bilateral or unilateral hearing loss will be included. For this review, a child is defined as age birth to 18 years. In most healthcare systems, pediatric care targets children from birth to adolescence (defined as less than 19 years), which is concordant with the Medical Subject Heading (MeSH) definition (https://www.ncbi.nlm.nih.gov/mesh/). The age limit is because only the childhood onset of hearing loss is targeted by the research question. We will include articles on children and adults only if results are presented separately for children; we will accept an upper range of the study group of approximately age 21 years (young adults), when the majority of participants are children.
Exposure
Studies must report data on risk factors associated with permanent hearing loss. Examples of specific risk factors are congenital cytomegalovirus (cCMV), toxoplasmosis, admission to a neonatal intensive care unit, ototoxic medications, and a family history of hearing loss [21]. Risk factors may be present at birth (e.g, cCMV, family history) or can present later in infancy or childhood (e.g., hyperbilirubinemia, meningitis). Hearing loss associated with these risk factors may have congenital onset, late onset, or be progressive in nature.
Risk factors associated with temporary conductive hearing loss will be excluded. Articles reporting “infants at risk” will not be eligible; the specific risk factor(s) must be identified.
Comparators
For studies that include a comparison group, the comparator will be either (1) no risk factors or (2) a comparison between one risk factor and other factors. Due to the nature of studies in this field, it is anticipated that most studies will not include a comparison group.
Outcome
Primary outcome
The primary outcome is permanent bilateral or unilateral hearing loss in childhood. Permanent hearing loss refers to sensorineural hearing loss or conductive loss that is structural in nature and expected to last for 6 months or more [24]. Therefore, studies reporting outcomes as sensorineural hearing loss, auditory neuropathy spectrum disorder, permanent conductive hearing loss or mixed loss will be included. Definitions of hearing loss (i.e., the severity of hearing loss) will be accepted as defined by authors. Our interest in this review is only on peripheral hearing loss; therefore, studies reporting auditory processing disorders (or central auditory processing) will be excluded.
We will categorize the outcomes according to the onset of hearing loss:
-
i)
Congenital and early onset is defined as hearing loss present at birth or diagnosed within the first 3 months of life. This definition is consistent with the JCIH (2007) benchmark of diagnosis by 3 months of age [21].
-
ii)
Late onset is defined as the occurrence of hearing loss, typically after 3 months of age, after a normal hearing screen or confirmation of normal hearing through audiologic assessment.
The flexibility in these definitions allows for known variations in the classification of onset of hearing loss in the literature. Studies that report exclusively on temporary conductive hearing loss or change in hearing following surgical procedures (e.g., middle ear surgery, cochlear implants) are not targeted by this review.
The diagnosis of hearing loss must be confirmed by appropriate assessment methods at the time of the study. Results from automated tests used for hearing screening (such as automated otoacoustic emissions or automated auditory brainstem responses) will be excluded. Outcomes based solely on parental report of hearing loss will also be excluded. Hearing loss defined as “requiring hearing aids/amplification” will be accepted as a permanent hearing loss only if assessed by audiologists or specialized professionals, based on appropriate tests.
Secondary outcome
The secondary outcome of interest for this review is progressive hearing loss. Any amount of progressive hearing loss as defined by the authors will be included. All other inclusion criteria detailed under the primary outcome will apply.
Search method and information sources
The search strategy was developed in collaboration with a health sciences librarian experienced in systematic reviews. The following databases will be searched: MEDLINE including Epub Ahead of Print, In-Process & Other Non-Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) (1946 to July 30 2018), and Embase (1946 to July 30 2018) using the Ovid interface. CINAHL will be searched from inception on July 23, 2018 (database coverage dates not stated). Searches will be limited to the cohort, case control, or randomized controlled trials using existing filters. We will consider exposure of all risk factors outlined in the provincial surveillance protocol for the Ontario IHP [24]. We will also include general terms such as surveillance, risk factor, or risk indicator to capture risk factors that may not be included on the Ontario IHP list. The search strategy is presented in Additional file 2.
Restrictions will apply to language and publication date. We will consider papers published only in English since 1990, that include birth cohorts ≥ 1985. Articles in other languages will be screened only at the broad screening level. The date limit was set to correspond broadly with the implementation of UNHS programs.
Selection process and data management
The study selection involves three stages, managed within covidence (covidence systematic review software, http://www.covidence.org). Prior to the title and abstract screening, a screening form will be developed based on the inclusion and exclusion criteria and will be tested among reviewers with a subset of articles. Titles and abstracts will be assessed by two independent reviewers (BV and EF or DN) for potential relevance; the reviewers will resolve any conflicts (e.g., one reviewer selected “yes” or “maybe” and the other selected “no”) and involve a third reviewer if required. At the second stage, two independent reviewers (BV and EF or DN) will screen all potentially relevant full-text articles (all articles tagged as “yes” or “maybe” during the previous stage). Disagreements will be resolved by consensus or a third member of the research team.
All processes will ensure that bias is minimized when deciding whether to include or exclude certain studies, based on the application of objective inclusion/exclusion criteria, independent reviewers, and conflicts resolved through a third reviewer.
A PRISMA flow chart will present the number of studies included/excluded in each stage of the selection process. Main reasons for exclusion during the full-text screening stage will be documented.
Data extraction
Using study-specific data forms, pre-determined data will be extracted for each study. Data extracted will include:
-
1)
Study characteristics (author names and contact information, year, institution, country, language, and source of funding)
-
2)
Study design
-
3)
Population characteristics (e.g., sample size, age at diagnosis, degree of hearing loss, and permanent bilateral/unilateral loss)
-
4)
Details of control or comparison groups (if available according to the study design)
-
5)
Type of assessment
-
6)
Exposure—risk factors (including any author-specific definitions)
-
7)
Onset of hearing loss (e.g., proportion with congenital, early, late onset including author-specific definitions)
-
8)
Progressive loss (including author-specific definitions)
-
9)
Outcome data (including author-specific definitions of hearing loss)
One researcher will extract all information, which will be verified by a second reviewer. Discrepant findings will be resolved through consensus or a third reviewer when required.
Quality assessment
The risk of bias assessment will be conducted by one researcher and verified by a second. Randomized control trials (RCT) will be evaluated using the Cochrane Risk of Bias Tool (https://methods.cochrane.org/bias/resources/cochrane-risk-bias-tool). Other study designs will be assessed using the Qualitative Assessment Tool for Quantitative Studies (Effective Public Health Practice Project, https://merst.ca/ephpp/). This tool, developed at McMaster University, provides an overall methodological rating (weak, moderate, or strong) based on an appraisal of eight domains: selection bias, study design, confounders, blinding, data collection methods, withdrawals and dropouts, intervention integrity, and analysis.
Evidence synthesis
Study characteristics will be summarized narratively in summary tables in the report; data will be presented as frequencies and percentages, means and standard deviations, or medians and interquartile ranges, as appropriate. It is anticipated that a full meta-analysis will not be possible due to heterogeneity in research designs and variability in study and reporting methods. A narrative synthesis of the evidence will be conducted when quantitative pooling of data is not possible. We anticipate that results will be organized in tables according to individual risk factors (e.g., type or onset of risk factors, onset of hearing loss). Other classifications to be considered are the types of hearing loss (e.g., sensorineural, auditory neuropathy spectrum disorder, mixed and permanent conductive disorders) and age of onset (preschool versus school age).
The feasibility of meta-analysis will be determined based on the similarity of clinical population, same risk factors, type of outcomes, and study designs. If meta-analysis is possible for some outcomes, we will use a random effects model to aggregate results. For continuous data, we will compute mean or standardized mean differences with 95% confidence intervals (CIs). For dichotomous data, risk ratios with 95% CIs will be computed, where appropriate. Statistical heterogeneity will be evaluated using I2 statistics; for the interpretation of I2, a rough guide of low (0–25%), moderate (25–50%), substantial (50–75%), and considerable (75–100%) heterogeneity will be used [27]; potential reasons for heterogeneity will be explored. If there is considerable heterogeneity, (> 75%), we will not conduct a pooled analysis. If data permit, sensitivity analyses will be undertaken with respect to risk of bias; assessing of low risk of bias will be compared against high risk/unclear risk of bias, funnel plots will be generated to assess for publication bias if sufficient numbers of studies are available for meta-analysis [27].
Subgroup analyses
We will examine the following variables in subgroup analyses. Subgroups will be specific to age at onset (congenital and early infancy vs later childhood) and to specific conditions or treatments such as cCMV infection, meningitis, and pediatric cancers treated with ototoxic medication.
Quality of evidence
After data extraction and analysis, we will assess the quality and strength of the overall body of evidence using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) tool (http://www.gradeworkinggroup.org) [28]. To use GRADE, a summary of findings tables will be created. We will rate the evidence for both the primary outcome (presence of bilateral or unilateral hearing loss) and for the secondary outcome of progressive hearing loss. These outcomes will be assessed across the five domains included in GRADE: risk of bias, consistency, directness, precision, and publication bias. Based on these assessments, we will rate the quality of the evidence as high (very confident that the outcome is related to the risk factor), moderate (moderately confident that outcome is related to risk factor), or low (limited confidence in the outcome/risk factor association), or very low (little confidence in the outcome/risk factor association). Consensus will be reached by the research team comprised of researchers and clinicians, and the quality of evidence will guide the strength of the recommendations.
Discussion
The primary objective of this review is to collect and synthesize information on risk factors associated with congenital, early, or late onset permanent hearing loss in children. It is widely acknowledged that children with congenital or neonatal risk factors need to be tested during the neonatal period and be closely monitored for late onset hearing loss. Our question is of importance because most newborn hearing screening and surveillance programs are based on the 2007 JCIH factors. This list was released more than 10 years ago, and its development process was not as accurate and transparent as in a systematic review. In addition, this list is modified in several programs suggesting that it does not apply to their context or practice or it is not followed in its entirety [29,30,31,32].
We intentionally developed a search strategy and included numerous study designs to capture as many risk factors as possible. Based on other reviews, it is expected that the majority of studies will be descriptive and include small samples or cohorts of children with hearing loss. Our aim is to provide recommendations for policy makers and clinics based on the best available evidence. Consequently, limiting this review to high-quality methodological designs such as randomized controlled trials or large cohorts would limit the evidence for decision-making. Because it is intended to gather evidence for clinical practitioners and policymakers, we are convinced that all types of data are useful, on the condition that their methodological strengths and weaknesses are assessed, and integrated into the results.
The selection of publication date for inclusion in this systematic review was a balanced decision. It has to be long enough to include well-known risk factors that have not been widely investigated recently, such as congenital rubella syndrome. On the other hand, it has to integrate the latest knowledge due to the evolution of healthcare and treatments over time. For example, care for premature newborns in neonatal intensive care units has changed and practice in the administration of ototoxic drugs is better controlled, which has a positive impact on acquired hearing loss. Consequently, the systematic review aims to integrate both knowledge that has not been recently investigated and new scientific data on the possible association between risk factors and hearing loss. Only risk factors included in the review articles will be addressed, that is, risk factors not included in articles covered by our comprehensive search strategy will not be considered for hearing loss in children.
We anticipate that the results from this systematic review will guide decision-making in the Ontario IHP and will be applicable to UNHS programs worldwide.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on request.
Abbreviations
- cCMV:
-
Congenital cytomegalovirus
- EHDI:
-
Early hearing detection and intervention
- IHP:
-
Infant Hearing Program
- JCIH:
-
Joint Committee on Infant Hearing
- PRISMA-P:
-
Preferred Reporting Items for Systematic review and Meta-Analysis Protocols
- UNHS:
-
Universal newborn hearing screening
References
Dalzell L, Orlando M, MacDonald M, Berg A, Bradley M, Cacace A, et al. The New York State universal newborn hearing screening demonstration project: ages of hearing loss identification, hearing aid fitting, and enrollment in early intervention. Ear Hear. 2000;21(2):118–30.
Kaye CI, Accurso F, La Franchi S, Lane PA, Hope N, Sonya P, et al. Newborn screening fact sheets. Pediatrics. 2006;118(3):e934–63.
Wood SA, Sutton GJ, Davis AC. Performance and characteristics of the Newborn Hearing Screening Programme in England: the first seven years. Int J Audiol. 2015;54(6):353–8.
Moeller MP. Early intervention and language development in children who are deaf and hard of hearing. Pediatrics. 2000;106(3):E43.
Yoshinaga-Itano C. Levels of evidence: universal newborn hearing screening (UNHS) and early hearing detection and intervention systems (EHDI). J Commun Disord. 2004;37(5):451–65.
Fulcher A, Purcell AA, Baker E, Munro N. Listen up: children with early identified hearing loss achieve age-appropriate speech/language outcomes by 3 years-of-age. Int J Pediatr Otorhinolaryngol. 2012;76(12):1785–94.
Stika CJ, Eisenberg LS, Johnson KC, Henning SC, Colson BG, Ganguly DH, et al. Developmental outcomes of early-identified children who are hard of hearing at 12 to 18 months of age. Early Hum Dev. 2015;91(1):47–55.
Halpin KS, Smith KY, Widen JE, Chertoff ME. Effects of universal newborn hearing screening on an early intervention program for children with hearing loss, birth to 3 yr of age. J Am Acad Audiol. 2010;21(3):169–75.
Sininger YS, Martinez A, Eisenberg L, Christensen E, Grimes A, Hu J. Newborn hearing screening speeds diagnosis and access to intervention by 20-25 months. J Am Acad Audiol. 2009;20(1):49–57.
Ching TYC, Dillon H, Button L, et al. Age at Intervention for Permanent Hearing Loss and 5-Year Language Outcomes Pediatrics. 2017;140(3):e20164274.
Wood SA, Davis AC, Sutton GJ. Effectiveness of targeted surveillance to identify moderate to profound permanent childhood hearing impairment in babies with risk factors who pass newborn screening. Int J Audiol. 2013;52(6):394–9.
Holzinger D, Weishaupt A, Fellinger P, Beitel C, Fellinger J. Prevalence of 2.2 per mille of significant hearing loss at school age suggests rescreening after NHS. Int J Pediatr Otorhinolaryngol. 2016;87:121–5.
van Noort-van der Spek IL, Goedegebure A, Hartwig NG, Kornelisse RF, MJP F, Weisglas-Kuperus N. Normal neonatal hearing screening did not preclude sensorineural hearing loss in two-year-old very preterm infants. Acta Paediatr. 2017;106(10):1569–75.
Fortnum HM, Summerfield AQ, Marshall DH, Davis AC, Bamford JM. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ. 2001;323(7312):536–40.
Watkin PM, Baldwin M. Identifying deafness in early childhood: requirements after the newborn hearing screen. Arch Dis Child. 2011;96(1):62–6.
Weichbold V, Nekahm-Heis D, Welzl-Mueller K. Universal newborn hearing screening and postnatal hearing loss. Pediatrics. 2006;117(4):e631–6.
Barreira-Nielsen C, Fitzpatrick E, Hashem S, Whittingham J, Barrowman N, Aglipay M. Progressive hearing loss in early childhood. Ear Hear. 2016;37(5):e311–21.
Dahl HH, Ching TY, Hutchison W, Hou S, Seeto M, Sjahalam-King J. Etiology and audiological outcomes at 3 years for 364 children in Australia. PLoS One. 2013;8(3):e59624.
Dedhia K, Kitsko D, Sabo D, Chi DH. Children with sensorineural hearing loss after passing the newborn hearing screen. JAMA Otolaryngol Head Neck Surg. 2013;139(2):119–23.
Harlor AD Jr, Bower C. Hearing assessment in infants and children: recommendations beyond neonatal screening. Pediatrics. 2009;124(4):1252–63.
Joint Committee on Infant Hearing. Year 2007 position statement: principles and guidelines for early hearing detection and intervention programs. Pediatrics. 2007;120(4):898–921.
World Health Organization. Newborn and infant hearing screening. Current issues and guiding principles for action. Genova: World Health Organization; 2010.
Muse C, Harrison J, Yoshinaga-Itano C, Grimes A, Brookhouser PE, Epstein S, et al. Supplement to the JCIH 2007 position statement: principles and guidelines for early intervention after confirmation that a child is deaf or hard of hearing. Pediatrics. 2013;131(4):e1324–49.
Ontario Ministry of Children and Youth Services Ontario Infant Hearing Program. Ontario Infant Hearing Program: a guidance document. 2017.
Beswick R, Driscoll C, Kei J. Monitoring for postnatal hearing loss using risk factors: a systematic literature review. Ear Hear. 2012;33(6):745–56.
Moher D, Stewart L, Shekelle P. Implementing PRISMA-P: recommendations for prospective authors. Syst Rev. 2016;5:15.
Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ. 2011;343:d4002.
Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–6.
Gilbey P, Kraus C, Ghanayim R, Sharabi-Nov A, Bretler S. Universal newborn hearing screening in Zefat, Israel: the first two years. Int J Pediatr Otorhinolaryngol. 2013;77(1):97–100.
Ohl C, Dornier L, Czajka C, Chobaut JC, Tavernier L. Newborn hearing screening on infants at risk. Int J Pediatr Otorhinolaryngol. 2009;73(12):1691–5.
Turchetta R, Conti G, Marsella P, Orlando MP, Picciotti PM, Frezza S, et al. Universal newborn hearing screening in the Lazio region, Italy. Ital J Pediatr. 2018;44(1):104.
Vos B, Lagasse R, Leveque A. The organisation of universal newborn hearing screening in the Wallonia-Brussels federation. B-ENT. 2013;9(Suppl 21):9–15.
Acknowledgements
The authors would like to acknowledge the contribution of Dr. Margaret Sampson, Manager at Library Services, CHEO, Ottawa, for her assistance in developing the search strategy and for conducting the literature searches in the databases.
Funding
Funding from the Ministry of Children, Community and Social Services to Western University for its work in support of the Ontario Infant Hearing Program.
Author information
Authors and Affiliations
Contributions
BV, DN, MP, MB, and EMF conceived the protocol. BV drafted the manuscript. EMF, DN, MP, and MB reviewed the manuscript. EMF is the guarantor of the review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
PRISMA-P 2015 Checklist. (DOCX 30 kb)
Additional file 2:
Search strategy to be used. (PDF 330 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Vos, B., Noll, D., Pigeon, M. et al. Risk factors for hearing loss in children: a systematic literature review and meta-analysis protocol. Syst Rev 8, 172 (2019). https://doi.org/10.1186/s13643-019-1073-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13643-019-1073-x