
Abstract
Heart regeneration is a fascinating and complex biological process. Decades of intensive studies have revealed a sophisticated molecular network regulating cardiac regeneration in the zebrafish and neonatal mouse heart. Here, we review both the classical and recent literature on the molecular and cellular mechanisms underlying heart regeneration, with a particular focus on how injury triggers the cell-cycle re-entry of quiescent cardiomyocytes to replenish their massive loss after myocardial infarction or ventricular resection. We highlight several important signaling pathways for cardiomyocyte proliferation and propose a working model of how these injury-induced signals promote cardiomyocyte proliferation. Thus, this concise review provides up-to-date research progresses on heart regeneration for investigators in the field of regeneration biology.
Similar content being viewed by others
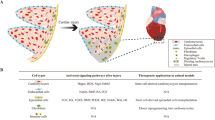

Background
Myocardial infarction (MI) is a devastating disease worldwide. At present, nearly 40 million patients are suffering from heart failure. Due to the lack of an adequate blood supply, ~ 25% of total cardiomyocytes (CMs) undergo apoptosis and necrosis within a few hours after MI (Murry et al. 2006). Since the regenerative capacity is poor, this massive loss of CMs eventually leads to cardiac fibrosis and heart failure. The main clinical therapies for heart failure include drug intervention, left ventricular assist device implantation, and heart transplantation (Heallen et al. 2019). However, current drug and assist device therapies cannot reverse the CM loss while donor hearts for transplantation are limited. It remains challenging to find effective therapeutic methods for heart failure. Over the past decades, cardiac regeneration has become one of the most exciting research topics in the search for innovative interventions and therapies for heart failure, consisting of induced pluripotent stem cell- and embryonic stem cell-derived CM transplantation, cardiac tissue engineering, stimulating endogenous CM proliferation, and reprogramming non-myocytes into CMs (Tzahor and Poss 2017; Sadek and Olson 2020).
After two decades of debate, it is well accepted that the so-called adult cardiac stem cells (CSCs), including c-Kit+ CSCs, make no or minimal contributions to cardiac repair, while recent studies do not support the existence of adult CSCs (Li et al., 2018a; Sultana et al. 2015; van Berlo et al. 2014). Besides, many investigators have paid attention to studying regenerative environments or niches, such as cardiac fibrosis, inflammation, and coronary vessel regeneration (Zeisberg and Kalluri 2010; Swirski and Nahrendorf 2013; Bardot and Dubois 2019; Kramann et al. 2015; Bajpai et al. 2018). Some lower vertebrates, such as zebrafish and axolotl, have a strong regenerative ability in the adult heart and other organs (Poss et al. 2002; Cano-Martinez et al. 2010). But in mammals, efficient cardiac regeneration only occurs in newborns, and this ability is lost a few days after birth. Adult mammalian CMs renew at a very low rate (Kajstura et al. 2010; Bergmann et al. 2009), and newly-generated CMs are likely derived from existing CMs (Ali et al. 2014; Senyo et al. 2013). Re-activating the regenerative potential in adult CMs may be a new alternative to replenish the lost CMs after MI. There are some excellent review articles regarding CM transplantation, cardiac tissue engineering, and reprogramming fibroblasts into CMs (Laflamme and Murry 2011; Vunjak-Novakovic et al. 2011; Srivastava and DeWitt 2016; Keepers et al. 2020). In this brief review, we focus on the current understanding of the molecular and cellular mechanisms underlying CM proliferation and heart regeneration.
Heart regeneration in model organisms
Regenerative capacity is quite different among organs and species. Some invertebrates, such as Hydra and planarians, can regenerate the whole body (Tanaka and Reddien 2011; Duncan and Sanchez, 2019). It was initially reported that ventricular injury induces CM proliferation but forms cardiac fibrosis in the adult newt, and later confirmed by others to support cardiac regeneration with minimal fibrosis (Oberpriller and Oberpriller 1971; Oberpriller and Oberpriller 1974; Witman et al. 2011; Mercer et al. 2013). The zebrafish has become a well-recognized model organism for studying embryonic development, organ function, and behavior, as well as organ regeneration including the heart (Poss et al. 2002; Raya et al. 2003). The zebrafish heart can fully regenerate after up to 20% ventricular resection (Poss et al. 2002). Others have reported a cryoinjury model of adult zebrafish heart that better mimics MI in mammals including human beings (Chablais et al. 2011; González-Rosa et al. 2011; Schnabel et al. 2011). In addition, genetic ablation of CMs in the adult zebrafish heart leads to an inflammatory response, endocardial and epicardial activation, and myocardial proliferation that is similar to the resection and cryoinjury models (Wang et al. 2011).
Although the adult mammalian heart has limited regenerative capacity, the mouse heart has full capacity to regenerate after ventricular resection at postnatal days 1-6 (P1-6), but loses this capacity by P7 (Porrello et al., 2011a). Others have argued that regeneration of the neonatal mouse heart is limited after apical resection, where the regenerative capacity is dependent on the surgical method, the size of the resection, and the evaluation method (Bryant et al. 2015; Andersen et al. 2016). Like the zebrafish heart, genetic ablation of CMs in the fetal mouse heart triggers a proliferative response in CMs and restores the morphology of the injured heart (Sturzu et al. 2015). In the cryoinjury model, the regenerative capacity of the neonatal mouse heart is dependent on the severity of injury. The neonatal heart can fully regenerate after non-transmural cryoinjury, but not after transmural cryoinjury which results in more CM loss and more severe fibrosis (Darehzereshki et al. 2015). Unlike other types of injury models, cryoinjury does not trigger significant numbers of proliferating CMs but frequently causes angiogenesis and epicardium activation (Darehzereshki et al. 2015; Malek et al. 2017). The neonatal mouse heart can also fully regenerate after left anterior descending coronary artery ligation (Mahmoud et al. 2014; Blom et al. 2016; Sereti et al. 2018). Also, the regenerative response of the neonatal heart has been demonstrated in large mammals such as pigs, with full regeneration without fibrosis at P1 and P2, and gradual loss of regenerative capacity from P3 to P14 (Zhu et al., 2018a; Ye et al. 2018). Even the neonatal human heart can recover from MI (Haubner et al. 2016). The heart regeneration models share the common feature of injury-induced CM proliferation and the resolution of fibrosis. Together, both the zebrafish and the neonatal mammalian heart have a full capacity to regenerate after different types of injury although the injury-induced responses are not identical. Compared with CMs that are terminally differentiated and quiescent in the adult zebrafish and newt heart, neonatal mouse CMs are immature and easily gain cell-cycle re-entry, thus it is better to consider the neonatal mouse heart as an injury-induced developmental model.
Myocardial regeneration capacity varies among species and changes dramatically within a week after birth in rodents. It is interesting to look at these differences from a developmental and evolutionary perspective. There are significant differences in heart structure, blood pressure, body temperature, oxygen content in the living environment, energy metabolism, and cardiac cell structure during individual development and evolution. From fish to mammals, the oxygen content of their living environment increases dramatically. This same change occurs right after birth in mammals. There is evidence that low-oxygen environments can stimulate regenerative potential (Kimura et al. 2015; Nakada et al. 2017). Hyperoxic environments may also affect the energy metabolism of CMs (Nakada et al. 2017). There are also significant differences in the structure of CM during evolution. Adult mammalian CMs have a more complete sarcomere structure. It has been shown that the CM proliferation is greatly related to the depolymerization of the sarcomeres (Ahuja et al. 2004). Others have found that the proportion of diploid CMs, which is thought to represent the regenerative capacity of CMs, is negatively correlated with metabolic rate in vertebrates, as well as body temperature in mammals. Also, they found that plasma thyroxine (T4) levels are inversely correlated with the proportion of diploid CMs. Inhibition of thyroid hormone signaling enhanced CM proliferation and regeneration (Hirose et al. 2019).
Cellular mechanisms underlying heart regeneration
After ventricular resection in zebrafish, a clot forms rapidly, followed by fibrin and collagen deposition from 2 to 9 days post-amputation (dpa). During the course of regeneration, newly-formed myofibers gradually replace the collagen and the fibrin clot to achieve a fully functional heart by ~ 60 dpa (Poss et al. 2002). Lineage tracing has revealed that the lost CMs are replenished by pre-existing cardiomyocytes in the adult zebrafish heart (Jopling et al. 2010; Kikuchi et al. 2010). Ventricular CMs can be transdifferentiated and regenerated from atrial CMs after injury, suggesting that the embryonic heart has greater plasticity (Zhang et al. 2013). Using the Rainbow-based lineage mapping method, others have revealed that the cortical layer contributes to the formation of new CMs during heart development and regeneration (Gupta and Poss 2012). A Cre-LoxP-based lineage tracing study of tbx5a-expressing CMs has found that trabecular CMs can generate cortical CMs for myocardial regeneration after injury (Sanchez-Iranzo et al., 2018a). However, the molecular and cellular changes in CMs during heart regeneration remain incompletely understood. It is now conceivable that they undergo sarcomeric disassembly with disorganized actin and myosin filaments and larger intercellular spaces during regeneration (Jopling et al. 2010). Some embryonic factors such as gata4 are re-expressed in CMs affected by injury (Jopling et al. 2010). The loss of Gata4 function blocks myocardial regeneration and is accompanied by severe scars (Gupta et al. 2013). In addition, a pool of undifferentiated CMs are formed in the injured, wound-healing tissues, and embryonic cardiac transcription factors and myosin heavy chain 7 are re-activated in these cells (Lepilina et al. 2006; Sallin et al. 2015). Another study has reported that the transcriptional profiling of CMs during regeneration is similar to that during heart development (Aguirre et al. 2014). Single-cell RNA-seq has confirmed that CMs in the border zone of the injured heart undergo de-differentiation and have transcriptional profiling similar to embryonic CMs (Honkoop et al. 2019). Together, heart regeneration in zebrafish naturally engages in CM de-differentiation and proliferation.
The regeneration process in the neonatal mouse heart is similar to that in zebrafish. After partial apical resection, a blood clot forms and later is replaced by newly-formed cardiac tissue in the injury site. Lineage tracing has shown that newly-formed CMs come from pre-existing CMs (Porrello et al., 2011a; Sereti et al. 2018; Porrello et al. 2013). Consistently, others have also demonstrated that non-CMs contribute to newly-formed CMs in developing embryos but not in the adult heart during homeostasis or after injury by using a dual genetic lineage tracing strategy (Li et al. 2019). Therefore, it is generally assumed that newly-formed CMs are from pre-existing CMs but not cardiac stem cells or cardiac progenitor cells in the adult heart. Like zebrafish, Gata4 is expressed in CMs in the neonatal mouse heart, and loss of Gata4 in CMs increases the scar size while its overexpression leads to improved regeneration in the neonatal heart after cryoinjury, probably through sarcomere disassembly and de-differentiation (Malek et al. 2017). Multicellular transcriptional analysis has shown that the neonatal heart retains a permissive embryonic developmental state, which may allow CMs to re-enter the cell cycle and continue to proliferate after injury (Quaife-Ryan et al. 2017). Single-nucleus RNA-seq analysis suggests that a sub-group of CMs preferentially enters the cell cycle and mitosis after injury (Cui et al. 2020). Together, new CMs are regenerated from CM de-differentiation and proliferation in the zebrafish and neonatal mouse heart.
The difference in cardiac regeneration across species and age groups may be related to the degree of CM polyploidization. Fetal or neonatal CMs are mostly mononuclear with a diploid genome in mammals and zebrafish. After birth, DNA can replicate, however, the ability of karyokinesis or cytokinesis varies between species. In rat CMs, after birth, DNA replicates but with no cytokinesis, increases binucleated CMs from day 4, and reaching 90% on day 12 (Li et al. 1996). In humans, CM DNA replicates without karyokinesis or cytokinesis, resulting in mononuclear CMs with tetraploid or higher DNA content (Laflamme and Murry 2011). The proportion of polyploid CMs in human hearts increased significantly from age 10 to 20 years (Mollova et al. 2013). Unlike mammals, zebrafish CMs do not undergo polyploidization, and more than 95% of CMs are mononuclear and diploid. There is evidence that diploid CMs have greater regenerative potential. Consistently, genetically modified zebrafish hearts containing 50% of polyploid CMs fail to regenerate after injury (Gonzalez-Rosa et al. 2018). Tnni3k knockout mice show more mononuclear diploid CMs and improved CM proliferation while Tnni3k overexpression in zebrafish leads to implicated cardiac regeneration (Patterson et al. 2017).
Besides CM proliferation after injury, other cardiac cells are also activated to participate in regeneration (Table 1). In zebrafish, cardiac injury stimulates the epicardium to promote myocardial proliferation, and the activated epicardium mainly forms epicardial cells, subepicardial cells, and perivascular cells, but rarely endothelial cells and CMs (Kikuchi et al., 2011a). The activated epicardium after injury also promotes angiogenesis and contributes to the formation of cardiac fibroblasts, which is necessary for cardiomyocyte proliferation (Gonzalez-Rosa et al. 2012; Sanchez-Iranzo et al., 2018b). During neonatal mouse heart regeneration, the epicardium is also activated after injury (Porrello et al., 2011a). Although embryonic epicardial cells are pluripotent, investigators have revealed that Tbx18+ epicardial cells only give rise to minimal numbers of CMs in the neonatal heart after injury (Cai et al. 2019). Adult epicardial cells are unable to produce CMs, but regulate cardiac regeneration via paracrine effects (Zhou et al. 2011). Similarly, the endocardium is activated within hours post-injury in zebrafish. Raldh2, the enzyme for synthesizing retinoid acids, is quickly induced in the endocardium and epicardium, and retinoic acid signaling is essential for heart regeneration (Kikuchi et al., 2011b).
Regeneration of coronary vessels is also critical for achieving heart regeneration. New vessels are formed in the injured area, and neovascularization provides oxygen and blood for the hypoxic infarcted region and is required for heart regeneration in zebrafish (Lepilina et al. 2006; Marin-Juez et al. 2016). As for the neonatal mouse heart, arterial endothelial cells migrate from capillaries to the infarcted region and form collateral arteries, which are essential for heart regeneration (Das et al. 2019). Besides, immune cells and nerve cells also contribute to zebrafish and neonatal mouse heart regeneration through complex interactions among different types of heart cells (Aurora et al. 2014; Han et al. 2015; Kikuchi 2020; Mahmoud et al. 2015; White et al. 2015; Lai et al. 2017).
Recently, several important studies have shown that the lymphatic system is also essential for cardiac regeneration and repair. The main function of the lymphatic vessels is to drain lymph and transport inflammatory cells. The abnormal lymphatic function lead to myocardial disease, arrhythmias, atherosclerosis, and other diseases (Norman and Riley 2016). Klotz and colleagues find that vascular Endothelial Growth Factor C (VEGF-C) induce a stronger lymphangiogenic response and result in improved cardiac function after myocardial infarction (Klotz et al. 2015). The extracellular protein reelin (RELN) is recently identified as a lympho-endothelial secreted factor that enhances cardiac regeneration in neonatal mice and improves heart function after myocardial infarction (Liu et al. 2020). Extensive lymphangiogenesis is also observed in zebrafish hearts after cryoinjury. Lymphatic vessels are essential for zebrafish cardiac regeneration, and genetically modified fish lines with lymphatic defects fail to regenerate after cryoinjury (Harrison et al. 2019; Gancz et al. 2019). Together, various types of cells in the injured heart coordinate and interact to achieve precise tissue regeneration, but many of these intercellular mechanisms are not well understood (Table 1).
Molecular mechanisms regulating cardiomyocyte proliferation
It is now well-recognized that injury-induced CM proliferation is the major cellular mechanism for heart regeneration in the zebrafish and neonatal mouse. Intensive studies have shown that a number of signaling pathways are involved in this complex process, in which the epicardium, endocardium/endothelium, fibroblasts, and leukocytes actively interact with CMs. We thus provide below a brief review of several essential signaling pathways that regulate CM proliferation.
Cell cycle regulators
Mammalian CMs are generally considered to be in cell-cycle arrest, often accompanied by a stronger expression of cell-cycle inhibitors (p21, p27, and Rb) and weaker expression of cell-cycle activators (cyclins and CDKs) (Tamamori-Adachi et al. 2008; Sdek et al. 2011; Tane et al. 2014). Cell-cycle regulatory proteins show dynamic expression patterns during rat and mouse cardiac development. For example, cyclin A2, cyclin B1, CDK2, and CDK6 are significantly down-regulated in the heart at 7 weeks compared to embryonic day 18 (E18) (Liu et al. 2010). In the mouse heart, the expression levels of cyclins (cyclins A, B1, D1, and E) and CDKs (CDKs 1, 2, and 4) decrease from E12 or E16 until P0, and then increase with a peak around P5 (Ikenishi et al. 2012). CDK inhibitors such as p21, p27, and p57 are important negative regulators of the cell cycle for CMs. The expression levels of p21 and p27 increase during the development of both the rat and human heart. p57 is only expressed in the rat heart from E12 to E15, but remains detectable from the embryonic to the adult human heart (Ikenishi et al. 2012). The pocket proteins (Rb, p107, and p130) are expressed in CMs with quite different dynamics. For example, the expression level of Rb increases in the heart during late gestation and postnatal stages, and it plays a vital role in the cell-cycle exit of CMs (MacLellan et al. 2005).
To promote adult CM cell-cycle re-entry, investigators have attempted to carry out ectopic expression of cell-cycle regulatory genes in CMs in vitro and the mouse heart in vivo. Transgenic overexpression of cyclin D2 results in increased DNA synthesis by CMs in the adult mouse heart, but overexpression of other cyclin D subfamily members, such as cyclins D1 and D3, appears to have little or no effect on the cell-cycle progression of CMs (Pasumarthi et al. 2005). A series of studies has shown that forced expression of cyclin A2 results in enhanced left ventricular systolic function both in mouse and pig (Chaudhry et al. 2004; Cheng et al. 2007; Shapiro et al. 2014; Woo et al. 2006). The cyclin B1-CDC2 complex is the key regulator of the G2/M-phase transition in CMs, and its overexpression re-initiates cell division of adult cardiomyocytes in vitro (Bicknell et al. 2004). Combined knockdown of CDK inhibitors (p21 and p27) is also able to promote adult CM proliferation with evident cytokinesis via increasing the expression of cyclins A and E (Tane et al. 2014; Di Stefano et al. 2011). As the key mediators of cell-cycle progression, E2F transcription factors have been well studied to address their roles in regulating cell-cycle progression in CMs. Overexpression of E2F1 and E2F2 in adult CMs increases DNA synthesis, but leads to apoptosis (Agah et al. 1997; Ebelt et al. 2008). Recently, Srivastava and colleagues have identified four cell-cycle regulatory genes (CDK1, CCNB, CDK4, and CCND) that are sufficient to induce the proliferation of mouse, rat, and human CMs, and adenoviral-mediated expression of these genes significantly improves cardiac function after MI in mice (Mohamed et al. 2018). Although forced expression of some cell-cycle regulators stimulates DNA synthesis, most of them have little effect on inducing CM cytokinesis, and some even have inhibitory effects. For instance, forced expression of cyclin G1 increases DNA synthesis but inhibits cytokinesis in neonatal CMs (Liu et al. 2010). Besides, forced expression of cell-cycle regulators in CMs induces apoptosis in mice (Agah et al. 1997; von Harsdorf et al. 1999; Kirshenbaum et al. 1996). Therefore, the recent work suggests that the spatio-temporal dose-dependent overexpression of cell-cycle regulators is essential for the cell-cycle re-entry of CMs (Mohamed et al. 2018).
Hippo signaling pathway
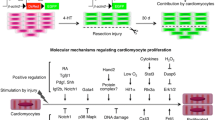
Hippo signaling is a highly-conserved pathway that regulates organ size. It regulates embryonic heart development in both mouse and zebrafish (Fukui et al. 2014; Bornhorst et al. 2019; Artap et al. 2018). Its function in heart regeneration was first reported by Martin’s lab (Heallen et al. 2013). Some key components of this pathway, such as MST, LATS, and Salvador (Salv), regulate CM proliferation. Genetic deletion of either MST or LATS induces CMs to re-enter the cell cycle (Heallen et al. 2011). Either knockdown of Salv by shRNA or Salv knockout improves systolic cardiac function after MI in mice (Heallen et al. 2011; Leach et al. 2017). YAP is a transcription coactivator and a downstream component of the Hippo pathway. Cardiac-specific knockout of YAP results in embryonic lethality (von Gise et al. 2012; Xin et al. 2011). Intriguingly, activation of YAP1 extends the regenerative window in the neonatal mouse heart beyond the first postnatal week, and improves cardiac function after injury, mostly by upregulating cell-cycle genes (Lin et al. 2014; Xin et al. 2013). PP1 and PP2A have been reported to activate YAP via de-phosphorylating p-YAP and translocating it into the nucleus (Haemmerle et al. 2017; Schlegelmilch et al. 2011). Administration of Agrin after MI leads to enhanced CM proliferation in mice (Bassat et al. 2017). Mechanistically, phosphorylation of YAP by Hippo enhances its interaction with dystroglycan 1 (DAG1), a component of the dystrophin glycoprotein complex (DGC), and thus inhibits the nuclear translocation of YAP (Morikawa et al. 2017). Agrin interferes with the YAP-DGC/DAG1 interaction, thus leading to YAP release, translocation into the nucleus, and cell-cycle progression by interaction with TEAD transcription factors (Bassat et al. 2017). A recent study further revealed that constitutive activation of YAP switches the chromatin to a more primitive, fetal-like, and proliferative state, making it easier for adult CMs to re-enter the cell cycle (Monroe et al. 2019). These studies suggest that inhibition of Hippo can be invoked as a strategy to promote CM regeneration for treating heart failure. However, others have found that Yap mostly regulates cell matrix and inflammatory gene networks and contributes to scar formation apart from CM proliferation during zebrafish heart regeneration (Flinn et al. 2019). In addition, Pitx2 has been identified as a transcription factor enriched in the regenerating Hippo-deficient mouse heart and is associated with the oxidative stress response. The Pitx2-deficient neonatal mouse heart fails to regenerate after apex resection, which probably affects mitochondrial function and adipose composition (Li et al., 2018b; Tao et al. 2016). Together, Hippo signaling negatively regulates CM proliferation via fine-tuning the YAP-DGC interaction, chromatin accessibility, and mitochondrial function (Fig. 1).

Summary of signaling pathways regulating cardiomyocyte proliferation. Hippo/YAP signaling is inactive when p-YAP is bound to the dystrophin glycoprotein complex (DGC). Binding of Agrin to the DGC leads to the translocation of p-YAP to the cytoplasm, phosphorylation of YAP is regulated by the kinase complex (Mst1/Lats2/Salv), and dephosphorylation of p-YAP by PP1/PP2A or others results in YAP activation and translocation to the nucleus. FGF signaling activates the MAPK pathway and induces the expression of Dusp6, a negative regulator of pERK, and Dusp6 protein is post-transcriptionally modified and degraded by H2O2. The PI3K-AKT signaling is regulated by NRG1/ErbB2/4, periostin/integrin, small molecule carbocyclin, and Wnt signaling pathways, which fine-tune β-catenin activity and its translocation into the nucleus. Binding of ligands such as Delta/Jagged to Notch receptors leads to Notch intracellular domain (NICD) activation and translocation to the nucleus. Together with injury-induced activation of chromatin remodeling factor Brg1/Dnmt3ab, the Yap/pERK-β-catenin/NICD activation in the nucleus regulates the expression of cell-cycle regulators including cyclins and cyclin inhibitors
Neuregulin/ErbB signaling pathway
Neuregulin1 (NRG1) is an EGF-like growth factor that binds with ErbB2 and ErbB4 receptors on the cell membrane of the CM. NRG1/ErbB signaling was first reported to promote CM proliferation and regeneration in mice, and was subsequently further shown to promote CM proliferation after MI or ventricular resection in the adult zebrafish and mice (Gemberling et al. 2015; Bersell et al. 2009; Polizzotti et al. 2015). During zebrafish heart regeneration, inhibition of ErbB receptors leads to a reduction of CM proliferation, while overexpression of Nrg1 increases it, suggesting that the Nrg1/ErbB signaling plays an important role in endogenous heart regeneration in zebrafish (Gemberling et al. 2015) (Fig. 1). Besides, others have found that the proliferative effect of vitamin D on CMs is dependent on Nrg1/Erbb2 signaling in zebrafish (Han et al. 2019). The conditional ablation of ERBB2 in embryonic or neonatal mouse CMs leads to reduced proliferation. Consistently, genetic activation of ERBB2 significantly improves cardiac function post-MI, indicating that this pathway can be exploited as a viable target for heart regeneration (D'Uva et al. 2015). To explore the potential therapeutic application of human recombinant NRG1 proteins (rNRG1), others have found that administration of rNRG1 immediately after cryoinjury is critical for promoting CM proliferation and regeneration using the P1 mouse heart, and rNRG1 has a similar effect of inducing proliferation in cultured CMs from pediatric patients (Polizzotti et al. 2015). Furthermore, a recent study has found that Nrg1/Erbb2 signaling induces metabolic reprogramming in the regenerating heart by switching oxidative phosphorylation to glycolysis in regenerating CMs (Honkoop et al. 2019). However, others have shown that rNRG does not induce CM DNA synthesis in either normal or infarcted adult mice (Reuter et al. 2014). Two clinical trials have shown dose-limited toxicity and a mild beneficial effect of rNRG1 in ~ 40 heart failure patients (Gao et al. 2010; Lenihan et al. 2016), therefore it remains to be determined whether rNRG1 has an effective clinical outcome for treating pediatric and/or adult heart failure patients.
Notch signaling pathway
The Notch pathway plays a key role in the heart development of mammalian embryos. Mice lacking Notch receptors or ligands show early embryonic lethality mainly due to cardiovascular abnormalities (Penton et al. 2012). In addition to its function in embryonic heart development and cell lineage specification, Notch is also critical for CM proliferation (Collesi et al. 2008; Campa et al. 2008; Croquelois et al. 2008). During early postnatal development, Notch signaling is up-regulated to reactivate the cell cycle by inducing the expression and nuclear localization of Cyclin D1 (Campa et al. 2008). In zebrafish, the Notch pathway has been widely reported to promote CM proliferation (Raya et al. 2003; Zhang et al. 2013). Conditional inhibition of Notch signaling via the ectopic expression of Notch inhibitor dominant-negative mastermind-like in endothelial cells (including the endocardium) decreases CM proliferation (Zhao et al. 2019; Zhao et al. 2014) (Fig. 1). Others have shown that flow-responsive klf2a and klf2b are critical for regulating endocardial Notch signaling including increased notch1b expression after embryonic ventricular CM ablation (Galvez-Santisteban et al. 2019; Li et al. 2020), and endothelial Brg1 fine-tunes Notch signaling during adult zebrafish heart regeneration (Xiao and Hou et al., unpublished data). In addition, adeno-associated virus delivery of Notch1 or its ligand Jagged1 induces neonatal mouse CM proliferation, in which Notch activation increases the transcription of Notch1-targeted genes via establishing an open chromatin conformation (Felician et al. 2014). However, the effect of Notch signaling on the adult mouse heart after MI is very limited. DNA methylation analysis has shown that the promoters of Notch target genes are more highly methylated in the adult mouse CM than in the neonatal heart, thus leading to transcription repression (Felician et al. 2014). Together, these studies suggest an essential role of endothelial and endocardial Notch signaling in regulating myocardial proliferation and regeneration.
Wnt signaling pathway
Wnt pathways include the classical Wnt/β-catenin pathway, the atypical planar cell polarity pathway, and the non-classical Wnt/Ca2+ pathway (Nusse and Varmus 2012). Wnt signaling plays vital roles in cardiac development, especially in myogenesis (Gessert and Kuhl 2010). The classical Wnt/β-catenin pathway positively regulates cardiac specialization during the early stages of development, but plays an inhibitory role in the later stages. Wnt signaling also promotes the proliferation of progenitor cells in the second heart field of the mouse embryo (Bisson et al. 2015). In addition, both the classical and non-classical Wnt pathways play a role in injury-induced cardiac repair. The Wnt signaling pathways are relatively inactive in the adult mammalian heart, but are immediately activated upon injury (Hermans et al. 2012). Wnt ligands have various expression patterns during MI. Wnt1 is rapidly induced and persists for 2 days after MI, while Wnt4 and Wnt7a are transiently up-regulated in the later stages of MI (Duan et al. 2012). These Wnt ligands activate both the classical and non-classical Wnt pathways. Others have recently reported that Wnt signaling regulates adult CM proliferation, and the small molecule CGX132146 and cardiomogen, inhibitors of Wnt signaling, promote CM proliferation in the infarcted border zone (Yang et al. 2017; Xie et al. 2020). Consistent with this, cardiac regeneration in mice with conditional knockout of Lrp6, a co-receptor of Wnt ligands, improves at least partly via promoting CM proliferation (Wu et al. 2020). In zebrafish, a transgenic reporter of Wnt/β-catenin signaling, TOPdGFP, is upregulated in the injured areas after ventricular resection (Stoick-Cooper et al. 2007), and inhibition of Wnt signaling increases CM proliferation (Zhao et al. 2019; Xie et al. 2020) (Fig. 1). Together, these studies suggest a biphasic action of Wnt signaling where Wnt ligands and receptors are induced upon injury but turning down Wnt signaling is essential for CM proliferation and regeneration after ventricular resection in zebrafish and MI in mice. This discrepancy warrants further mechanistic studies.
MAPK signaling pathway
The MAPK signaling pathway is a classical kinase cascade pathway activated by growth factors such as fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF), and it regulates cell proliferation, differentiation, and survival, as well as playing a key role in cardiovascular diseases (Muslin 2008). Both the FGF and PDGF signaling pathways are required for zebrafish heart regeneration by regulating the epicardial epithelium-mesenchyme transition, angiogenesis, and CM proliferation (Lepilina et al. 2006; Lien et al. 2006). In mice, MAPK signaling regulates CM proliferation during heart regeneration. MKK/p38α has been demonstrated to negatively regulate proliferation in both the zebrafish and mouse. In zebrafish, overexpression of MKK6 impairs cardiogenesis and heart regeneration, while inactivation of p38α is a prerequisite for re-entry into the cell cycle (Jopling et al. 2012). In mice, overexpression of MKK3 or MKK6 leads to premature heart failure (Liao et al. 2001). Besides, the activation of p38 suppresses neonatal CM proliferation, while attenuation of p38 activity causes cardiac hypertrophy and increases mitosis in CMs (Engel et al. 2005; Braz et al. 2003). Dual specificity phosphatase 6 (Dusp6), an ERK phosphatase, is a negative feedback factor in FGF signaling. Dusp6 is transiently induced after ventricular resection in zebrafish, and overexpression of dusp6 impairs heart regeneration (Han et al. 2014), its deletion increases CM proliferation and angiogenesis, and decreases cardiac fibrosis after ventricular resection in zebrafish (Missinato et al. 2018). In the neonatal mouse heart, Dusp6 disruption leads to cardiac hypertrophy and increased CM proliferation (Maillet et al. 2008). In addition, MEK1/2 and ERK1/2 are upregulated after ventricular resection in zebrafish. Inhibition of MEK1/2 by ADZ6244 down-regulates pERK and thus decreases CM proliferation. Consistently, over-expression of dominant-negative MEK1K98M prevents Fli1+ endothelial cell migration into the injury site, angiogenesis, and cardiac regeneration (Liu and Zhong 2017). Thus, several components of the MAPK signaling pathway are responsive to injury and play essential roles in heart regeneration (Fig. 1).
TGFβ signaling pathway
TGFβ signaling is an important pathway to regulate tissue fibrosis including cardiac fibrosis. In zebrafish, TGFβ ligands (Tgfb1, b2, and b3) and receptors (Alk4, Alk5a, and Alk5b) are induced after cryoinjury. Small-molecule inhibition of TGFβ type I receptors suppresses pSmad3 and impairs heart regeneration by attenuating CM proliferation and enhancing scar formation (Chablais and Jazwinska 2012; Choi et al. 2013). Fstl1, a TGFβ family member, has been shown to promote CM re-entry into the cell cycle. Delivering Fstl1 by epicardial patch or myocardial injection into the mouse and pig heart promotes CM proliferation and improves cardiac function. However, this pro-proliferative effect can only be achieved when Fstl1 is derived from epicardium but not from CMs (Wei et al. 2015). Similarly, during adult cardiac valve regeneration, TGFβ signaling promotes the re-entry of endothelial and hematopoietic cells into the cell cycle (Bensimon-Brito et al. 2020). These studies suggest that TGFβ signaling is essential for promoting endothelial and CM proliferation in the adult zebrafish and mouse heart after injury.
Epigenetic regulation
Epigenetic regulation plays an important role in cardiac development and heart regeneration (Zhu et al., 2018b; Moore-Morris et al. 2018). It consists of multiple components, such as DNA methylation, histone modification (methylation, acetylation, and phosphorylation), chromatin remodeling complexes, and non-coding RNAs (microRNAs and long non-coding RNAs). During the past decade, major progress has been made, revealing the functions of microRNAs (miRNAs) in CM proliferation in the zebrafish and mouse. miRNAs are 21-23-base single-strand non-coding RNAs. The miR-15 family has been reported to regulate the cell cycle of CMs and cardiac remodeling in ischemic heart disease. A miR-15 family member, miR-195, is strongly induced in the P10 ventricle relative to P1, and knockdown of the miR-15 family in the neonatal mouse with locked nucleic-acid modified anti-miRNAs increases the number of mitotic CMs (Porrello et al., 2011b). Inhibition of the miR-15 family in the adult mouse and pig reduces the infarcted area after ischemia-reperfusion (Hullinger et al. 2012). Others have performed high-content fluorescence microscopy screening in neonatal rat CMs with a library of 875 miRNA mimics, and have identified a number of miRNAs including miR-199a, as important regulators of CM proliferation. AAV9-mediated overexpression of miR-199a promotes neonatal and adult rat CM proliferation as well as CM regeneration and repair after MI in mice (Eulalio et al. 2012). The same group has further demonstrated that over-expression of miR-199a improves cardiac repair after MI in pigs, but most of the treated pigs died suddenly due to arrhythmia (Gabisonia et al. 2019). Mechanistic studies have shown that miR-199a-3p directly binds to TAOK1, β-TRCP, and cofilin2 to inhibit Hippo signaling, thus leading to YAP activation and inhibition of actin depolymerization (Torrini et al. 2019). Similarly, overexpression of the miRNA cluster miR302-367 in the adult mouse heart down-regulates MST and LATS kinases in the Hippo signaling pathway, thus promoting CM proliferation (Tian et al. 2015). Recently, other microRNAs such as miR-19a/19b, miR-128, and miR-294 have also been reported to regulate CM proliferation in mice (Gao et al. 2019; Huang et al. 2018; Ponnusamy et al. 2019). Several miRNAs including miR-133, miR-101a, miR-99/100, let-7a/c, and miR-26a are critical for heart regeneration in zebrafish as reviewed previously (Zhu et al., 2018b).
In addition to microRNAs, other non-coding RNAs including long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) are also potential regulators of CM proliferation. Bioinformatics analysis has revealed that lncRNA CPR (Cardiomyocyte Proliferation Regulator) is a negative regulator of CM proliferation and regeneration (Ponnusamy et al. 2019). CircRNA is a special kind of noncoding RNA that has a closed circular structure. It is not affected by RNA exonucleases, and its expression is more stable. A recent study has revealed that cirNfix normally inhibits CM proliferation, and thus its knockdown promotes CM proliferation. Consistent with this, the super-enhancer region of cirNfix has binding sites for Meis1, a negative regulator of CM proliferation, and thus regulates the expression of cirNfix (Huang et al. 2019).
The SWI/SNF complex is an ATP-dependent chromatin remodeling complex that changes the accessibility of transcription factors to DNA. It contains different subunits, including the core ATPase subunit Brg1 or Brm. The SWI/SNF subunits play important roles in CM differentiation and development. During zebrafish heart regeneration, brg1, baf60c, and baf180 are induced after injury. Brg1 regulates CM proliferation by interacting with Dnmt3ab to increase methylation of the cdkn1c promoter (Xiao et al. 2016). The expression of Baf60c significantly increases during neonatal mouse heart regeneration, and knockdown of Baf60c suppresses CM proliferation (Nakamura et al. 2016). These studies suggest that injury-induced expression of the SWI/SNF complex modulates chromatin structure and gene transcription during heart regeneration. Others have reported that histone modification of H3K27me3 is essential for the silencing of structural genes, and conditional over-expression of H3.3K27M mutant variants decreases the K27me3 level and as a result, inhibits cardiac regeneration (Ben-Yair et al. 2019).
Transcription factors and regeneration enhancers
Some embryonic cardiac transcriptional factors are re-activated after ventricular resection in zebrafish (Lepilina et al. 2006). Transcriptional factors such as Gata4, Hand2, Stat3, and NF-kB have essential functions during heart regeneration as reviewed previously (Pronobis and Poss 2020). In the neonatal mouse heart, transcription factors Meis1, Gata4, and Tbx20 play important roles in CM proliferation by regulating the expression of cell-cycle genes. Conditional knockout of Meis1 in mouse CMs drives mature CMs to re-enter the cell cycle, thus prolonging the regeneration window beyond P7, while the overexpression of Meis1 inhibits heart regeneration in the neonatal mouse (Mahmoud et al. 2013). The cardiac transcription factor Gata4 regulates neonatal heart regeneration via the paracrine factor FGF16. Conditional knockout of Gata4 leads to the loss of regenerative ability in neonatal mice, and overexpression of Fgf16 via AAV9 in the Gata4-deficient heart partially rescues cardiac hypertrophy and improves heart function after injury (Yu et al. 2016). In addition, Tbx20 inhibits the expression of p21 while activates YAP, BMP, and Akt signaling, resulting in CM proliferation and improved cardiac function (Xiang et al. 2016). RE1 silencing transcription factor, a transcriptional inhibitor of neuronal genes, promotes CM proliferation via inhibiting p21 expression after injury (Zhang et al. 2017). Together, these studies suggest that embryonic cardiac transcriptional factors are re-deployed for cardiac regeneration after injury.
It remains to be addressed how injury triggers CM proliferation and heart regeneration in model systems such as the zebrafish and neonatal mouse heart. It is logically hypothesized that injury signals may trigger chromatin remodeling (such as an active or repressive state regulated by the SWI/SNF complex or histone modification) and cardiac transcription factors are then recruited to bind regulatory elements (such as promoters and enhancers). Recent global analyses of transcriptome and histone modifications (H3K4me3/H3K27ac) have identified early responsive regenerative enhancers in the zebrafish, killifish, and neonatal mouse heart (Quaife-Ryan et al. 2017; Kang et al. 2016; Wang et al. 2020). Comparative transcriptome profiling has revealed that a number of genes are activated during both heart and fin regeneration, among which lepb is strongly induced after injury in zebrafish (Kang et al. 2016). Further analyses have led to identifying the regulatory elements of lepb, which consist of a heart-specific enhancer and a fin-specific enhancer that are active in both the zebrafish and neonatal mouse heart and fins/digits. By creating a biotinylatable H3.3 histone variant in CMs, Poss and colleagues have elegantly performed the histone profiling of putative enhancers with H3.3 occupancies during heart regeneration, and have thus isolated several enhancers that are active in the injured heart (Goldman et al. 2017). Their most recent work applying CRISPR-based deletion of enhancers in zebrafish has further demonstrated that some of the regeneration enhancers are responsible for adjacent gene expression including lepb, although others may regulate distant gene expression (Thompson et al. 2020). By taking an evolutionary approach from killifish to zebrafish, other investigators have identified 49 putative enhancers during fin regeneration by integrating transcriptome and H3K27ac profiling (Wang et al. 2020). Interestingly, a large fraction of these enhancers is activated in the blastema cells of the regenerating fin, suggesting their potential roles in fin regeneration. They have further found that the killifish inhba regeneration enhancer K-IEN and the zebrafish inhba enhancer Z-IEN are active in blastema cells, the essential regenerative cells, while human H-IEN is active in non-regenerative cells. Bioinformatics analysis has revealed multiple AP-1 binding sites in these regeneration enhancers, consistent with recent studies on the potential function of AP-1 family members in initiating organ regeneration (Gehrke et al. 2019; Beisaw et al. 2020). Together, the exciting progress on regeneration enhancers, combined with the identification of regenerative transcription factors, will facilitate our understanding of the complex networks regulating heart regeneration (Table 2).
Perspective on research directions
Inducing CM proliferation has become a promising strategy to develop treatments for heart failure. A large number of studies have shown that cell-cycle regulatory factors, growth factor signaling pathways, miRNAs, transcription factors, and epigenetic modifications stimulate the proliferation of endogenous CMs. In general, injury signals trigger quiescent CMs (mononuclear, polyploid, or multinuclear CMs) into cell-cycle re-entry, via de-differentiation, morphological changes in mitochondria, and reduced cell adhesion, for which reprogramming and mitogenic signals such as Hippo, NRG1/Erbb2/4, FGF, and other signaling pathways are required (Fig. 2). The field has accumulated a mass of data showing that a number of genes and non-coding RNAs are required for CM proliferation, but it remains largely unknown what genetic and epigenetic signals are sufficient to promote CM proliferation and regeneration in non-regenerating hearts such as the adult mammalian heart. To address these unresolved questions, it is necessary to carry out unbiased genetic and/or chemical screens to identify genes and small molecules that promote CM proliferation, such as high-content chemical screens, CRISPR-based genetic screens, and TET-ON based genetic screens (Ma et al. 2017). Recently, a series of studies have succeeded in chemical screening for a variety of biological factors in the mouse and zebrafish (Han et al. 2019; Choi et al. 2013; Magadum et al. 2017). With the rapid development of genomic technology, such as RNA-seq, ATAC-Seq, ChIP-seq, and single-cell sequencing, future studies will continue to decipher the networks that regulate heart regeneration, particularly by investigating the expression and function of putative regenerative enhancers as well as their interactions with transcription factors. Although several genetic and epigenetic factors have been determined to function in CM proliferation, it remains largely unknown how they are coordinated to drive quiescent CMs into cell-cycle re-entry. It has been reported that Hippo/YAP signaling not only interacts with the β-catenin pathway, but also with the PI3K/Akt pathway. Also, NRG1/ErbB2/4 is able to activate both the PI3K/Akt and YAP signaling pathways (Lin et al. 2015; Sudol 2014). Thus, it is very important to address the interplay and mechanisms among the signal pathways that regulate CM proliferation. On the other hand, the current delivery methods for genes and small molecules are mainly systemic delivery, epicardial patches, or direct intramyocardial injection. These delivery methods can also stimulate the proliferation of non-CMs. Therefore, how to improve delivery methods to fine-tune drug release and specifically target CMs awaits further investigation.

Working model of injury-induced cardiomyocyte proliferation. Injury signals trigger the cell-cycle re-entry of CMs, leading to the formation of mononuclear, polynuclear, or polyploidy CMs. De-differentiation signals together with extracellular matrix (ECM) re-organization then regulate the generation of putative de-differentiated CMs, which have evident changes in mitochondrial morphology, disassembled sarcomeres, and reduced cell adhesion. Finally, reprogramming signals and mitogens drive the formation of putative progenitors and cell division
References
Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, et al. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. 1997;100:2722–8.
Aguirre A, Montserrat N, Zacchigna S, Nivet E, Hishida T, Krause MN, et al. In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell. 2014;15:589–604.
Ahuja P, Perriard E, Perriard JC, Ehler E. Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J Cell Sci. 2004;117:3295–306.
Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. 2014;111:8850–5.
Andersen DC, Jensen CH, Baun C, Hvidsten S, Zebrowski DC, Engel FB, et al. Persistent scarring and dilated cardiomyopathy suggest incomplete regeneration of the apex resected neonatal mouse myocardium--a 180 days follow up study. J Mol Cell Cardiol. 2016;90:47–52.
Artap S, Manderfield LJ, Smith CL, Poleshko A, Aghajanian H, See K, et al. Endocardial hippo signaling regulates myocardial growth and cardiogenesis. Dev Biol. 2018;440:22–30.
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382–92.
Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24:1234–45.
Bardot ES, Dubois NC. A watershed finding for heart regeneration. Cell. 2019;176:947–9.
Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch UK, Yifa O, et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–84.
Beauchemin M, Smith A, Yin VP. Dynamic microRNA-101a and Fosab expression controls zebrafish heart regeneration. Development. 2015;142:4026–37.
Beisaw A, Kuenne C, Guenther S, Dallmann J, Wu CC, Bentsen M, et al. AP-1 contributes to chromatin accessibility to promote sarcomere disassembly and Cardiomyocyte protrusion during Zebrafish heart regeneration. Circ Res. 2020;126:1760–78.
Bensimon-Brito A, Ramkumar S, Boezio GLM, Guenther S, Kuenne C, Helker CSM, et al. TGF-β signaling promotes tissue formation during cardiac valve regeneration in adult Zebrafish. Dev Cell. 2020;52:9–20.e7.
Ben-Yair R, Butty VL, Busby M, Qiu Y, Levine SS, Goren A, et al. H3K27me3-mediated silencing of structural genes is required for zebrafish heart regeneration. Development. 2019;146:dev178632.
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, et al. Evidence for Cardiomyocyte renewal in humans. Science. 2009;324:98–102.
Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70.
Bicknell KA, Coxon CH, Brooks G. Forced expression of the cyclin B1-CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem J. 2004;382:411–6.
Bisson JA, Mills B, Paul Helt JC, Zwaka TP, Cohen ED. Wnt5a and Wnt11 inhibit the canonical Wnt pathway and promote cardiac progenitor development via the Caspase-dependent degradation of AKT. Dev Biol. 2015;398:80–96.
Blom JN, Lu X, Arnold P, Feng Q. Myocardial infarction in neonatal mice, a model of cardiac regeneration. J Vis Exp. 2016;111:54100.
Bornhorst D, Xia P, Nakajima H, Dingare C, Herzog W, Lecaudey V, et al. Biomechanical signaling within the developing zebrafish heart attunes endocardial growth to myocardial chamber dimensions. Nat Commun. 2019;10:4113.
Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–86.
Bryant DM, O'Meara CC, Ho NN, Gannon J, Cai L, Lee RT. A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol. 2015;79:315–8.
Cai W, Tan J, Yan J, Zhang L, Cai X, Wang H, et al. Limited regeneration potential with minimal Epicardial progenitor conversions in the neonatal mouse heart after injury. Cell Rep. 2019;28:190–201.e3.
Campa VM, Gutierrez-Lanza R, Cerignoli F, Diaz-Trelles R, Nelson B, Tsuji T, et al. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol. 2008;183:129–41.
Cano-Martinez A, Vargas-Gonzalez A, Guarner-Lans V, Prado-Zayago E, Leon-Oleda M, Nieto-Lima B. Functional and structural regeneration in the axolotl heart (Ambystoma mexicanum) after partial ventricular amputation. Arch Cardiol Mex. 2010;80:79–86.
Chablais F, Jazwinska A. The regenerative capacity of the zebrafish heart is dependent on TGFβ signaling. Development. 2012;139:1921–30.
Chablais F, Veit J, Rainer G, Jazwinska A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev Biol. 2011;11:21.
Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem. 2004;279:35858–66.
Cheng RK, Asai T, Tang H, Dashoush NH, Kara RJ, Costa KD, et al. Cyclin A2 induces cardiac regeneration after myocardial infarction and prevents heart failure. Circ Res. 2007;100:1741–8.
Choi WY, Gemberling M, Wang J, Holdway JE, Shen MC, Karlstrom RO, et al. In vivo monitoring of cardiomyocyte proliferation to identify chemical modifiers of heart regeneration. Development. 2013;140:660–6.
Collesi C, Zentilin L, Sinagra G, Giacca M. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J Cell Biol. 2008;183:117–28.
Croquelois A, Domenighetti AA, Nemir M, Lepore M, Rosenblatt-Velin N, Radtke F, et al. Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J Exp Med. 2008;205:3173–85.
Cui M, Wang Z, Chen K, Shah AM, Tan W, Duan L, et al. Dynamic transcriptional responses to injury of regenerative and non-regenerative Cardiomyocytes revealed by single-nucleus RNA sequencing. Dev Cell. 2020;53:102–16 e8.
Darehzereshki A, Rubin N, Gamba L, Kim J, Fraser J, Huang Y, et al. Differential regenerative capacity of neonatal mouse hearts after cryoinjury. Dev Biol. 2015;399:91–9.
Das S, Goldstone AB, Wang H, Farry J, D'Amato G, Paulsen MJ, et al. A unique collateral artery development program promotes neonatal heart regeneration. Cell. 2019;176:1128–42.e18.
Di Stefano V, Giacca M, Capogrossi MC, Crescenzi M, Martelli F. Knockdown of cyclin-dependent kinase inhibitors induces cardiomyocyte re-entry in the cell cycle. J Biol Chem. 2011;286:8644–54.
Dogra D, Ahuja S, Kim HT, Rasouli SJ, Stainier DYR, Reischauer S. Opposite effects of Activin type 2 receptor ligands on cardiomyocyte proliferation during development and repair. Nat Commun. 2017;8:1902.
Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012;31:429–42.
Duncan EM, Sanchez AA. Regulation of genomic output and (Pluri) potency in regeneration. Annu Rev Genet. 2019;53:327–46.
D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–38.
Ebelt H, Zhang Y, Kampke A, Xu J, Schlitt A, Buerke M, et al. E2F2 expression induces proliferation of terminally differentiated cardiomyocytes in vivo. Cardiovasc Res. 2008;80:219–26.
Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–87.
Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–81.
Fang Y, Gupta V, Karra R, Holdway JE, Kikuchi K, Poss KD. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2013;110:13416–21.
Felician G, Collesi C, Lusic M, Martinelli V, Ferro MD, Zentilin L, et al. Epigenetic modification at notch responsive promoters blunts efficacy of inducing notch pathway reactivation after myocardial infarction. Circ Res. 2014;115:636–49.
Flinn MA, Jeffery BE, O'Meara CC, Link BA. Yap is required for scar formation but not myocyte proliferation during heart regeneration in zebrafish. Cardiovasc Res. 2019;115:570–7.
Fukui H, Terai K, Nakajima H, Chiba A, Fukuhara S, Mochizuki N. S1P-Yap1 signaling regulates endoderm formation required for cardiac precursor cell migration in zebrafish. Dev Cell. 2014;31:128–36.
Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019;569:418–22.
Galvez-Santisteban M, Chen D, Zhang R, Serrano R, Nguyen C, Zhao L, et al. Hemodynamic-mediated endocardial signaling controls in vivo myocardial reprogramming. Elife. 2019;8:e44816.
Gancz D, Raftrey BC, Perlmoter G, Marin-Juez R, Semo J, Matsuoka RL, et al. Distinct origins and molecular mechanisms contribute to lymphatic formation during cardiac growth and regeneration. Elife. 2019;8:e44153.
Gao F, Kataoka M, Liu N, Liang T, Huang ZP, Gu F, et al. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat Commun. 2019;10:1802.
Gao R, Zhang J, Cheng L, Wu X, Dong W, Yang X, et al. A phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. 2010;55:1907–14.
Gehrke AR, Neverett E, Luo YJ, Brandt A, Ricci L, Hulett RE, et al. Acoel genome reveals the regulatory landscape of whole-body regeneration. Science. 2019;363:eaau6173.
Gemberling M, Karra R, Dickson AL, Poss KD. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife. 2015;4:e05871.
Gessert S, Kuhl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ Res. 2010;107:186–99.
Goldman JA, Kuzu G, Lee N, Karasik J, Gemberling M, Foglia MJ, et al. Resolving heart regeneration by replacement histone profiling. Dev Cell. 2017;40:392–404 e5.
González-Rosa JM, Martín V, Peralta M, Torres M, Mercader N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development. 2011;138:1663–74.
Gonzalez-Rosa JM, Peralta M, Mercader N. Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev Biol. 2012;370:173–86.
Gonzalez-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, et al. Myocardial Polyploidization creates a barrier to heart regeneration in Zebrafish. Dev Cell. 2018;44:433–46 e7.
Gupta V, Gemberling M, Karra R, Rosenfeld GE, Evans T, Poss KD. An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr Biol. 2013;23:1221–7.
Gupta V, Poss KD. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature. 2012;484:479–84.
Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. 2017;8:310.
Han C, Nie Y, Lian H, Liu R, He F, Huang H, et al. Acute inflammation stimulates a regenerative response in the neonatal mouse heart. Cell Res. 2015;25:1137–51.
Han P, Zhou XH, Chang N, Xiao CL, Yan S, Ren H, et al. Hydrogen peroxide primes heart regeneration with a derepression mechanism. Cell Res. 2014;24:1091–107.
Han Y, Chen A, Umansky KB, Oonk KA, Choi WY, Dickson AL, et al. Vitamin D stimulates Cardiomyocyte proliferation and controls organ size and regeneration in Zebrafish. Dev Cell. 2019;48:853–63.e5.
Harrison MR, Feng X, Mo G, Aguayo A, Villafuerte J, Yoshida T, et al. Late developing cardiac lymphatic vasculature supports adult zebrafish heart function and regeneration. Elife. 2019;8:e42762.
Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, et al. Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res. 2016;118:216–21.
He L, Huang X, Kanisicak O, Li Y, Wang Y, Li Y, et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J Clin Invest. 2017;127:2968–81.
Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, et al. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683–90.
Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61.
Heallen TR, Kadow ZA, Kim JH, Wang J, Martin JF. Stimulating Cardiogenesis as a treatment for heart failure. Circ Res. 2019;124:1647–57.
Hermans KC, Daskalopoulos EP, Blankesteijn WM. Interventions in Wnt signaling as a novel therapeutic approach to improve myocardial infarct healing. Fibrogenesis Tissue Repair. 2012;5:16.
Hirose K, Payumo AY, Cutie S, Hoang A, Zhang H, Guyot R, et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science. 2019;364:184–8.
Honkoop H, de Bakker DEM, Aharonov A, Kruse F, Shakked A, Nguyen PD, et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. 2019;8:e50163.
Huang S, Li X, Zheng H, Si X, Li B, Wei G, et al. Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation. 2019;139:2857–76.
Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, et al. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat Commun. 2018;9:700.
Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2012;110:71–81.
Ikenishi A, Okayama H, Iwamoto N, Yoshitome S, Tane S, Nakamura K, et al. Cell cycle regulation in mouse heart during embryonic and postnatal stages. Develop Growth Differ. 2012;54:731–8.
Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua BJ. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9.
Jopling C, Suñe G, Morera C, Izpisua Belmonte JC. p38α MAPK regulates myocardial regeneration in zebrafish. Cell Cycle. 2012;11:1195–201.
Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogórek B, et al. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–15.
Kang J, Hu J, Karra R, Dickson AL, Tornini VA, Nachtrab G, et al. Modulation of tissue repair by regeneration enhancer elements. Nature. 2016;532:201–6.
Karra R, Foglia MJ, Choi WY, Belliveau C, DeBenedittis P, Poss KD. Vegfaa instructs cardiac muscle hyperplasia in adult zebrafish. Proc Natl Acad Sci U S A. 2018;115:8805–10.
Karra R, Knecht AK, Kikuchi K, Poss KD. Myocardial NF-kappaB activation is essential for zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2015;112:13255–60.
Keepers B, Liu J, Qian L. What's in a cardiomyocyte - and how do we make one through reprogramming? Biochim Biophys Acta Mol Cell Res. 2020;1867:118464.
Kikuchi K. New function of zebrafish regulatory T cells in organ regeneration. Curr Opin Immunol. 2020;63:7–13.
Kikuchi K, Gupta V, Wang J, Holdway JE, Wills AA, Fang Y, et al. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development. 2011a;138:2895–902.
Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, et al. Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature. 2010;464:601–5.
Kikuchi K, Holdway Jennifer E, Major Robert J, Blum N, Dahn Randall D, Begemann G, et al. Retinoic acid production by endocardium and Epicardium is an injury response essential for Zebrafish heart regeneration. Dev Cell. 2011b;20:397–404.
Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM, et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature. 2015;523:226–30.
Kirshenbaum LA, Abdellatif M, Chakraborty S, Schneider MD. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol. 1996;179:402–11.
Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dube KN, et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522:62–7.
Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66.
Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–35.
Lai SL, Marin-Juez R, Moura PL, Kuenne C, Lai J, Tsedeke AT, et al. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. Elife. 2017;6:e25605.
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111:16029–34.
Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. 2017;550:260–4.
Lenihan DJ, Anderson SA, Lenneman CG, Brittain E, Muldowney JAS 3rd, Mendes L, et al. A phase I, single ascending dose study of Cimaglermin Alfa (Neuregulin 1β3) in patients with systolic dysfunction and heart failure. JACC Basic Transl Sci. 2016;1:576–86.
Lepilina A, Coon A, Kikuchi K, Holdway J, Roberts R, Burns C, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127:607–19.
Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–46.
Li L, Tao G, Hill MC, Zhang M, Morikawa Y, Martin JF. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development. 2018b;145.
Li X, Lu Q, Peng Y, Geng F, Shao X, Zhou H, et al. Primary cilia mediate Klf2-dependant notch activation in regenerating heart. Protein Cell. 2020;11:433–45.
Li Y, He L, Huang X, Bhaloo SI, Zhao H, Zhang S, et al. Genetic lineage tracing of Nonmyocyte population by dual Recombinases. Circulation. 2018a;138:793–805.
Li Y, Lv Z, He L, Huang X, Zhang S, Zhao H, et al. Genetic tracing identifies early segregation of the Cardiomyocyte and Nonmyocyte lineages. Circ Res. 2019;125:343–55.
Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–8.
Lien C, Schebesta M, Makino S, Weber G, Keating M. Gene expression analysis of zebrafish heart regeneration. PLoS Biol. 2006;4:e260.
Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–63.
Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, et al. Pi3kcb links hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116:35–45.
Liu P, Zhong TP. MAPK/ERK signalling is required for zebrafish cardiac regeneration. Biotechnol Lett. 2017;39:1069–77.
Liu X, De la Cruz E, Gu X, Balint L, Oxendine-Burns M, Terrones T, et al. Lymphoangiocrine signals promote cardiac growth and repair. Nature. 2020;588:705–11.
Liu Z, Yue S, Chen X, Kubin T, Braun T. Regulation of cardiomyocyte polyploidy and multinucleation by CyclinG1. Circ Res. 2010;106:1498–506.
Ma Z, Zhu P, Pang M, Guo L, Chang N, Zheng J, et al. A novel inducible mutagenesis screen enables to isolate and clone both embryonic and adult zebrafish mutants. Sci Rep. 2017;7:10381.
MacLellan WR, Garcia A, Oh H, Frenkel P, Jordan MC, Roos KP, et al. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Biol. 2005;25:2486–97.
Magadum A, Ding Y, He L, Kim T, Vasudevarao MD, Long Q, et al. Live cell screening platform identifies PPARdelta as a regulator of cardiomyocyte proliferation and cardiac repair. Cell Res. 2017;27:1002–19.
Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–53.
Mahmoud AI, O'Meara CC, Gemberling M, Zhao L, Bryant DM, Zheng R, et al. Nerves regulate Cardiomyocyte proliferation and heart regeneration. Dev Cell. 2015;34:387–99.
Mahmoud AI, Porrello ER, Kimura W, Olson EN, Sadek HA. Surgical models for cardiac regeneration in neonatal mice. Nat Protoc. 2014;9:305–11.
Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF, Molkentin JD. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. 2008;283:31246–55.
Malek MM, Kattih B, Grund A, Froese N, Korf-Klingebiel M, Gigina A, et al. The transcription factor GATA4 promotes myocardial regeneration in neonatal mice. EMBO Mol Med. 2017;9:265–79.
Marin-Juez R, Marass M, Gauvrit S, Rossi A, Lai SL, Materna SC, et al. Fast revascularization of the injured area is essential to support zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2016;113:11237–42.
Mercer SE, Odelberg SJ, Simon HG. A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration. Dev Biol. 2013;382:457–69.
Missinato MA, Saydmohammed M, Zuppo DA, Rao KS, Opie GW, Kuhn B, et al. Dusp6 attenuates Ras/MAPK signaling to limit zebrafish heart regeneration. Development. 2018;145:dev157206.
Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, et al. Regulation of cell cycle to stimulate adult Cardiomyocyte proliferation and cardiac regeneration. Cell. 2018;173:104–16 e12.
Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110:1446–51.
Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, et al. YAP partially reprograms chromatin accessibility to directly induce adult Cardiogenesis in vivo. Dev Cell. 2019;48:765–79.e7.
Moore-Morris T, van Vliet PP, Andelfinger G, Puceat M. Role of epigenetics in cardiac development and congenital diseases. Physiol Rev. 2018;98:2453–75.
Morikawa Y, Heallen T, Leach J, Xiao Y, Martin JF. Dystrophin-glycoprotein complex sequesters yap to inhibit cardiomyocyte proliferation. Nature. 2017;547:227–31.
Murry CE, Reinecke H, Pabon LM. Regeneration gaps: observations on stem cells and cardiac repair. J Am Coll Cardiol. 2006;47:1777–85.
Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond). 2008;115:203–18.
Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–7.
Nakamura R, Koshiba-Takeuchi K, Tsuchiya M, Kojima M, Miyazawa A, Ito K, et al. Expression analysis of Baf60c during heart regeneration in axolotls and neonatal mice. Develop Growth Differ. 2016;58:367–82.
Norman S, Riley PR. Anatomy and development of the cardiac lymphatic vasculature: its role in injury and disease. Clin Anat. 2016;29:305–15.
Nusse R, Varmus H. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J. 2012;31:2670–84.
Oberpriller J, Oberpriller JC. Mitosis in adult newt ventricle. J Cell Biol. 1971;49:560–3.
Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187:249–53.
Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–8.
Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet. 2017;49:1346–53.
Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. 2012;23:450–7.
Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, et al. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. 2015;7:281ra45.
Ponnusamy M, Liu F, Zhang YH, Li RB, Zhai M, Liu F, et al. Long noncoding RNA CPR (Cardiomyocyte proliferation regulator) regulates Cardiomyocyte proliferation and cardiac repair. Circulation. 2019;139:2668–84.
Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011b;109:670–9.
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011a;331:1078–80.
Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187–92.
Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–90.
Pronobis MI, Poss KD. Signals for cardiomyocyte proliferation during zebrafish heart regeneration. Curr Opin Physiol. 2020;14:78–85.
Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, et al. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017;136:1123–39.
Raya A, Koth CM, Buscher D, Kawakami Y, Itoh T, Raya RM, et al. Activation of notch signaling pathway precedes heart regeneration in zebrafish. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11889–95.
Reuter S, Soonpaa MH, Firulli AB, Chang AN, Field LJ. Recombinant neuregulin 1 does not activate cardiomyocyte DNA synthesis in normal or infarcted adult mice. PLoS One. 2014;9:e115871.
Sadek H, Olson EN. Toward the goal of human heart regeneration. Cell Stem Cell. 2020;26:7–16.
Sallin P, de Preux CA, Duruz V, Pfefferli C, Jazwinska A. A dual epimorphic and compensatory mode of heart regeneration in zebrafish. Dev Biol. 2015;399:27–40.
Sanchez-Iranzo H, Galardi-Castilla M, Minguillon C, Sanz-Morejon A, Gonzalez-Rosa JM, Felker A, et al. Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat Commun. 2018a;9:428.
Sanchez-Iranzo H, Galardi-Castilla M, Sanz-Morejon A, Gonzalez-Rosa JM, Costa R, Ernst A, et al. Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc Natl Acad Sci U S A. 2018b;115:4188–93.
Schindler YL, Garske KM, Wang J, Firulli BA, Firulli AB, Poss KD, et al. Hand2 elevates cardiomyocyte production during zebrafish heart development and regeneration. Development. 2014;141:3112–22.
Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 2011;144:782–95.
Schnabel K, Wu C-C, Kurth T, Weidinger G. Regeneration of Cryoinjury induced necrotic heart lesions in Zebrafish is associated with Epicardial activation and Cardiomyocyte proliferation. PLoS One. 2011;6:e18503.
Sdek P, Zhao P, Wang Y, Huang CJ, Ko CY, Butler PC, et al. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol. 2011;194:407–23.
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–6.
Sereti KI, Nguyen NB, Kamran P, Zhao P, Ranjbarvaziri S, Park S, et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat Commun. 2018;9:754.
Shapiro SD, Ranjan AK, Kawase Y, Cheng RK, Kara RJ, Bhattacharya R, et al. Cyclin A2 induces cardiac regeneration after myocardial infarction through cytokinesis of adult cardiomyocytes. Sci Transl Med. 2014;6:224ra27.
Srivastava D, DeWitt N. In vivo cellular reprogramming: the next generation. Cell. 2016;166:1386–96.
Stoick-Cooper CL, Weidinger G, Riehle KJ, Hubbert C, Major MB, Fausto N, et al. Distinct Wnt signaling pathways have opposing roles in appendage regeneration. Development. 2007;134:479–89.
Sturzu AC, Rajarajan K, Passer D, Plonowska K, Riley A, Tan TC, et al. Fetal mammalian heart generates a robust compensatory response to cell loss. Circulation. 2015;132:109–21.
Sudol M. Neuregulin 1-activated ERBB4 as a "dedicated" receptor for the hippo-YAP pathway. Sci Signal. 2014;7:pe29.
Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, et al. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat Commun. 2015;6:8701.
Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–6.
Tamamori-Adachi M, Goto I, Yamada K, Kitajima S. Differential regulation of cyclin D1 and D2 in protecting against cardiomyocyte proliferation. Cell Cycle. 2008;7:3768–74.
Tanaka EM, Reddien PW. The cellular basis for animal regeneration. Dev Cell. 2011;21:172–85.
Tane S, Ikenishi A, Okayama H, Iwamoto N, Nakayama KI, Takeuchi T. CDK inhibitors, p21(Cip1) and p27(Kip1), participate in cell cycle exit of mammalian cardiomyocytes. Biochem Biophys Res Commun. 2014;443:1105–9.
Tang J, Zhang H, He L, Huang X, Li Y, Pu W, et al. Genetic fate mapping defines the vascular potential of Endocardial cells in the adult heart. Circ Res. 2018;122:984–93.
Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–23.
Thompson JD, Ou J, Lee N, Shin K, Cigliola V, Song L, et al. Identification and requirements of enhancers that direct gene expression during zebrafish fin regeneration. Development. 2020;147:dev191262.
Tian Y, Liu Y, Wang T, Zhou N, Kong J, Chen L, et al. A microRNA-hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med. 2015;7:279ra38.
Torrini C, Cubero RJ, Dirkx E, Braga L, Ali H, Prosdocimo G, et al. Common regulatory pathways mediate activity of MicroRNAs inducing Cardiomyocyte proliferation. Cell Rep. 2019;27:2759–71.e5.
Tzahor E, Poss KD. Cardiac regeneration strategies: staying young at heart. Science. 2017;356:1035–9.
van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, et al. C-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–41.
von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, et al. YAP1, the nuclear target of hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–9.
von Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R. E2F-1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclin-dependent kinases in the presence of insulin-like growth factor I. Circ Res. 1999;85:128–36.
Vunjak-Novakovic G, Lui KO, Tandon N, Chien KR. Bioengineering heart muscle: a paradigm for regenerative medicine. Annu Rev Biomed Eng. 2011;13:245–67.
Wang J, Panáková D, Kikuchi K, Holdway JE, Gemberling M, Burris JS, et al. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development. 2011;138:3421–30.
Wang W, Hu CK, Zeng A, Alegre D, Hu D, Gotting K, et al. Changes in regeneration-responsive enhancers shape regenerative capacities in vertebrates. Science. 2020;369:eaaz3090.
Wei K, Serpooshan V, Hurtado C, Diez-Cunado M, Zhao M, Maruyama S, et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature. 2015;525:479–85.
White IA, Gordon J, Balkan W, Hare JM. Sympathetic Reinnervation is required for mammalian cardiac regeneration. Circ Res. 2015;117:990–4.
Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol. 2011;354:67–76.
Woo YJ, Panlilio CM, Cheng RK, Liao GP, Atluri P, Hsu VM, et al. Therapeutic delivery of cyclin A2 induces myocardial regeneration and enhances cardiac function in ischemic heart failure. Circulation. 2006;114:I206–13.
Wu Y, Zhou L, Liu H, Duan R, Zhou H, Zhang F, et al. LRP6 downregulation promotes cardiomyocyte proliferation and heart regeneration. Cell Res. 2020. https://doi.org/10.1038/s41422-020-00411-7.
Xiang FL, Guo M, Yutzey KE. Overexpression of Tbx20 in adult Cardiomyocytes promotes proliferation and improves cardiac function after myocardial infarction. Circulation. 2016;133:1081–92.
Xiao C, Gao L, Hou Y, Xu C, Chang N, Wang F, et al. Chromatin-remodelling factor Brg1 regulates myocardial proliferation and regeneration in zebrafish. Nat Commun. 2016;7:13787.
Xie S, Fu W, Yu G, Hu X, Lai KS, Peng X, et al. Discovering small molecules as Wnt inhibitors that promote heart regeneration and injury repair. J Mol Cell Biol. 2020;12:42–54.
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110:13839–44.
Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, et al. Regulation of insulin-like growth factor signaling by yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70.
Yang D, Fu W, Li L, Xia X, Liao Q, Yue R, et al. Therapeutic effect of a novel Wnt pathway inhibitor on cardiac regeneration after myocardial infarction. Clin Sci (Lond). 2017;131:2919–32.
Ye L, D'Agostino G, Loo SJ, Wang CX, Su LP, Tan SH, et al. Early regenerative capacity in the porcine heart. Circulation. 2018;138:2798–808.
Yin VP, Lepilina A, Smith A, Poss KD. Regulation of zebrafish heart regeneration by miR-133. Dev Biol. 2012;365:319–27.
Yu W, Huang X, Tian X, Zhang H, He L, Wang Y, et al. GATA4 regulates Fgf16 to promote heart repair after injury. Development. 2016;143:936–49.
Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107:1304–12.
Zhang D, Wang Y, Lu P, Wang P, Yuan X, Yan J, et al. REST regulates the cell cycle for cardiac development and regeneration. Nat Commun. 2017;8:1979.
Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, et al. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature. 2013;498:497–501.
Zhao L, Ben-Yair R, Burns CE, Burns CG. Endocardial notch signaling promotes Cardiomyocyte proliferation in the regenerating Zebrafish heart through Wnt pathway antagonism. Cell Rep. 2019;26:546–54 e5.
Zhao L, Borikova AL, Ben-Yair R, Guner-Ataman B, MacRae CA, Lee RT, et al. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proceed Nat Acad Sci. 2014;111:1403–8.
Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121:1894–904.
Zhu W, Zhang E, Zhao M, Chong Z, Fan C, Tang Y, et al. Regenerative potential of neonatal porcine hearts. Circulation. 2018a;138:2809–16.
Zhu X, Xiao C, Xiong JW. Epigenetic regulation of organ regeneration in Zebrafish. J Cardiovasc Dev Dis. 2018b;5:57.
Acknowledgments
The authors thank Dr. IC Bruce for reading and editing the manuscript.
Funding
This paper was supported by grants from the National Key Research & Development Program of China (2018YFA080051) and the National Natural Science Foundation of China (31730061 and 81870198).
Author information
Authors and Affiliations
Contributions
LZ, JD, ZW, and QZ wrote the paper, XZ and JWX edited and revised the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, L., Du, J., Wang, Z. et al. Molecular regulation of myocardial proliferation and regeneration. Cell Regen 10, 13 (2021). https://doi.org/10.1186/s13619-021-00075-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13619-021-00075-7