
Abstract
Background
Previous studies have shown that bone morphogenetic protein 9 (BMP9) is almost exclusively produced in the liver and reaches tissues throughout the body as a secreted protein. However, the mechanism of BMP9 action and its role in aging-associated liver injury and inflammation are still unclear.
Results
Aging significantly aggravates acetaminophen (APAP)-induced acute liver injury (ALI). Increased expression of CCAAT/enhancer binding protein α (C/EBPα) and BMP9 was identified in aged livers and in hepatocytes and macrophages (MФs) isolated from aged mice. Further analysis revealed that excess BMP9 was directly related to APAP-induced hepatocyte injury and death, as evidenced by activated drosophila mothers against decapentaplegic protein 1/5/9 (SMAD1/5/9) signaling, an increased dead cell/total cell ratio, decreased levels of ATG3 and ATG7, blocked autophagy, increased senescence‐associated beta‐galactosidase (SA‐β‐Gal) activity, and a higher rate of senescence‐associated secretory phenotype (SASP) acquisition. In contrast, Bmp9 knockout (Bmp9−/−) partially alleviated the aforementioned manifestations of BMP9 overexpression. Moreover, BMP9 expression was found to be regulated by C/EBPα in vitro and in vivo. Notably, BMP9 also downregulated autophagy through its effect on autophagy-related genes (ATG3 and ATG7) in MΦs, which was associated with aggravated liver injury and SASP acquisition.
Conclusions
In summary, the present study highlights the crucial roles played by C/EBPα-BMP9 crosstalk and provides insights into the interrelationship between hepatocytes and MΦs during acute liver injury.
Similar content being viewed by others


Background
The liver plays a crucial role in the catabolism of nutrients and toxins and is considered to be a vital barrier against complex pathogenic factors entering the body from the external environment [1]. Physiologically, liver injury can be repaired by hepatocytes in young and healthy livers, as they contribute to the recovery of liver mass and function [2]. However, after excessive hepatocyte necrosis, the liver loses its regenerative capacity, leading to acute liver injury (ALI) and sometimes acute liver failure (ALF). The loss of liver regeneration capacity and injury resistance are the most prominent aging-associated alterations [3]. The aged liver undergoes extensive histological and molecular biological changes, such as altered hepatocyte morphology, reduced mitochondrial quantity and quality, and an increased proportion of multinucleated cells [4, 5]. Moreover, a typical feature of aging is a long-term chronic inflammatory response, and the aged liver produces a large amount of senescence‐associated secretory phenotype (SASP) factors, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), IL-6, and monocyte chemoattractant protein-1 (MCP-1) [6]. These alterations affect the morphology and normal physiological functions of the liver; therefore, the aged liver is sensitive to various factors that cause disease, including exogenous pathogenic factors, leading to a series of complex clinical problems.
Hepatocytes and abundant nonparenchymal cells constitute a complex intrahepatic microenvironment. The high rate of macrophage (MФ) recruitment to the liver is a hallmark feature of liver injury [7, 8], and the roles of these MФs depend on the recognition of relevant signals that drive their polarization in association with inflammation, such as cytokines, chemokines, and damage-associated molecular patterns (DAMPs) [9]. In the liver, an appropriate degree of inflammation helps to stimulate hepatocyte regeneration and self-repair, but excessive inflammation induces downstream proinflammatory cascades that promote hepatocyte injury and death [10]. Notably, MΦs can be reprogrammed to adapt to the body challenged by different immune environments, and interactions MΦs and hepatocytes undergo a plethora of interactions [11, 12]. Although innate immune responses represented by MΦs have been suggested to be essential in liver defense mechanisms, aging-associated responses of hepatic immune and nonimmune cells are not fully understood.
The bone morphogenetic protein (BMP) family is a subfamily of the transforming growth factor-beta (TGF-β) superfamily. BMP9, also known as growth differentiation factor 2 (GDF2), is a secretory protein in the BMP family and is expressed almost exclusively in the liver, where it acts in a paracrine and autocrine manner [13, 14]. In the liver, BMP9 is thought to inhibit mainly hepatocyte proliferation and to promote liver fibrosis [15]. Notably, BMP9 enhanced the proinflammatory phenotypic transformation of MΦs through an NF-κB-dependent pathway and promoted methionine- and choline-deficient diet-induced nonalcoholic steatohepatitis in mice [16]. Moreover, BMP2, a member of the BMP family, has been reported to promote colon cancer progression through the macroautophagy/autophagy pathway. However, few reports on the effect of BMP9 on autophagy in the liver are available, and the overall regulatory roles played by BMP9 in mouse liver aging, such as its effect on autophagy, hepatocyte survival after injury, and the regulation of the immune-inflammatory response, are not fully understood.
The present study revealed a C/EBPα-BMP9 axis in the liver. We investigated the cause of the increased BMP9 level in the mouse liver during aging and the potential related mechanisms in hepatocytes and MΦs. The aged liver produces excess BMP9, which downregulates ATG3 and ATG7 expression, inhibits autophagy, promotes hepatocyte injury, and drives MΦ proinflammatory activation, further aggravating liver injury. Moreover, BMP9 deletion relieved autophagy suppression, reduced proinflammatory SASP production, and attenuated acetaminophen (APAP)-induced acute liver injury (APAP-ALI) in vivo and in vitro. In addition, the expression of BMP9 was regulated by C/EBPα, which was abundantly expressed in the aged liver, possibly explaining the reason for the great increase in BMP9 expression in the aged liver.
Results
Aging accelerates APAP-ALI and liver inflammation
We first examined the effect of aging on APAP-ALI. The analysis revealed that aging significantly aggravates liver injury induced by APAP, as manifested by increased serum ALT and AST levels (Fig. 1A) and increases in necrotic tissue areas, as indicated by H&E staining (Fig. 1B). Moreover, the number of the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labelling (TUNEL) positive cells was significantly increased in liver tissue sections of aged mice (Fig. 1C). In addition, increased MΦ infiltration was found in the livers of aged mice after APAP-ALI had been induced (Fig. 1D). Consistent with these observations, aging promoted the expression of proinflammatory cytokines, such as SASP components (Il1b, Il6, and Tnfa), and decreased the expression of the anti-inflammatory cytokine Il10 in the liver (Fig. 1E). The severity of APAP-induced liver injury was dose dependent. In a survival study, the mortality was significantly increased in the aged mice that received a higher dose of APAP (500 mg/kg) (Fig. 1F). These results indicate that aging greatly exacerbates APAP-ALI and this increase in ALI severity is accompanied by increased infiltration of MΦs and higher proinflammatory cytokine expression.

Aging accelerates APAP-ALI and inflammation. Young and aged mice were administered APAP (300 mg/kg) or PBS and analyzed 24 h later. A Serum ALT and AST levels of the young and aged mice treated with PBS or APAP. B Representative images showing H&E staining liver slices from young and aged mice treated with PBS or APAP. C Representative images showing TUNEL staining in the liver slices from young and aged mice treated with PBS or APAP. Quantitative data are shown in the right panel. D Representative images showing IHC staining for F4/80 in liver slices from young and aged mice treated with PBS or APAP. Quantitative data are shown in the right panel. E mRNA expression (Il1b, Il6, Tnfa, and Il10) in the livers from young and aged mice treated with PBS or APAP as measured by RT‒qPCR. The average target gene/Gapdh ratios of different experimental groups relative to the control group are reported. F Survival curves of young and aged mice treated with APAP when the dose of APAP was increased to 500 mg/kg. *p < 0.05, **p < 0.01, and ***p < 0.001
BMP9 promotes hepatocyte injury and senescence
RT‒qPCR- and immunoblot-based measurements revealed that the expression of both Bmp9 and Cebpa was increased in the liver tissue of aged mice (Fig. 2A, B). Since BMP9 is a secreted protein and enriched in the aged liver, we used mouse BMP9 recombinant protein (Rm-BMP9) to investigate the effect of BMP9 on APAP-treated hepatocytes. With prolonged APAP treatment in vitro, the ratio of dead-to-live hepatocytes after Rm-BMP9 treatment was significantly higher than that after APAP treatment alone, indicating that BMP9 might play a direct role in promoting hepatocyte injury induced by APAP (Fig. 2C). We further explored the mechanism by which BMP9 aggravates hepatocyte injury in vitro. Increased phosphorylation of SMAD1/5/9, representing the activation of BMP signaling, suggested that BMP9 may have activated downstream signaling cascades in hepatocytes after APAP-induced injury (Fig. 2D). Additionally, the activation of SMAD1/5/9 was increased in the livers of the aged mice, and the phosphorylation rates of SMAD1/5/9 were decreased in the livers of aged Bmp9-knockout (Bmp9−/−) mice (Additional file 1: Figure S1A).

BMP9 promotes hepatocyte injury and senescence. A, B mRNA (A) and protein (B) expression in the livers from young and aged mice. (C–E) Primary hepatocytes with or without Rm-BMP9 addition were treated with APAP, and 1 h, 3 h, 6 h, 12 h and 24 h later, we measured the proportion of APAP-induced hepatocyte death to normal growing primary hepatocytes (C), the protein expression levels of SMAD1/5/9 and P-SMAD1/5/9 (D), and the protein expression levels of ATG3, ATG7, p62 and LC3 I/II (E). F, G Rm-BMP9 was injected into the tail vein, and the APAP-ALI model (300 mg/kg) was established 1 h later. Serum and liver tissue were collected from each group of mice 24 h after model establishment. We measured the serum ALT and AST levels (F), and representative images showing H&E staining (G) in each group are shown. H, I APAP-ALI model (300 mg/kg) was established with young and aged WT mice, as well as with young and aged Bmp9−/− mice. Serum and liver tissue were collected from each group of mice 24 h later. We measured the serum ALT and AST levels (H), and representative images showing H&E staining (I) in each group are shown. The average target gene/Gapdh ratios of different experimental groups to the control group are reported. *p < 0.05, **p < 0.01, and ***p < 0.001
Autophagy-related 3 (ATG3, with E2-like enzyme activity) and ATG7 (with E1-like enzyme activity) are key molecules in autophagy, mediating the transformation of microtubule-associated protein 1 light chain 3 II (MAP1LC3 II/LC3 II) and autophagosome maturation [17, 18]. Notably, Rm-BMP9 downregulated ATG3 and ATG7 expression and significantly decreased the transformation of LC3 I to LC3 II in APAP-injured hepatocytes, results that were consistent with the accumulation of SQSTM1/p62 (Fig. 2E).
These results illustrate the aggravating effect of BMP9 on aging-associated APAP-ALI in vitro, and therefore, we further investigated the effect of BMP9 on APAP-ALI in vivo by injecting Rm-BMP9 into mice. The serum ALT/AST levels (Fig. 2F) and H&E staining results (Fig. 2G) indicated significantly increased APAP-ALI severity, and the number of TUNEL- stained cells was increased (Additional file 1: Figure S1B). Then, we found that Rm-BMP9 promoted the expression of Ilb, Il6 and Tnfa in the mouse livers (Additional file 1: Figure S1C). Consistent with the results obtained from exogenous supplementation of BMP9 in mice, Bmp9 deletion reduced the severity of the liver injury in both young and aged mice (Fig. 2H, I). Additionally, the number of TUNEL-stained cells in the livers of young and aged mice treated with APAP was significantly decreased in the Bmp9−/− groups (Additional file 1: Figure S1D). RT‒qPCR assays revealed that Bmp9 deletion reduced the expression of proinflammatory factors (Ilb, Il6 and Tnfa) in liver tissue (Additional file 1: Figure S1E).
These results suggest that BMP9 inhibits hepatocyte autophagy through ATG3 and ATG7, thereby regulating the severity of APAP-ALI. The high level of BMP9 protein associated with aging may be an important factor in the aggravation of APAP-ALI during aging.
The expression of BMP9 is regulated by C/EBPα in vitro
Because the cell type that expresses BMP9 has not been clarified in previous studies, we sought to visualize the expression and distribution of BMP9 in aged livers. Therefore, we performed immunofluorescence (IF) staining for F4/80 and BMP9 in liver tissues of young and aged mice. In uninjured livers, BMP9 was mainly localized to F4/80-positive cells, and BMP9 expression was higher in aged mouse livers (Fig. 3A). In mice with APAP-ALI, BMP9 accumulated in injured area, BMP9 expression localized to F4/80-positive cells was increased, and BMP9 expression, in general, was identified in hepatocytes (Fig. 3A). Then, we isolated hepatocytes and hepatic MΦs from young and aged mice. We found that BMP9 expression was markedly upregulated in both the hepatocytes and hepatic MΦs from the aged mice (Fig. 3B, C). Notably, we also detected increased C/EBPα and decreased ATG3 and ATG7 levels in the isolated hepatocytes and hepatic MΦs from the aged mice (Fig. 3B, C).

BMP9 expression is regulated by C/EBPα in vitro. A IF staining for BMP9 and F4/80 in liver slices from young and aged mice treated with PBS or APAP (300 mg/kg) for 24 h. B, C Primary hepatocytes and MΦs were isolated from young and aged mice by in situ perfusion of mouse livers. Protein expression levels of BMP9, C/EBPα, ATG3 and ATG7 in mouse primary hepatocytes (B) and hepatic MΦs (C). D–G To study the regulation of BMP9 by C/EBPα in MΦs, we altered Cebpa expression in iBMDMs. D Protein expression levels of C/EBPα and BMP9 after overexpressing Cebpa. E Protein expression levels of C/EBPα and BMP9 after inhibiting Cebpa with or without treatment with the APAP-treated AML-12 cell supernatant (Sup). F Protein expression levels of CD206 and iNOS after overexpressing Cebpa with or without Sup treatment. G mRNA expression of related cytokines (Il1b, Il6 and Tnfa) after overexpressing Cebpa with or without Sup treatment. (H, I) To study the regulation of BMP9 by C/EBPα in hepatocytes, we altered Cebpa expression in AML-12 cells. H Protein expression levels of C/EBPα and BMP9 after overexpressing Cebpa. I Protein expression levels of C/EBPα and BMP9 after inhibiting Cebpa with or without Sup treatment. The average target gene/Gapdh ratios in different experimental groups relative to the control group. *p < 0.05, **p < 0.01, and ***p < 0.001
C/EBPα is an important transcription factor that is a determinant in multiple physiological functions. High expression of C/EBPα and BMP9 was detected in both senescent hepatocytes and MΦs; however, whether BMP9 was regulated by C/EBPα remained unclear. Concomitant with Cebpa overexpression (OE), BMP9 expression was increased in the immortalized murine bone marrow-derived MΦ (iBMDM) cell line (Fig. 3D). Similarly, Cebpa expression in iBMDMs that was downregulated by siRNA reduced BMP9 expression (Fig. 3E). BMDMs derived from young and aged mice were then stimulated with APAP-treated AML-12 cell supernatant (Sup) to mimic DAMP signaling induced by hepatocyte death. Moreover, enhanced Cebpa expression promoted iNOS expression and decreased CD206 expression in the iBMDMs stimulated by Sup (Fig. 3F). Following Cebpa overexpression, the mRNA expression of Il1b, Il6, and Tnfa induced by Sup was increased in the iBMDMs (Fig. 3G). In addition, Cebpa overexpression increased the BMP9 level in AML-12 cells (Fig. 3H), and downregulated Cebpa expression correspondingly led to reduced BMP9 expression (Fig. 3I).
These results suggest that the expression of BMP9 is driven by C/EBPα signaling in MΦs and hepatocytes during aging, which might be a key intrinsic driver of aggravated APAP-ALI associated with aging.
The C/EBPα-BMP9 axis in hepatocytes promotes APAP-ALI progression
Given the link between C/EBPα and BMP9, the effect of C/EBPα on BMP9 expression in hepatocytes and APAP-ALI in vivo was investigated by overexpressing Cebpa via AAV2/8. Both the mRNA and protein expression levels of Bmp9 were upregulated after Cebpa was overexpressed in the liver 2 weeks after Cebpa AAV2/8 injection into the tail vein (Additional file 1: Figure S2A, B). Notably, IF and IHC assays confirmed successful Cebpa overexpression (Additional file 1: Figure S2C). After Cebpa was overexpressed in hepatocytes, the APAP-ALI model was established. Increased serum ALT and AST levels suggested that Cebpa overexpression aggravated APAP-ALI (Fig. 4A), paralleling the images showing H&E staining (Fig. 4B) and the increase in the number of TUNEL-stained cells (Fig. 4C). Notably, Cebpa overexpression in hepatocytes induced the mRNA expression of Il1b, Il6 and Tnfa in the liver (Fig. 4D). Moreover, we also detected increased Cxcl1, Cxcl13 and Mcp1 and decreased Arg1 and Il10 expression levels (Additional file 1: Figure S3A, B). For a survival study, we increased the dose of APAP administered (500 mg/kg) to the mice and found that Cebpa overexpression in the liver significantly increased the mortality of the mice treated with high-dose APAP (Additional file 1: Figure S3C).

The C/EBPα-BMP9 axis in the liver promotes APAP-ALI progression. APAP-ALI (300 mg/kg) was established 2 weeks after confirmation that Cebpa was successfully overexpressed by Cebpa-overexpressing AAV2/8 injection into the tail vein of the mice, and serum and liver tissue were collected from each group of mice after 24 h. A Serum ALT and AST levels. B Representative images showing H&E staining. C Representative images showing TUNEL staining images. D mRNA expression of Il1b, Il6 and Tnfa. E Representative images showing IHC staining for BMP9. F Representative images showing IHC staining for P-SMAD1/5/9. G Protein expression levels of C/EBPα, BMP9, SMAD1 and P-SMAD1/5/9. H Protein expression levels of ATG3, ATG7, LC3 I/II and p62. I Representative images showing IF staining for BMP9 and F4/80 in the liver slices of each group. The average target gene/Gapdh ratios different experimental groups relative to the control group. *p < 0.05, **p < 0.01, and ***p < 0.001
IHC assay of liver sections showed that the expression of BMP9 was upregulated after Cebpa was overexpressed (Fig. 4E). Moreover, the rate of SMAD1/5/9 phosphorylation was increased (Fig. 4F). These results indicated that Cebpa overexpression promoted BMP9 production and activated BMP signaling. Next, we sought to identify the proteins that potentially control APAP-ALI progression in the liver after Cebpa is overexpressed. By performing immunoblotting, we confirmed that Cebpa was successfully overexpressed and that the expression level of BMP9 and SMAD1/5/9 phosphorylation rates were increased (Fig. 4G). Furthermore, we detected the downregulation of ATG3 and ATG7 expression in the liver, which was accompanied by blocked autophagy (Fig. 4H). In addition, we also detected increased Bmp9 expression after successful Cebpa overexpression, followed by decreased Atg3 and Atg7 mRNA levels (Additional file 1: Figure S3D).
IF staining for F4/80 and BMP9 in liver sections revealed that Cebpa overexpression increased the expression of BMP9 localized to F4/80-positive cells in the liver (Fig. 4I). Similar to our observations of aged livers, increased expression of BMP9 in F4/80-negative cells was observed in livers after APAP treatment, suggesting the diversity and complexity of BMP9 sources in the liver (Fig. 4I).
BMP9 inhibits ATG3 and ATG7 expression to block autophagy in MΦs
Our results thus far confirmed that BMP9 is abundantly expressed in hepatic MΦs and directly aggravates APAP-induced injury by inhibiting hepatocyte autophagy. Rapamycin (RAPA) has been extensively reported to activate autophagy and reduce senescence [19, 20]. Therefore, we investigated the roles of aging and BMP9 on MΦs treated with RAPA. The LC3 I-to-LC3 II conversion rate was reduced, and BMP9 was highly expressed in primary hepatic MΦs from aged mice treated with RAPA and/or bafilomycin (Ba) (Additional file 1: Figure S4). Next, after infecting Ad-mCherry-GFP-LC3 adenovirus and expressing mCherry-CFP-tagged LC3 in BMDMs, we confirmed that autophagy in aged mouse-derived MΦs was hindered compared with that in the young group (Fig. 5A). Dual IF staining for LC3 and BMP9 (two colors) was then performed with BMDMs derived from aged mice, and we found that the BMP9 level increased while LC3 level decreased in cells from the aged mice, as indicated through the assessment of high-resolution images (Fig. 5B). Furthermore, the number of mCherry puncta was increased in cells treated with RAPA after Bmp9 was deleted (Fig. 5C).

BMP9 inhibits ATG3 and ATG7 and thus blocks autophagy in MΦs. A BMDMs derived from the bone marrow of young and aged mice were infected with Ad-mCherry-GFP-LC3 (MOI = 20) on the fifth day of culture when the cells were not fully mature and then treated with rapamycin on the seventh day of culture. Representative confocal laser microscopy images of each group. Quantitative data are shown in the right panel. B BMDMs from young and aged mice were treated with rapamycin on day 7. Representative images showing IF staining for LC3-I/II (red) and BMP9 incorporation (green) with DAPI counterstaining (blue) in each group. Quantitative data are shown in the right panel. C BMDMs derived from the bone marrow of WT and Bmp9−/− mice were infected with Ad-mCherry-GFP-LC3 (MOI = 20) on the fifth day of culture when the cells were not fully mature and then treated with rapamycin on the seventh day of culture. Representative confocal laser microscopy images of each group. Quantitative data are shown in the right panel. D Protein expression levels of ATG3, ATG7, LC3 I/II and p62 in BMDMs from WT and Bmp9.−/− mice with or without APAP-treated AML-12 cell supernatant (Sup). E Protein expression levels of ATG3, ATG7, LC3 I/II and p62 were in BMDMs with or without Rm-BMP9 and/or Sup treatment. *p < 0.05, **p < 0.01, and ***p < 0.001
Autophagy was inhibited in the short term after exposure to Sup (described above), which may be an important driver of MΦs proinflammatory action during the acute phase of liver injury (Fig. 5D, E). After BMP9 was depleted, ATG3 and ATG7 expression in MΦs was upregulated, the LC3 I-to-LC3 II conversion rate was increased, and the p62 level was decreased, indicating that autophagy had been activated (Fig. 5D). Additionally, Rm-BMP9 addition resulted in downregulated ATG3 and ATG7 expression, a decreased LC3 I-to-LC3 II conversion rate, and increased p62 accumulation in the MΦs (Fig. 5E).
In previous studies, a decline in autophagy promoted M1-type MΦ polarization during ALI, inducing proinflammatory and injury-promoting effects [21]. Importantly, BMP9 can directly promote APAP-induced hepatocyte death; therefore, the aging-associated excess accumulation of BMP9 may be an essential driver of aggravated liver injury.
BMP9 regulates the senescence and immunophenotype acquisition of MΦs
After etoposide (1 μg/ml) treatment for 2 days, the mRNA levels of p21 and p53 in iBMDMs were increased, and the expression of the SASP components Il1b, Il6 and Tnfa was upregulated (Additional file 1: Figure S5A). Similar to the BMP9 promotion of etoposide-induced hepatocyte senescence, BMP9 promoted etoposide-induced senescence phenotype acquisition by iBMDMs (Additional file 1: Figure S5B). In hepatic MΦs isolated from Bmp9−/− mice, Il1b, Il6, Tnfa and iNos expression levels were reduced after etoposide treatment alone and/or with APAP-treated AML-12 cell supernatant treatment (Additional file 1: Figure S5C).
Considering these results, we isolated primary hepatic MΦs from young WT and Bmp9−/− mice and aged WT and Bmp9−/− mice. SA-β-gal staining revealed that Bmp9 deletion reduced the number of MΦs that acquired the senescence phenotype (Fig. 6A). Next, after these young and aged cells were treated with Sup, the expression levels of Il1b, Il6 and Tnfa were decreased in the Bmp9−/− group (Fig. 6B). Moreover, cytokine (IL-1β, IL-6, TNF-α and IL-10) levels in the supernatants of the cells subjected to different treatments were measured by ELISAs. The results showed that Bmp9 deletion reduced the secretion of IL-1β, IL-6 and TNF-α (proinflammatory cytokines) and increased the production of IL-10 (a cytokine that suppresses inflammation and promotes injury repair), while increases in the exogenous Rm-BMP9 level significantly increased the levels of secreted IL-1β, IL-6 and TNF-α and reduced the concentration of IL-10 (Fig. 6C). The expression and intracellular distribution of iNOS and CD206 were then determined by IF staining, and the results showed that aging promoted iNOS production and decreased CD206 levels, and excess BMP9 in cells further enhanced iNOS production and reduced CD206 levels, while inhibiting Bmp9 expression led to the opposite outcomes (Additional file 1: Figure S6A).

BMP9 regulates the senescence and immunophenotype acquisition of MΦs. A SA-β-gal staining of BMDMs derived from young and aged WT mice and young and aged Bmp9.−/− mice. B mRNA expression levels of Il1b, Il6 and Tnfa in each group. C Cytokine levels (IL-1β, IL-6, TNF-α and IL-10) in the supernatants of cells in each group as measured by ELISAs. To further study the effect of macrophage-derived BMP9 on liver injury, an F4/80-specific AAV2/8-overexpressing plasmid vector (AAV2/8-F4/80-Bmp9) was constructed. APAP-ALI (300 mg/kg) was established for 2 weeks after AAV2/8-F4/80-Bmp9 and AAV2/8-F4/80-CTRL injection. Serum and liver tissue were collected from each group of mice 24 h later. D, E Serum ALT and AST levels (D) and representative images showing H&E staining (E) of liver slices of each group. F Representative images showing TUNEL staining. G mRNA expression levels of Il1b, Il6 and Tnfa. The average target gene/Gapdh ratios of different experimental groups relative to the control group. *p < 0.05, **p < 0.01, and ***p < 0.001
To further study the effect of MΦ-derived BMP9 on APAP-ALI, an F4/80-specific Bmp9-overexpressing AAV2/8 virus was constructed (AAV2/8-F4/80-Bmp9) [22]. APAP-ALI was established 2 weeks via tail vein injection of AAV2/8-F4/80-Bmp9. IF staining of isolated hepatic MΦs showed that BMP9 expression in the AAV2/8-F4/80-Bmp9 group was significantly upregulated, confirming the successful overexpression of MΦ-specific BMP9 (Additional file 1: Figure S6B). As shown by serum ALT/AST levels and H&E staining, APAP-ALI severity was significantly increased in the F4/80-specific Bmp9-overexpressing group (Fig. 6D, E). Additionally, the number of TUNEL-stained cells in the livers of the F4/80-specific Bmp9-overexpressing mice treated with APAP were significantly increased (Fig. 6F). Moreover, the mRNA expression of Il1b, Il6 and Tnfa was upregulated in the livers of the F4/80-specific Bmp9-overexpressing mice (Fig. 6G).
These data suggest that BMP9 plays a key role in the senescence of MΦs and that MΦ-derived BMP9 clearly drives the progression of APAP-ALI.
Discussion
C/EBPα is involved in the regulation of myeloid cell differentiation, proliferation, metabolism and immune function [23] and has been reported to be involved in regulating the fate of all myeloid cells [24]. C/EBPα is required for maintaining the integrity of hematopoietic stem cells and plays a central role in the early stage of myeloid cell differentiation and maturation [25,26,27]. Importantly, the reprogramming of mature myeloid cells derived from bone marrow hematopoietic stem cells plays an indispensable role in regulating multiple physiological functions. C/EBPα in combination with PU1 or C/EBPβ reverses the acquisition of a lymphoid phenotype by B- or T-cell progenitors in vitro, and these progenitors ultimately differentiate into monocytes/granulocytes [28]. Treatment with therapeutic saRNA overexpressing C/EBPα (MTL-C/EBPα) in hepatic cancer patients indicated that the number of monocytic myeloid-derived suppressor cells (M-MDSCs) and M2 tumor-associated MΦs (TAMs) were significantly reduced [29], suggesting that C/EBPα attenuates the negative immunoregulatory function of MΦs after MΦ reprogramming. This outcome favors MΦ polarization s into tumor-killing M1 cells, whereas normal liver repair capacity is impaired after liver injury.
Our results in this study enabled us to identify a previously unknown BMP-involved mechanism of liver inflammation and injury in which C/EBPα regulates BMP9 expression and BMP9 further drives liver inflammation and injury via its effects on both hepatocytes and MΦs. No previous study had explored the function and mechanism of BMP9 in senescence-associated APAP-ALI; therefore, the similarities and differences between hepatocytes and MΦs have been unclear to date. However, previous studies have shown that the constitutively low level of BMP9 expression helps stabilize hepatocyte function and that excess BMP9 promotes liver injury and fibrosis [15]. Despite these findings, the precise role and mechanism of BMP9 in aging and APAP-ALI remain unclear. BMP9 is mainly expressed and secreted by the liver, and it is then redistributed throughout the body, where it exerts effects [13, 14]. Some studies have suggested that BMP9 is expressed mainly in hepatic nonparenchymal cells, such as hepatic stellate cells and KCs [13, 30], and other studies have shown that BMP9 is also expressed in hepatocytes [31]. Our results showed that BMP9 was constitutively expressed to a certain extent in MΦs in the resting state and that its expression in other cells (non-MΦs), including hepatocytes, was evident after APAP-ALI had been induced.
After BMPs induce receptor hetero-oligomerization, type I receptors are activated via the phosphorylation of type II receptors. These activated receptor complexes phosphorylate SMAD proteins (R-SMADs: SMAD1, SMAD5 and SMAD8/9), forming complexes with SMAD4 [30]. An R-SMAD complex enters the nucleus and regulates target gene expression by binding to a regulatory element and recruiting transcriptional corepressors and/or activation complexes to these promoters. In this study, enhanced BMP9 activated downstream SMAD1/5/9 phosphorylation, further mediating the downregulation of ATG3 and ATG7 and leading to autophagy suppression. Moreover, the study provides direct evidence suggesting that excess BMP9 leads to autophagy inhibition and is associated with a tendency toward increased APAP-ALI severity in aged mice. The degree of liver injury and the rate of proinflammatory phenotype acquisition were exacerbated when additional Rm-BMP9 was injected into APAP-ALI model mice and in livers treated with Cebpa-overexpressing AAV2/8 or F4/80-specific Bmp9-overexpressing AAV2/8. These results show that redundant BMP9 action promotes proinflammatory responses and liver injury. Moreover, severe liver injury and inflammatory responses were alleviated in Bmp9-ko mice treated with APAP.
By transporting cargo to be degraded to lysosomes, autophagosomes effectively removes excess or dysfunctional organelles and unwanted proteins, establishing favorable conditions for cell survival and maintenance of function. Two ubiquitin-like coupling systems, ATG12-ATG5 and LC3-phosphatidyl ethanolamine (PE) conjugates, are closely related to autophagosome formation. ATG12 is activated by ATG7 (an E1-like enzyme) ubiquitination, then transferred to ATG10 (an E2-like enzyme), and ultimately is attached to ATG5 to form a ATG5-ATG12 conjugate [17, 32]. This conjugate recruits ATG16L1 to form a multimeric complex that can enhance the formation of the LC3-PE conjugate [17]. ATG4 cleaves LC3 to generate LC3-I, which binds to PE through a ubiquitin-like reaction involving ATG7 and ATG3 (with E2-like enzyme activity) to form lipidated LC3 (LC3-II), which binds to the autophagosome membrane [18].
Among the multiple molecular changes associated with aging, altered autophagy has become a hallmark of senescence in different species, and autophagy deficiency during aging is one of the most common drivers of tissue homeostasis dysregulation [33]. Similarly, the regulation of inflammation is inextricably linked to autophagy, as impaired autophagy in immune cells leads to uncontrolled inflammation and is recognized as a major driver of aging-related tissue damage [34]. In previous studies, autophagy has been identified as a key mechanism that coordinates the metabolic and differentiation states of innate immune cells. When autophagic flux is inhibited, MΦs acquire a proinflammatory phenotype and promote tissue damage [33], and autophagy interventions can often achieve better therapeutic efficacy in aging-related diseases [19, 20]. Therefore, improving the resistance of elderly patients to liver injury has important clinical application value for the diagnosis and treatment of aging-related liver diseases.
Exploring the mechanism of senescence-associated MΦ reprogramming may help to identify its roles in senescence-associated liver injury, and hence, the development of personalized treatment regimens for liver diseases may lead to positive results. For example, intracellular oxidants inhibit ATG3 and ATG7 by inducing their oxidation, inhibiting autophagy and thereby promoting aging progression [35]. In this study, we revealed that BMP9 expression was increased in hepatocytes and MΦs in the livers of aged mice, while both ATG3 and ATG7 expression was decreased, disrupting autophagy. In cultured hepatocytes and MΦs, excessive levels of BMP9 downregulated the expression of ATG3 and ATG7, inhibiting autophagic flux. Further studies have indicated that when Cebpa was overexpressed in the liver, ATG3 and ATG7 were downregulated and autophagic flux was reduced, aggravating liver inflammation and injury.
Conclusions
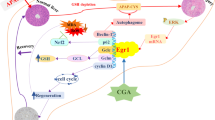
In conclusion, the excessive production of aging-associated C/EBPα upregulates BMP9 expression, which plays a central role in the aging-associated aggravation after APAP-ALI via its paracrine and autocrine activity. On the one hand, BMP9 directly accelerates APAP-induced hepatocyte death, and on the other hand, it promotes the proinflammatory phenotype acquisition of MΦs (Fig. 7). Mechanistically, BMP9 promotes the phosphorylation of the downstream transcription factors SMAD 1/5/9, which in turn leads to a decrease in ATG3 and ATG7 expression, which is associated with autophagy blockade. Our study revealed a crucial function for the C/EBPα-BMP9 axis in APAP-ALI and provided insight into acute liver injury. These results help in understanding the interrelatedness of different cells involved in acute liver injury and indicate new therapeutic directions. However, additional work is needed to identify how BMP9 interacts with regulatory signaling pathways in multiple cell types in the liver during aging and the common and unique effects of BMP9 on the senescence of other cells and tissues across the body.

Model depicting the aggravation of ALI during aging through ectopic C/EBPα-BMP9 crosstalk
Materials and methods
Animal experiments
Young (6–10 weeks) and old-aged (> 16 months) [36] male C57BL/6N mice were used in experiments. Bmp9−/− mice were purchased from GemPharmatech Co., Ltd. The Bmp9−/− mice with a C57BL/6 background were established by CRISPR‒Cas9, and the effects of treatments on KO mice were compared to those of WT littermates. All the uses of the mice were approved by the Laboratory Animal Ethics Committee of the University of Science and Technology of China. The mice were housed in an environmentally controlled, pathogen-free isolation facility under a 12-h light–dark cycle with food and water freely available. The mice were fasted overnight (i.e., for approximately 15 h) before the administration of 300 mg/kg APAP (30 μl/g body weight) or vehicle (warm PBS) via i.p. injection [37]. Liver injury was assessed via the determination of serum alanine aminotransferase (ALT) activity and hematoxylin and eosin (H&E) staining of liver Sects. [38].
Cells
Mouse primary hepatocytes and MΦs were isolated from mice fed ad libitum via a combination of magnetic activated cell sorting and collagenase‐based density gradient centrifugation [39, 40]. The isolated hepatocytes were seeded on collagen-coated plates in Williams' E medium supplemented with 10% fetal bovine serum (FBS), 2 mm L-glutamine, 1% penicillin (100 U/ml)-streptomycin (100 μg/ml) (P/S), 100 nm dexamethasone, and 5% insulin transferrin selenium (ITS). A mouse hepatocyte cell line (AML12) was cultured in DMEM supplemented with 10% FBS, 5% ITS, 2 mm L-glutamine, 1% P/S, and 100 nm dexamethasone. Unless otherwise indicated, all cells were cultured at 37 °C in 95% ambient air and 5% CO2. Primary BMDMs were cultured for 6 days in DMEM supplemented with 10% FBS, m-CSF (20 ng/ml), and 1% P/S. iBMDMs were cultured in DMEM supplemented with 10% FBS and 1% P/S.
Antibodies and reagents
The antibodies used in this study are listed in the Supplementary Information. Murine m-CSF (PeproTech, 315–02-100), Rm-BMP9 (R&D Systems, 5566-BP), RAPA (Sigma–Aldrich, V900930), and etoposide (Sigma–Aldrich, E1383) were obtained from commercially available sources.
Recombinant AAV2/8 was generated using pAAV-CR and pHelper in HEK293 cells as described previously [41]. AAV2/8 plasmid vectors overexpressing Cebpa were produced and purchased from Hanbio Tech (Shanghai, China). An empty vector was constructed at the same time. AAV2/8 plasmid vectors overexpressing Bmp9 under the control of the MФ-specific AAV2/8-F4/80-Bmp9 promoter were produced and purchased from Hanbio Tech (Shanghai, China). An empty vector under the control of the MФ-specific AAV2/8-F4/80 promoter was constructed at the same time. AAV2/8-F4/80-Bmp9 titers of 1.5 × 1013 vector genomes (vg)/mL and AAV2/8-F4/80 titers of 1.5 × 1013 vg/mL were used in the study.
Immunohistochemistry, immunofluorescence, and immunoblotting
The immunohistochemistry, immunofluorescence and immunoblotting experiments are described in detail in the Supplementary Information.
Plasmids, siRNAs, and Rt-qPCR
Cell lines (the AML12 and iBMDM cell lines) were transfected with overexpression (OE) plasmids (OE-Bmp9 and OE-Cebpa) (Shanghai GeneChem Co., Ltd.), or small interfering RNAs (siRNAs) (Bmp9 siRNA and siCebpa siRNA) (Tsingke Biotechnology Co., Ltd.) using Lipofectamine 3000 (Invitrogen, L3000). Total RNA was extracted using TRIzol (Invitrogen). We synthesized cDNA from 1 μg of total RNA following the manufacturer’s protocol (Takara, RR036A). Then, RT–PCR was performed with a premixed kit (Takara, RR820A) according to the manufacturer’s instructions. The sequences of the primer pairs are listed in Additional file 1: Table S1.
Ad-mCherry-GFP-LC3
Ad-mCherry-GFP-LC3 (Beyotime Biotechnology, C3011), which is an adenovirus expressing a mCherry-GFP-LC3 fusion protein, was used for autophagy detection after infection of cells or tissues as described previously [42]. BMDMs were infected with Ad-mCherry-GFP-LC3 (MOI = 20) according to the manufacturer's instructions on the fifth day of culture, when the cells were not fully mature, and then treated with LPS or Torin 1 (1 µM) on the seventh day of culture.
Statistical analysis
Statistical analysis was performed with Prism 8.2 (GraphPad Software). Specifically, one-way analysis of variance (ANOVA) was performed for comparisons among groups. The differences in survival based on survival curves were determined via log-rank test. Differences for which P < 0.05 were considered to be statistically significant.
Availability of data and materials
All data and material will be made available on request.
References
Forbes SJ, Newsome PN. Liver regeneration - mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol. 2016;13:473–85.
Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol. 2013;3:485–513.
Nie YZ, Zheng YW, Taniguchi H. Improving the repopulation capacity of elderly human hepatocytes by decoding aging-associated hepatocyte plasticity. Hepatology. 2022. https://doi.org/10.1002/hep.32443.
Pinto C, Ninfole E, Gaggiano L, Benedetti A, Marzioni M, Maroni L. Aging and the biological response to liver injury. Semin Liver Dis. 2020;40:225–32.
Gan L, Chitturi S, Farrell GC. Mechanisms and implications of age-related changes in the liver: nonalcoholic fatty liver disease in the elderly. Curr Gerontol Geriatr Res. 2011;2011: 831536.
Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75–95.
Sun X, Wu J, Liu L, Chen Y, Tang Y, Liu S, Chen H, et al. Transcriptional switch of hepatocytes initiates macrophage recruitment and T cell suppression in endotoxemia. J Hepatol. 2022. https://doi.org/10.1016/j.jhep.2022.02.028.
Huang X, Yao Y, Hou X, Wei L, Rao Y, Su Y, Zheng G, et al. Macrophage SCAP contributes to metaflammation and lean NAFLD by activating STING-NF-kappaB signaling pathway. Cell Mol Gastroenterol Hepatol. 2022. https://doi.org/10.1016/j.jcmgh.2022.03.006.
Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang XM, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019. https://doi.org/10.1126/science.aau0964.
Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–94.
Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, Graham MJ, Han X, et al. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab. 2019;29(383–398): e387.
Yang Y, Fan S, Chen Q, Lu Y, Zhu Y, Chen X, Xia L, et al. Acute exposure to gold nanoparticles aggravates lipopolysaccharide-induced liver injury by amplifying apoptosis via ROS-mediated macrophage-hepatocyte crosstalk. J Nanobiotechnol. 2022;20:37.
Miller AF, Harvey SA, Thies RS, Olson MS. Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. J Biol Chem. 2000;275:17937–45.
Bidart M, Ricard N, Levet S, Samson M, Mallet C, David L, Subileau M, et al. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell Mol Life Sci. 2012;69:313–24.
Breitkopf-Heinlein K, Meyer C, Konig C, Gaitantzi H, Addante A, Thomas M, Wiercinska E, et al. BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut. 2017;66:939–54.
Jiang Q, Li Q, Liu B, Li G, Riedemann G, Gaitantzi H, Breitkopf-Heinlein K, et al. BMP9 promotes methionine- and choline-deficient diet-induced nonalcoholic steatohepatitis in non-obese mice by enhancing NF-kappaB dependent macrophage polarization. Int Immunopharmacol. 2021;96: 107591.
Wible DJ, Chao HP, Tang DG, Bratton SB. ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov. 2019;5:42.
Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–22.
Schinaman JM, Rana A, Ja WW, Clark RI, Walker DW. Rapamycin modulates tissue aging and lifespan independently of the gut microbiota in Drosophila. Sci Rep. 2019;9:7824.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5.
Ji J, Xue TF, Guo XD, Yang J, Guo RB, Wang J, Huang JY, et al. Antagonizing peroxisome proliferator-activated receptor gamma facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell. 2018;17: e12774.
Zhao HY, Zhang YY, Xing T, Tang SQ, Wen Q, Lyu ZS, Lv M, et al. M2 macrophages, but not M1 macrophages, support megakaryopoiesis by upregulating PI3K-AKT pathway activity. Signal Transduct Target Ther. 2021;6:234.
Avellino R, Delwel R. Expression and regulation of C/EBPalpha in normal myelopoiesis and in malignant transformation. Blood. 2017;129:2083–91.
Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, Jaitin D, et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat Cell Biol. 2018;20:836–46.
van Oevelen C, Collombet S, Vicent G, Hoogenkamp M, Lepoivre C, Badeaux A, Bussmann L, et al. C/EBPalpha activates pre-existing and De Novo macrophage enhancers during induced Pre-B cell transdifferentiation and myelopoiesis. Stem Cell Reports. 2015;5:232–47.
Pundhir S, Bratt Lauridsen FK, Schuster MB, Jakobsen JS, Ge Y, Schoof EM, Rapin N, et al. Enhancer and transcription factor dynamics during myeloid differentiation reveal an early differentiation block in cebpa null progenitors. Cell Rep. 2018;23:2744–57.
Hasemann MS, Lauridsen FK, Waage J, Jakobsen JS, Frank AK, Schuster MB, Rapin N, et al. C/EBPalpha is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014;10: e1004079.
Di Tullio A, Vu Manh TP, Schubert A, Castellano G, Mansson R, Graf T. CCAAT/enhancer binding protein alpha (C/EBP(alpha))-induced transdifferentiation of pre-B cells into macrophages involves no overt retrodifferentiation. Proc Natl Acad Sci U S A. 2011;108:17016–21.
Hashimoto A, Sarker D, Reebye V, Jarvis S, Sodergren MH, Kossenkov A, Sanseviero E, et al. Upregulation of C/EBPalpha inhibits suppressive activity of myeloid cells and potentiates antitumor response in mice and patients with cancer. Clin Cancer Res. 2021;27:5961–78.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84.
Yang Z, Li P, Shang Q, Wang Y, He J, Ge S, Jia R, et al. CRISPR-mediated BMP9 ablation promotes liver steatosis via the down-regulation of PPARalpha expression. Sci Adv. 2020. https://doi.org/10.1126/sciadv.abc5022.
Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–52.
Aman Y, Schmauck-Medina T, Hansen M, Morimoto RI, Simon AK, Bjedov I, Palikaras K, et al. Autophagy in healthy aging and disease. Nat Aging. 2021;1:634–50.
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576–90.
Frudd K, Burgoyne T, Burgoyne JR. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun. 2018;9:95.
Ramirez T, Li YM, Yin S, Xu MJ, Feng D, Zhou Z, Zang M, et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J Hepatol. 2017;66:601–9.
Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, et al. Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of Mmp12. J Hepatol. 2017;66:787–97.
Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–32.
Berry MN, Grivell AR, Grivell MB, Phillips JW. Isolated hepatocytes–past, present and future. Cell Biol Toxicol. 1997;13:223–33.
Alpini G, Phillips JO, Vroman B, LaRusso NF. Recent advances in the isolation of liver cells. Hepatology. 1994;20:494–514.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–78.
Liu R, Cui J, Sun Y, Xu W, Wang Z, Wu M, Dong H, et al. Autophagy deficiency promotes M1 macrophage polarization to exacerbate acute liver injury via ATG5 repression during aging. Cell Death Discov. 2021;7:397.
Acknowledgements
We thank Prof. Hua Wang and Prof. Xiaoying Liu for discussion and suggestions.
Funding
This research is funded by the National Natural Science Foundation of China (81870450).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. SY and RL designed the research. RL, WTX and HZ performed the experiments. RL and ZJD analyzed the data. SY and RL wrote the manuscript. HKD participated in the experimental discussions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Laboratory Animal Ethics Committee of Anhui Medical University. All animal experiments were performed according to procedures approved by the Laboratory Animal Ethics Committee of Anhui Medical University.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primer sequences used for real-time RT-PCR. Figure S1. Supplementary figure related to Figure 2. (A-B) Rm-BMP9 was injected into the tail vein, the APAP-ALI model was established 1 h later, and 24 h later, serum and liver tissue were collected from each group of mice. Representative images showing TUNEL staining (A) and the mRNA expression levels of related cytokines (Il1b, Il6 and Tnfa) (B) in each group. (C-D) An APAP-ALI model was established with young and aged WT mice and with young and aged Bmp9-/- mice. Representative images showing TUNEL staining (C) and the mRNA expression levels of related cytokines (Il1b, Il6 and Tnfa) (D) in each group. (E) IF staining for P-SMAD1/5/9 in young and aged WT mice and aged Bmp9-/- mice. The average target gene/Gapdh ratios of different experimental groups relative to the control group. GAPDH was used as the loading control for immunoblotting. *p < 0.05, **p < 0.01, and ***p < 0.001. Figure S2. BMP9 expression is regulated by C/EBPα in vivo. (A-C) Cebpa-overexpressing AAV2/8 was injected into the mouse tail vein 2 weeks after the models were established. (A) IF and IHC staining for C/EBPα in liver slices. (B) mRNA expression levels of Cebpa and Bmp9. (C) Protein expression levels of C/EBPα and BMP9. The average target gene/Gapdh ratios of different experimental groups relative to the control group. GAPDH was used as the loading control for immunoblotting. *p < 0.05, **p < 0.01, and ***p < 0.001. Figure S3. Supplementary figures related to Figure 4. (A-B) Additional mRNA expression level measurements (Cxcl1, Cxcl13, Mcp1, Arg1 and Il10). (C) Survival curves of mice in the Cebpa-overexpressing and control groups when the dose of APAP treatment was increased to 500 mg/kg. (D) mRNA expression level measurements (Cebpa, Bmp9, Atg3 and Atg7). The average target gene/Gapdh ratios of different experimental groups relative to the control group. *p < 0.05, **p < 0.01, and ***p < 0.001. Figure S4. (A) LC3 I/II and BMP9 levels in primary hepatic MΦs from young and aged mice treated with APAP-treated AML-12 cell supernatant (Sup) and/or RAPA and/or BA. GAPDH was used as the loading control for immunoblotting. Figure S5. BMP9 increases etoposide-induced macrophage senescence and proinflammatory cytokine expression. (A) mRNA expression levels of p21, p53, Il1b, Il6 and Tnfa. (B) SA-β-gal staining for iBMDMs after control treatment, etoposide treatment for 1 day, etoposide treatment for 2 days, or etoposide plus Rm-BMP9 treatment for 2 days. (C) mRNA expression levels of iNos, Il1b, Il6 and Tnfa in hepatic MΦs isolated from Bmp9-/- and WT mice. The average target gene/Gapdh ratios of different experimental groups relative to the control group. *p < 0.05, **p < 0.01, and ***p < 0.001. Figure S6. (A) Representative images showing IF staining for iNOS (red) and CD206 incorporation (green) with DAPI counterstaining (blue) in each iBMDM group. (B) Representative images showing BMP9 IF staining of primary MΦs isolated from mouse livers treated after AAV-F4/80 or AAV-F4/80-Bmp9 injection.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, R., Xu, W., Zhu, H. et al. Aging aggravates acetaminophen-induced acute liver injury and inflammation through inordinate C/EBPα-BMP9 crosstalk. Cell Biosci 13, 61 (2023). https://doi.org/10.1186/s13578-023-01014-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01014-6