
Abstract
Pancreatic cancer is highly lethal malignant tumor with characterised rapid progression, invasiveness and resistance to radiochemotherapy. Transforming growth factor-β (TGF-β) signaling plays a dual role in both pro-tumorigenic and tumor suppressive of pancreatic cancer, depending on tumor stage and microenvironment. TGF-β signaling components alteration are common in pancreatic cancer, and its leading role in tumor formation and metastases has received increased attention. Many therapies have investigated to target TGF-β signaling in the preclinical and clinical setting. In this review, we highlight the dual roles of TGF-β and touch upon the perspectives on therapeutic target of TGF-β signaling in pancreatic cancer.
Similar content being viewed by others

Background
Pancreatic cancer is the fifth leading cause of death by cancer in the world [1]. The major histological subtype is pancreatic ductal adenocarcinoma, which comprises 90% of all pancreatic cancers. Pancreatic cancer is highly aggressive malignancy with an increasing incidence, which features rapid progression, invasiveness and resistance to radiochemotherapy [2]. At present the 5-year survival for pancreatic cancer is only 6% and the median survival from diagnosis is about 6 months [3]. Moreover, pancreatic cancer is expected to rise to the second leading cause of cancer-associated mortality by 2030 according to incidence’s prediction [4]. Current treatment choices available for pancreatic cancer show no significant improvement in overcoming the invasion and metastasis in the recent decades [5]. The key to improving is to control their local invasion, and distant metastasis, and these features underscore the pressing need to develop new therapeutic strategies specifically [6].
Transforming growth factor-β (TGF-β) plays an important role in regulating numerous normal cellular, physiological, and developmental processes. More evidence is emerging that TGF-β has a potential influence on the tumorigenic process. Deregulation of TGF-β signaling is involved in the pathophysiology of pancreatic cancer [7]. The insensitivity to growth inhibitory pathways is one of the hallmarks of cancer. Cancer genes consist of oncogenes and tumor-suppressor genes, but a growing number of them play a dual role and defy these categories. TGF-β signaling is one of the 12 core signaling pathways involved in pancreatic cancer. Mutation in at least one of the TGF-β signaling genes occurs in 100% of the pancreatic cancer. The action of TGF-β in pancreatic cancer is now attracting considerable attention. The role of TGF-β during pancreatic cancer initiation and progression is complex and somewhat paradoxical. TGF-β plays a tumor suppressor in early-stage pancreatic cancer by promoting apoptosis and inhibiting epithelial cell cycle progression, but plays a tumor promoter in late-stage by genomic instability, neoangiogenesis, immune evasion, cell motility, and metastasis [8].
In this review, we discuss recent insights into the regulation of TGF-β signaling and focus more on dual roles of TGF-β in pancreatic cancer. We also highlight knowledge on TGF-β signaling in cancer stem cells and tumor microenvironment of pancreatic cancer. We finally touch upon the perspectives on therapeutic target of TGF-β signaling in pancreatic cancer.
TGF-β signaling pathways
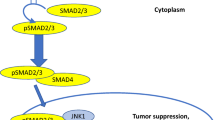
Recently, researchers are doing wide studies on the TGF-β signaling pathways. Three isoforms endow with the TGF-β of mammals, namely TGF-β1, TGF-β2, and TGF-β3 [9]. Each TGF-β is differentially expressed and activated during development and upon various cellular stresses [10]. Of these, TGF-β1 is the most abundant isoform in humans. In general, TGF-β1 expression is elevated by signals that promote cell growth and proliferation, whereas TGF-β2 and TGF-β3 are induced by differentiation and growth arrest signals. TGF-β signaling starts with activation and releasing of the TGFβ1. The TGF-β type I (TGFβRI) and type II receptors (TGFβRII) form heterotetrameric complexes at the cell surface and bind the dimeric ligands (Fig. 1). The functional receptor complex regulates the activation of downstream Smad and non‐Smad pathways. In Smad pathway, the activated TGFβRI/TGFβRII phosphorylates the Smad2 and Smad3 proteins, which modulate transcription in association with Smad4. The activated Smad complex translocates to the nucleus and binds to specific DNA sequence motifs called Smad-binding elements (SBEs). Upon binding, pSmad2/3-Smad4 complexes interact with additional transcriptional regulators to transactivate TGFβ-dependent genes (Fig. 1).

Overview of the TGF-β signaling pathway. TGF-β signaling is transduced through two pathways of Smad (canonical) and non-Smad (non-canonical). In Smad pathway, cell surface complexes of TGFβRI and TGFβRII phosphorylate upon TGF-β ligand binding and activate Smad2 and Smad3. Smad4 and activated Smad2/Smad3 form a Smads complex, and then interacts with other transcription factors to regulate transcription of target genes. TGF-β signaling also activates non-Smad pathways, including PI3K-Akt, Ras-Erk, p38, JNK, and GTPases
TGFβ-mediated tumor cell-autonomous and host-tumor interactions in cancer progression are also controlled by non-Smad pathways. Besides this ‘canonical’ signaling pathway, TGF-β signaling can also be transduced through the non-canonical Smad-independent pathways, including phosphatidylinositol-3 kinase (PI3K)/protein kinase B (AKT) pathway, JNK/p38 pathway, mitogen-activated protein kinase (MAPK) pathway, and Rho GTPases [11, 12] (Fig. 1). MED12, a transcriptional MEDIATOR complex protein, is mutated in cancers and found to interfere with maturation of TGF-βRII in the Golgi [13, 14]. MED12 loss therefore leads to activation of TGB-β signaling [15]. Consequently, TGF-β signaling causes activation of MEK/ERK signaling and restores the reduced MAPK pathway activation by tyrosine kinase inhibitors [15].
TGF-β alterations in pancreatic cancer
Pancreatic cancer is a genetic disease characterized by somatic mutations of multiple genes [16]. The expression of TGF-β obviously increases in pancreatic cancer, and overexpression of TGF-β is associate with venous invasion, advanced tumor stages, progressive disease, shorter patient survival duration, and liver metastases [17,18,19,20,21]. Studies have shown that TGF-β signaling components often become genetically inactivated in pancreatic cancer and disabling TGF-β signaling may be a critical event in pancreatic cancer progression. Pancreatic cancer has detected loss of function or truncating mutations of TGFβRI, TGFβRII, Smad2, and Smad4 genes [22, 23]. Smad7, an inhibitory Smad family member, are proved overexpression and enhances tumorigenicity in human pancreatic cancer [24]. TGFβRII mutations are involved in 4–7% of pancreatic cancers [25,26,27], while mutations in TGFβRI are found in 2% of them [26,27,28,29]. Furthermore, 60% of pancreatic cancer is observed to lost 18q21 chromosome that harbors the Smad4 gene [28, 30]. Smad4 acts as a central mediator in the TGF-β signaling, and its inactivation is relatively specific for pancreatic cancer [11, 31,32,33,34]. KRAS mutation, which is necessary for carcinogenesis and subsequent cancer maintenance, is found in approximately 90% of all pancreatic ductal adenocarcinomas [35]. But, KRAS mutation alone is not sufficient for malignant transformation [36]. The data from whole-genome sequencing analyses demonstrated that the common co-mutations detected in pancreatic cancer are SMAD4, KRAS, MED12, TP53, and CDKN2A [37]. Mutations in tumor suppressors, such as SMAD4, SMAD4, and CDKN2A, are required for carcinogenesis in addition KRAS mutation [38]. So, losing the normal signaling of Smad4 may promote KRAS-driven malignant transformation of pancreatic duct cells [39].
TGF-β in pancreatic cancer initiation and progression
TGF-β signaling function in pancreatic cancer appears complex and it is clearly evident that TGF-β acts in both an anti- and pro-tumorigenic activities. TGF-β exerts suppressive effects on tumor-promoting inflammation and on early stage of carcinogenesis, whereas during advance stage TGF-β acquires pro-oncogenic and pro-metastatic roles, which are associated with observable increase in the locally secreted TGF-β level [40,41,42].
Tumor-suppressor role of TGF-β
TGF-β exhibits potent growth inhibitory effect in early stage of pancreatic cancer by promoting apoptosis and inhibiting cell cycle progression through G1 arrest [43]. Hezel et al. [44] found that TGF-β acts in a common tumor suppressor pathway whose pharmacologic inactivation promotes pancreatic cancer progression. TGF-β inhibits pancreatic cancer growth by decreasing VEGF and increasing thrombospondin-1, and perturbations of TGF-β signaling pathway during tumor progression relieves this inhibition [45]. Singh et al. [46] reported that TGF-β can inhibit pancreatic cancer cells growth in a p53-independent manner. Indeed, pancreatic cancer progression requires shutting down the tumor-suppressive effects of TGF-β signaling through mutation Smad transcription factors (Smad2, Smad4) [41].
Tumor-promoter role of TGF-β
During advanced stage of carcinogenesis, TGF-β promotes invasion and metastasis of pancreatic cancer. TGF-β can promote stromal ‘‘activation’’, and induce angiogenesis, while attenuating a productive anti-tumor immune response [47, 48]. TGF-β ligands are commonly overexpressed in pancreatic cancer, and can promote epithelial-to-mesenchymal transition (EMT) and invasion in cell lines [49, 50]. TGF-β is one of the best known inducers of EMT-inducing transcription factors such as Snail, Slug, Twist, or Zeb1 [43]. However, David et al. demonstrate that TGF-β drives tumor suppression in pancreatic cancer cells by promoting EMT-linked remodeling of the transcription factor landscape, which converts TGFβ-induced Sox4 from an enforcer of tumorigenesis in the epithelial state into a promoter of apoptosis after EMT [51]. TGF-β induces an EMT generally considered as a pro-tumorigenic event. However, in TGFβ-sensitive pancreatic adenocarcinoma cells, EMT becomes lethal by converting TGFβ-induced Sox4 from an enforcer of tumorigenesis into a promoter of apoptosis [51] (Fig. 2). This study provides elegant mechanistic data to elucidate the dichotomous effects of TGF-β on pancreatic cancer cells [51] (Fig. 2).

The dual role of TGF-β in pancreatic cancer cell. (Left) TGFβ-mediated epithelial–mesenchymal transition (EMT) and apoptosis in Smad4-positive pancreatic cancer cell. TGF-β signaling induces EMT by induction SNAIL and repression KLF5. Alternatively, TGF-β signaling induces apoptosis in KLF5 absence cell by SOX4 inducing transcription of pro-apoptotic genes. (Right) TGF-β promotes tumor progression in Smad4-negative pancreatic cancer cell. TGF-β signaling induces SOX4 induction mediated through Smad2/3, and then SOX4 and KLF5 cooperate for tumorigenesis
TGF-β actions in cancer stem cells
Cancer stem cells, a subpopulation of cancer cells with stem cell characteristics, are widely believed responsible for tumor carcinogenesis, progression and recurrence [52]. Recent studies have demonstrated that cancer stem cells stay in quiescent status and resist to traditional chemo-therapy and radio-therapy [53]. Researchers isolated cancer stem cells from pancreatic cancer which were characterized with self-renewal, highly tumorigenic, and more differentiated progenies [54, 55]. Cancer stem cells are the root of cancer which cannot be killed by traditional methods. So, developing novel drugs or approaches to radically eliminate the origin of tumor cells will bring great effect on cancer therapy. The signal pathways contributing to self-renew is an important research direction of exploring the targeting drugs. Research increasingly suggests that TGF-β plays an important role in the occurrence and development of pancreatic cancer stem cells. TGF-β signaling has been confirmed more widely role in the maintenance of pancreatic stem cells [56]. TGF-β signaling through Activin/Nodal activation is required for self-renewal and tumorigenicity of cancer stem cells in pancreatic cancer [57].
TGF-β actions in tumor microenvironment
Pancreatic cancer displays greater prominent desmoplastic stromal reaction, though the cancer itself is the epithelial component [58]. The expression of TGF-β shows high level in pancreatic cancer tissue [59]. The microenvironment of pancreatic cancer features a pronounced stromal reaction composed of collagen-rich extracellular matrix, pancreatic stellate cells, and inflammatory cells [60, 61]. Tumor microenvironment plays a significant role in tumor initiation and development, and it can influence the interaction between pancreatic cancer cells and TGF-β [62]. Pancreatic stellate cells are responsible for excess extracellular matrix production in pancreatic cancer. TGF-β, as a potent activator, mediates the interaction between pancreatic stellate cells and cancer cells [63]. Growth factors produced and released by stroma to pancreatic cancer cells result with the reactive stroma [64]. These stromal elements in addition to TGF-β signaling participation in autocrine and paracrine produce a modified extracellular matrix that can accelerates growth and metastasis of pancreatic cancer cells [64, 65]. In sum, TGF-β has the dual role at the microenvironment level of pancreatic cancer. TGF-β is initially utilized to prevent occurrence and proliferation of pancreatic cancer in precancerous and early stage, but it is ultimately used to promote pancreatic cancer progression in advanced stage of carcinogenesis.
Therapeutic perspectives
TGF-β signaling is considered a prominent treatment target for pancreatic cancer in oncology [66]. Several relevant therapeutic approaches have been investigated in the preclinical and clinical setting and have shown efficacy [40, 67,68,69,70,71]. Ligand, ligand-receptor binding and intracellular signal transduction are the three levels of the therapeutic strategies to disrupt TGF-β signaling, such as TGF-β antisense RNA molecules, TGF-β blocking antibodies, neutralizing antibodies to the TGFβRII, and TGFβRI kinase small molecule inhibitors [40, 69, 70].
Therapeutic strategy on the ligand level
RNA interference (RNAi) has been applied to restrain the synthesis of TGF-β by regulation of TGFβ-coding genes expression. The short interfering RNA (siRNA) and the micro interfering RNA (miRNA) are mainly two types of antisense RNA molecules. Trabedersen (AP 12009), a TGF-β2 antisense RNA molecule, significantly reduced tumor growth, angiogenesis and lymph node metastasis in a metastatic pancreatic cancer mouse model [68, 72, 73].
Therapeutic strategy on the ligand-receptor interaction level
Natural TGF-β inhibitors, monoclonal blocking antibodies and soluble TGF-β receptors are mainly compounds of intervention on the ligand–receptor level. A soluble TGFβRII protein that blocks cellular responsiveness to TGF-β1 could reduce pancreatic cancer cell metastasis by the expression decrease of metastasis-associated genes in an orthotopic mouse model [67, 71]. Murakami et al. [74] described the efficacy of SB431542, a TGFβRI inhibitor, in a human pancreatic-cancer orthotopic mouse model by color-coded intravital imaging. The result of study demonstrated that color-coded intravital imaging readily detect the selective anti-stromal-cell targeting of SB431542.
Therapeutic strategy on the intracellular signaling level
Most of inhibitors on the intracellular signaling level target the kinase of TGF-β receptors. But others are peptide aptamers targeting Smads interaction with TGF-β receptors. SD-208, an inhibitor of TGFβRI kinase, reduced pancreatic cancer growth and metastasis in vivo and reduced fibrosis in the tumor microenvironment [72, 75]. SD-093, a selective inhibitor of TGFβRI kinase, strongly reduced the motility and invasiveness of the pancreatic cancer cells in vitro [76, 77]. LY2109761, a dual inhibitor of TGFβRI/II kinase, significantly reduces the tumor burden, abdominal metastases, and improves survival of metastatic pancreatic cancer in a murine model [68]. Galunisertib (LY2157299), an inhibitor of TGFβRI kinase, has been demonstrated an acceptable tolerability and safety profile in Japanese patients with advanced pancreatic and lung cancers in a phase 1 clinical study [78]. However, Oyanagi et al. [79] reported that galunisertib (LY2157299) can promote the invasion in collagen matrix of pancreatic carcinoma cells through hepatocyte growth factor produced by fibroblast. Gore et al. [80] reported that combinatorial targeting of TGFβRI with LY2157299 and EGFR/HER2 with lapatinib suppresses lymphangiogenesis and metastasis in a syngeneic orthotopic pancreatic cancer model. Therefore, under some pathological conditions, the inhibitors TGF-β signaling may contribute to development of cancer [79]. To face the metastasis dissemination challenge of cancer patients, the therapeutic strategy of intervention TGF-β signaling has been approached over the years. The inhibitors of TGF-β signaling have been shown effective in a number of studies of pancreatic cancer patients. Yet, developmental work requires to further efforts in novel type of inhibitors,e.g. substrate-mimicking drugs.
Conclusions
TGF-β signaling has kind of a dual role of promotion and inhibition in pancreatic cancer depending on different cancer stage and microenvironment. The alteration of TGF-β signaling components in pancreatic cancer is common and pronounced, and its leading role in cancer formation and metastases is arousing more attention. TGF-β also conducts a pivotal role of caner stem cells and tumor microenvironment in pancreatic cancer. TGF-β signaling targeted therapies have been investigated in the preclinical and clinical setting and have shown efficacy in pancreatic cancer. This novel strategy may be lead to the identification of improved outcomes for lethal pancreatic cancer.
Abbreviations
- TGF-β:
-
transforming growth factor-β
- EMT:
-
epithelial-to-mesenchymal transition
References
Li HY, Cui ZM, Chen J, Guo XZ, Li YY. Pancreatic cancer: diagnosis and treatments. Tumour Biol. 2015;36(3):1375–84.
Parikh PY, Lillemoe KD. Surgical management of pancreatic cancer–distal pancreatectomy. Semin Oncol. 2015;42(1):110–22.
Teague A, Lim KH, Wang-Gillam A. Advanced pancreatic adenocarcinoma: a review of current treatment strategies and developing therapies. Ther Adv Med Oncol. 2015;7(2):68–84.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21.
Mian OY, Ram AN, Tuli R, Herman JM. Management options in locally advanced pancreatic cancer. Curr Oncol Rep. 2014;16(6):388.
Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144(6):1316–26.
Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415–24.
Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal. 2017;15(1):19.
Roberts AB, Flanders KC, Heine UI, Jakowlew S, Kondaiah P, Kim SJ, Sporn MB. Transforming growth factor-beta: multifunctional regulator of differentiation and development. Philos Trans R Soc Lond B Biol Sci. 1990;327(1239):145–54.
Roberts AB, Kim SJ, Noma T, Glick AB, Lafyatis R, Lechleider R, Jakowlew SB, Geiser A, O’Reilly MA, Danielpour D, et al. Multiple forms of TGF-beta: distinct promoters and differential expression. Ciba Found Symp. 1991;157:7–15 (discussion 15–28).
Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11(3):229–43.
Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19(1):128–39.
Siraj AK, Masoodi T, Bu R, Pratheeshkumar P, Al-Sanea N, Ashari LH, Abduljabbar A, Alhomoud S, Al-Dayel F, Alkuraya FS, et al. MED12 is recurrently mutated in Middle Eastern colorectal cancer. Gut. 2017. doi:10.1136/gutjnl-2016-313334.
Yokouchi H, Nishihara H, Harada T, Ishida T, Yamazaki S, Kikuchi H, Oizumi S, Uramoto H, Tanaka F, Harada M, et al. Immunohistochemical profiling of receptor tyrosine kinases, MED12, and TGF-betaRII of surgically resected small cell lung cancer, and the potential of c-kit as a prognostic marker. Oncotarget. 2017;8(24):39711–26.
Huang S, Holzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, Garnett M, Grernrum W, Sun C, Prahallad A, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell. 2012;151(5):937–50.
Goggins M, Kern SE, Offerhaus JA, Hruban RH. Progress in cancer genetics: lessons from pancreatic cancer. Ann Oncol. 1999;10(Suppl 4):4–8.
Teraoka H, Sawada T, Yamashita Y, Nakata B, Ohira M, Ishikawa T, Nishino H, Hirakawa K. TGF-beta1 promotes liver metastasis of pancreatic cancer by modulating the capacity of cellular invasion. Int J Oncol. 2001;19(4):709–15.
Culhaci N, Sagol O, Karademir S, Astarcioglu H, Astarcioglu I, Soyturk M, Oztop I, Obuz F. Expression of transforming growth factor-beta-1 and p27Kip1 in pancreatic adenocarcinomas: relation with cell-cycle-associated proteins and clinicopathologic characteristics. BMC Cancer. 2005;5:98.
Wagner M, Kleeff J, Friess H, Buchler MW, Korc M. Enhanced expression of the type II transforming growth factor-beta receptor is associated with decreased survival in human pancreatic cancer. Pancreas. 1999;19(4):370–6.
Friess H, Yamanaka Y, Buchler M, Ebert M, Beger HG, Gold LI, Korc M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105(6):1846–56.
Truty MJ, Urrutia R. Basics of TGF-beta and pancreatic cancer. Pancreatology. 2007;7(5–6):423–35.
Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997;57(13):2578–80.
Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, et al. DPC4 gene in various tumor types. Cancer Res. 1996;56(11):2527–30.
Kleeff J, Ishiwata T, Maruyama H, Friess H, Truong P, Buchler MW, Falb D, Korc M. The TGF-beta signaling inhibitor Smad7 enhances tumorigenicity in pancreatic cancer. Oncogene. 1999;18(39):5363–72.
Lin X, Feng XH. Abrogation of transforming growth factor-beta signaling in pancreatic cancer. World J Surg. 2005;29(3):312–6.
Hansel DE, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Annu Rev Genomics Hum Genet. 2003;4:237–56.
Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998;58(23):5329–32.
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–3.
Achyut BR, Yang L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology. 2011;141(4):1167–78.
Hahn SA, Hoque AT, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996;56(3):490–4.
Iacobuzio-Donahue CA, Song J, Parmiagiani G, Yeo CJ, Hruban RH, Kern SE. Missense mutations of MADH4: characterization of the mutational hot spot and functional consequences in human tumors. Clin Cancer Res. 2004;10(5):1597–604.
Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CV, Moses HL. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20(22):3147–60.
Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20(22):3130–46.
Vincent DF, Yan KP, Treilleux I, Gay F, Arfi V, Kaniewski B, Marie JC, Lepinasse F, Martel S, Goddard-Leon S, et al. Inactivation of TIF1gamma cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 2009;5(7):e1000575.
Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83.
Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg. 2007;14(3):224–32.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103(15):5947–52.
Leung L, Radulovich N, Zhu CQ, Wang D, To C, Ibrahimov E, Tsao MS. Loss of canonical Smad4 signaling promotes KRAS driven malignant transformation of human pancreatic duct epithelial cells and metastasis. PLoS ONE. 2013;8(12):e84366.
Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6(7):506–20.
Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19(1):89–102.
Massague J. TGFbeta in cancer. Cell. 2008;134(2):215–30.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Hezel AF, Deshpande V, Zimmerman SM, Contino G, Alagesan B, O’Dell MR, Rivera LB, Harper J, Lonning S, Brekken RA, et al. TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 2012;72(18):4840–5.
Schwarte-Waldhoff I, Volpert OV, Bouck NP, Sipos B, Hahn SA, Klein-Scory S, Luttges J, Kloppel G, Graeven U, Eilert-Micus C, et al. Smad4/DPC4-mediated tumor suppression through suppression of angiogenesis. Proc Natl Acad Sci USA. 2000;97(17):9624–9.
Singh B, Murphy RF, Ding XZ, Roginsky AB, Bell RH Jr, Adrian TE. On the role of transforming growth factor-beta in the growth inhibitory effects of retinoic acid in human pancreatic cancer cells. Mol Cancer. 2007;6:82.
Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, Fujii T, Komuro A, Kiyono K, Kaminishi M, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci USA. 2007;104(9):3460–5.
Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B, Grutzmann R, Pilarsky C, Ungefroren H, Saeger HD, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Can Res. 2007;67(17):8344–50.
Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36.
Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J Biol Chem. 2009;284(1):245–53.
David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massague J. TGF-beta tumor suppression through a lethal EMT. Cell. 2016;164(5):1015–30.
Xiao J, Mu J, Liu T, Xu H. Dig the root of cancer: targeting cancer stem cells therapy. J Med Discov. 2017;2(2):jmd17003.
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–68.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–7.
Kabashima A, Higuchi H, Takaishi H, Matsuzaki Y, Suzuki S, Izumiya M, Iizuka H, Sakai G, Hozawa S, Azuma T, et al. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. Int J Cancer. 2009;124(12):2771–9.
Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, Zagorac S, Alcala S, Rodriguez-Arabaolaza I, Ramirez JC, et al. Nodal/activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9(5):433–46.
Neuzillet C, de Gramont A, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. Perspectives of TGF-beta inhibition in pancreatic and hepatocellular carcinomas. Oncotarget. 2014;5(1):78–94.
Korc M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer. 2003;2:8.
Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194(4 Suppl):S84–6.
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76.
Cohen R, Neuzillet C, Tijeras-Raballand A, Faivre S, de Gramont A, Raymond E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget. 2015;6(19):16832–47.
Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29(3):179–87.
Vonlaufen A, Phillips PA, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 2008;68(19):7707–10.
Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–97.
Calone I, Souchelnytskyi S. Inhibition of TGFbeta signaling and its implications in anticancer treatments. Exp Oncol. 2012;34(1):9–16.
Rowland-Goldsmith MA, Maruyama H, Matsuda K, Idezawa T, Ralli M, Ralli S, Korc M. Soluble type II transforming growth factor-beta receptor attenuates expression of metastasis-associated genes and suppresses pancreatic cancer cell metastasis. Mol Cancer Ther. 2002;1(3):161–7.
Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G, Abbruzzese JL, Chiao PJ. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7(4):829–40.
Arteaga CL. Inhibition of TGFbeta signaling in cancer therapy. Curr Opin Genet Dev. 2006;16(1):30–7.
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–67.
Rowland-Goldsmith MA, Maruyama H, Kusama T, Ralli S, Korc M. Soluble type II transforming growth factor-beta (TGF-beta) receptor inhibits TGF-beta signaling in COLO-357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin Cancer Res. 2001;7(9):2931–40.
Gaspar NJ, Li L, Kapoun AM, Medicherla S, Reddy M, Li G, O’Young G, Quon D, Henson M, Damm DL, et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol Pharmacol. 2007;72(1):152–61.
Schlingensiepen KH, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ, Kielmanowicz M, Schneider A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011;102(6):1193–200.
Murakami T, Hiroshima Y, Miyake K, Hwang HK, Kiyuna T, DeLong JC, Lwin TM, Matsuyama R, Mori R, Kumamoto T, et al. Color-coded intravital imaging demonstrates a transforming growth factor-beta (TGF-beta) antagonist selectively targets stromal cells in a human pancreatic-cancer orthotopic mouse model. Cell Cycle. 2017;16(10):1008–14.
Medicherla S, Li L, Ma JY, Kapoun AM, Gaspar NJ, Liu YW, Mangadu R, O’Young G, Protter AA, Schreiner GF, et al. Antitumor activity of TGF-beta inhibitor is dependent on the microenvironment. Anticancer Res. 2007;27(6B):4149–57.
Ge R, Rajeev V, Ray P, Lattime E, Rittling S, Medicherla S, Protter A, Murphy A, Chakravarty J, Dugar S, et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type I receptor kinase in vivo. Clin Cancer Res. 2006;12(14 Pt 1):4315–30.
Subramanian G, Schwarz RE, Higgins L, McEnroe G, Chakravarty S, Dugar S, Reiss M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004;64(15):5200–11.
Fujiwara Y, Nokihara H, Yamada Y, Yamamoto N, Sunami K, Utsumi H, Asou H, Takahash IO, Ogasawara K, Gueorguieva I, et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;76(6):1143–52.
Oyanagi J, Kojima N, Sato H, Higashi S, Kikuchi K, Sakai K, Matsumoto K, Miyazaki K. Inhibition of transforming growth factor-beta signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp Cell Res. 2014;326(2):267–79.
Gore J, Imasuen-Williams IE, Conteh AM, Craven KE, Cheng M, Korc M. Combined targeting of TGF-beta, EGFR and HER2 suppresses lymphangiogenesis and metastasis in a pancreatic cancer model. Cancer Lett. 2016;379(1):143–53.
Authors’ contributions
The manuscript outline was planned by WS, GQT and ZQT. The draft manuscript was written by WS, GQT, YZ and BC. This manuscript was revised by JS. This manuscript was finalized by ZQT. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by Wuxi People’s Hospital Affiliated Nanjing Medical University.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shen, W., Tao, Gq., Zhang, Y. et al. TGF-β in pancreatic cancer initiation and progression: two sides of the same coin. Cell Biosci 7, 39 (2017). https://doi.org/10.1186/s13578-017-0168-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-017-0168-0