
Abstract
Nonsense-mediated RNA decay (NMD) was originally discovered as a cellular surveillance pathway that safeguards the quality of mRNA transcripts in eukaryotic cells. In its canonical function, NMD prevents translation of mutant mRNAs harboring premature termination codons (PTCs) by targeting them for degradation. However, recent studies have shown that NMD has a much broader role in gene expression by regulating the stability of many normal transcripts. In this review, we discuss the function of NMD in normal physiological processes, its dynamic regulation by developmental and environmental cues, and its association with human disease.
Similar content being viewed by others
Background
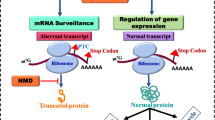
Nonsense-mediated RNA decay (NMD) is an essential RNA quality control and gene regulatory mechanism that is conserved among eukaryotes [1,2,3,4,5,6,7,8,9]. NMD safeguards the quality of the transcriptome and maintains cellular homeostasis by eliminating transcripts that harbor premature termination codons (PTCs). PTCs can arise from errors in nucleic acid metabolism, such as genetic mutations or defects in splicing or transcription. In addition, PTCs may also form during mRNA synthesis from normal gene structures, including from programmed recombination. In its canonical role, NMD prevents translation of transcripts that might produce C-terminally truncated proteins with reduced or aberrant function.
NMD also targets non-mutant transcripts, and its regulation of normal gene expression impacts a wide range of physiological processes including cell differentiation, response to stress and development of disease. Recent estimates suggest that NMD-mediated degradation affects up to 25% of transcripts, either directly or indirectly in certain cellular milieus [1, 10, 11]. Although the overall process of NMD is conserved for both mutant and non-mutant transcripts, the signals that trigger NMD or its inhibition vary according to the specific target and biological context.
In this review we focus on the function and impact of NMD on normal gene expression in mammals. We outline the NMD process and highlight the known roles for NMD in normal physiology, with a particular emphasis on its function as a gene regulatory mechanism and its dynamic regulation by environmental and developmental signals. We conclude with an overview of the impact of NMD dysregulation on human disease and discuss the potential of treating genetic and neurological disorders and cancer by manipulating NMD activity.
Overview of the NMD pathway
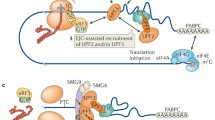
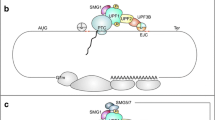
First discovered in yeast and then extensively studied in Caenorhabditis elegans, Drosophila, mouse, human cells, and other model systems, NMD is a RNA surveillance pathway that acts at the interface between transcription and translation [10, 12,13,14,15]. NMD must accurately distinguish a PTC from a normal stop codon on an mRNA and then recruit and activate enzymes to destroy the transcript. There are two main models to explain how transcripts are identified as targets for NMD. The exon-junction complex (EJC) model proposes that EJC—a large multi-protein assembly deposited ~20–24 bases upstream of an exon–exon junction as a result of pre-mRNA splicing—acts as a second signal to mark an upstream stop codon as a PTC [16,17,18,19,20,21,22,23,24,25,26,27,28,29]. During translation, ribosomes scan the mRNA and will pause at a stop codon. If an EJC is present more than 50–55 bases downstream of the stop codon, the protein kinase SMG1, its substrate Upf1, a ATPase/helicase, and eukaryotic polypeptide release factors eRF1 and eRF3 are then recruited to form a complex—the SURF complex—on the mRNA. Phosphorylation of UPF1 by SMG1 leads to the recruitment of SMG5, SMG6 and SMG7 via phospho-specific interactions [30, 31]. After recruitment, SMG5 and SMG7 promote RNA decapping and deadenylation by recruiting factors such as DCP1a and POP2, leading to the exposure of the transcript ends to cellular exonucleases [32,33,34,35,36,37,38,39]. SMG6, which has endonuclease activity, provides a second mechanism for initiation of mRNA decay by cleaving transcripts internally near the PTC, generating two unprotected RNA ends that are further degraded by cellular nucleases [40,41,42,43].
The second model for NMD posits that the abnormally long 3′ untranslated region (UTR) downstream of a PTC acts as a second signal to promote PTC recognition. While the molecular mechanism of this model is less well defined, it has been proposed that accumulation of UPF1 as well as other regulatory elements in the 3′ UTR mediates the recruitment of other NMD factors and the initiation of mRNA decay [44,45,46,47]. For additional information about the mechanisms of PTC recognition, readers are directed to a number of recent reviews [13, 48,49,50].
A debate remains about where NMD takes place in the cell. NMD inherently relies on the translation process, which normally occurs in the cytoplasm. However, some investigators have proposed that translation can also take place in the cell nucleus [51,52,53]. Several studies suggest that NMD is associated with the nucleus or nuclear fraction. For example, levels of PTC-containing triosephosphate isomerase (TPI) and mouse major urinary protein (MUP) transcripts were specifically reduced in the nuclear fraction [54,55,56,57]. NMD-mediated degradation of the TCRβ transcript can also take place in purified nuclei [58]. Several other nonsense reporter mRNAs also appear to be targeted by NMD in the nucleus [51, 59,60,61,62,63,64]. The idea of PTC recognition in the nucleus is also consistent with the existence of nonsense-associated altered splicing (NAS), a NMD-related nuclear pathway that also requires PTC recognition [65]. However, the claims of nuclear NMD and translation remain controversial as other lines of evidence suggest that NMD is primarily a cytoplasmic process [25]. Indeed, alternative interpretations of the nuclear NMD data are possible, including the possibility that NMD occurs during nuclear export, of which there is some evidence [66].
Role of NMD and its regulation in normal physiological processes
Bioinformatics analysis of EST databases and RNA sequencing data in cells where NMD is disrupted have clearly demonstrated that NMD has a widespread effect on gene expression [10, 67, 68]. This realization has led to the identification of numerous putative NMD target mRNAs, based on characteristics such as PTCs or long 3′ UTRs and an observed increase in the stability and/or levels of transcripts after NMD suppression [1, 4, 5, 11, 69,70,71,72,73,74,75].
Multiple mechanisms exist to generate PTCs in transcripts of normal genes (Fig. 1). Alternative splicing generates diversity in mRNA isoforms but can also lead to formation of PTCs that target transcripts for NMD. For instance, the RNA binding protein polypyrimidine tract binding protein 1 (PTBP1) can repress splicing of exon 11 of its own mRNA, leading to NMD of the transcript [76]. As such, PTBP1 negatively regulates its own expression. Human arginine–serine rich (SR) splicing factors have also been shown to be regulated by alternative splicing coupled to NMD [77, 78]. This so-called unproductive splicing and translation (RUST) represents an autoregulatory mechanism that controls the levels of splicing factors and other RNA binding proteins [77,78,79,80,81]. The use of alternative transcription initiation sites can also generate mRNA isoforms with a stop codon upstream of a splice junction, resulting in NMD [82]. Programmed ribosomal frameshifting (PRF)—which can potentially occur in up to 8–12% of genes—is another mechanism that can create a PTC, leading to NMD [83,84,85,86]. Stop codons that trigger NMD can also form if the primary coding region of an mRNA is preceded by an upstream open reading frame (uORF) [87, 88]. Transcriptome analysis indicates that long 3′ UTRs are among the most common features of NMD targets, although 3′ UTR length per se is not considered a reliable predictor of NMD of a given transcript [29, 44,45,46, 74, 88,89,90,91].
Transcripts encoding selenoproteins comprise another interesting class of NMD targets. A UGA codon normally signals a stop to translation but can be redefined to code for the amino acid selenocysteine in a high selenium environment [92]. If selenocysteine is incorporated in the last exon of a transcript it generally evades NMD, while if selenium is not abundant, these transcripts will be degraded via NMD if their stop codon resides upstream of an exon–exon junction [93, 94]. This regulatory mechanism enables cells to respond to alterations in levels of the essential trace element selenium. Therefore, while some NMD targets—such as those encoding selenoproteins or splicing factors—have been well characterized, the validation of other putative NMD targets is ongoing, as is the understanding of the consequences of NMD-induced regulation of gene expression.

NMD functions both in RNA surveillance and in gene regulation. Several features of mRNA transcripts can mark the transcripts as substrates for degradation by NMD. Green boxes, exons; lines, introns; potential splicing events are shown by blue or purple lines; genetic mutations and transcriptional aberrations are denoted with stars. The translation start site (ATG) is marked by arrows and stop codons are marked by red circles
There are many examples where normal physiological processes employ NMD to regulate gene expression (Table 1). As an example of the essential nature of NMD for normal cellular processes, knockouts of Smg1, Smg6, Upf1, or Upf2 have been shown to cause embryonic lethality in mice [1,2,3,4, 6]. NMD has also been shown to play a central role in the development and differentiation of specific cell types through regulation of gene expression.
During lymphocyte development, cells undergo a series of programmed genomic rearrangements to assemble immunoglobin and T cell receptor (TCR) genes. Two-thirds of these rearrangement events yield unproductive gene products harboring PTCs, whose clearance requires NMD [5, 95, 96]. Consistent with this observation, conditional ablation of NMD in T-cells significantly increased the abundance of these nonsense TCR transcripts, resulting in apoptotic cell death [5]. Interestingly, thymocyte development could be restored by introducing a complete TCRβ sequence that prevents the accumulation of nonsense counterparts, indicating that removal of the mutant transcripts by NMD is key to the survival of these cells [97]. However, conditional knockout of Upf2 had minimal effects on mature T cells, perhaps because T cells naturally downregulate NMD as part of the differentiation process [5]. In the myeloid lineage, the LMNB1 mRNA is specifically downregulated by NMD during granulopoiesis due to programmed intron retention. Importantly, this regulation of LMNB1 mRNA is required for normal differentiation of granulocytes [98].
Similar to the hematopoietic system, embryonic stem cells (ESCs) rely on NMD for their proliferation, while their differentiation is associated with downregulation of NMD activity [99, 100]. NMD influences stem cell differentiation by regulating the signaling of two key growth factors, TGFβ and BMP [100]. Ablation of NMD by SMG6 knockout in mouse ESCs prevented cellular differentiation, and re-expression of wild type but not mutant SMG6 restored proper differentiation [6]. Knockdown of other NMD factors caused a similar phenotype [6]. Prolonged, elevated expression of NMD-regulated pluripotency genes, such as c-Myc, underlies the inability of NMD-deficient ESCs to differentiate [6].
A number of studies have also revealed connections between NMD and proper development of the nervous system [90, 101,102,103,104,105,106,107,108,109]. In mammals, UPF3B expression is altered during brain development, and UPF3B mutants with impaired NMD function inhibit proper neurite outgrowth [105, 107]. Neuronal development is also compromised when UPF3B is downregulated with shRNA or when NMD is inhibited with the compound Amlexanox [105, 107]. NMD also functions to limit the expression of Robo3.2 in commissural neurons, which is required for proper axonal migration during development [109]. NMD components such as SMG1, UPF1 and UPF2 can localize to axonal growth cones in neurons, consistent with the idea that NMD modulates gene expression locally in these structures [109]. During the process of neurogenesis, a regulatory circuit is activated in differentiating neurons whereby expression of miR-128 targets mRNAs of several NMD factors for translational repression [90]. This results in NMD attenuation and upregulation of NMD target genes, many of which foster proper neuronal development [90].
Nonsense-mediated RNA decay also can impact gene expression in mature neurons, however. Ablation of the EJC factor eIF4AIII in mature neurons results in altered expression of critical factors such as ARC, leading to increased synaptic strength [108]. During seizure, when neuronal activity is aberrant, the RNA splicing protein NOVA regulates insertion of cryptic exons into the mRNA of a number of neuronal factors, leading to NMD of these transcripts [110]. Together, these findings highlight the importance of NMD in the development and function of the nervous system.
In developing muscle cells, NMD activity is attenuated as myoblasts differentiate to myotubes. During myogenesis, gene expression can be downregulated by NMD or by a related pathway, Staufen-mediated mRNA decay (SMD). SMD is increased in myoblasts due to the upregulation of the STAU1 (Staufen homolog) protein, which binds its cognate sites in the 3′ UTR of target mRNAs [111]. STAU1 competes with UPF1 for binding to UPF2, which functions in both NMD and SMD [112, 113]. This competition leads to inhibition of UPF2-dependent NMD and increased expression of the NMD target myogenin that promotes myogenesis [112].
Interactions of cells with external factors such as viruses can also be modulated by NMD. Robust NMD activity targets certain viral RNAs harboring NMD-inducing features to suppress expression of viral proteins and limit viral titer in host cells [114, 115]. However, some viruses possess mechanisms to co-opt the NMD process for their own benefit. For example, it has been found that the RNA-binding proteins tax and rex, expressed by the human T-cell leukemia virus type-1 (HTLV-1), stabilize both viral RNAs and host RNAs that would normally be targets for NMD [116, 117]. An element in the 3′ UTR of the Rous sarcoma virus also renders the viral RNA insensitive to host NMD, possibly by inhibiting the capacity of UPF1 to initiate NMD [118, 119]. As another example, hepatitis C infection triggers inactivation of NMD by binding and sequestering WIBG/PYM, a protein required for recycling of the EJC [120].
Recent studies suggest that NMD controls not only the levels of mRNAs, but also that of long non-coding RNAs (lncRNAs). While the majority of the genome is transcribed into RNA, only about 2% of the genome has been shown to code for proteins [121, 122]. LncRNAs are a prominent class of RNA molecules that have important roles in cellular processes, including modifying chromatin, regulating transcription, altering mRNA stability, and influencing translation [123, 124]. A subset of lncRNAs have been shown to be associated with the translation machinery—sometimes producing detectable micropeptides—and about 17% of lncRNAs were found to be targets of NMD [125,126,127,128,129,130]. While the biological significance of this regulation remains to be defined, the fact that so many lncRNA transcripts are targeted by NMD suggests that NMD plays a central role in regulating the functions of lncRNAs and their corresponding micropeptide products.
Dynamic regulation of NMD during cellular responses to stress
Cellular stress activates widespread changes in gene expression that allow cells to adapt to challenging conditions. One mechanism that enables this response is the inhibition of NMD (Table 1). Cellular stresses such as amino acid deprivation, hypoxia and endoplasmic reticulum (ER) stress induce phosphorylation of the translation initiation factor eIF2α, which in turn causes NMD repression and the stabilization and increased expression of critical stress response factors such as ATF4, ATF3, CHOP, and IRE1α [70, 73, 131,132,133,134,135,136]. NMD is also attenuated in response to an increase in intracellular calcium levels as well as persistent DNA damage [137 AN and ZY, unpublished]. By controlling the expression of specific genes, this dynamic regulation of NMD serves as an adaptive response to cope with cellular stress and promote survival. When the environmental insults are too severe, NMD also contributes to apoptosis. An early event during apoptosis is the cleavage of UPF1, which generates a dominant negative peptide fragment that stifles NMD activity [138]. The resulting reduction in NMD activity allows for the upregulation of several pro-apoptotic NMD target genes including GADD45α, GADD45β, BAK1, GAS5, DAP3, and DUSTP2, leading to cell death [138, 139]. GADD45α, which acts in the MAP kinase pathway, has also been proposed to be the key target that triggers apoptosis when NMD is disrupted in the absence of exogenous stress [140].
The observations that NMD is suppressed in response to a number of cellular stresses raises the question of how abnormal RNAs—which are often generated during gene expression—are dealt with during intervals of reduced NMD activity. One possibility is that the benefits of the expression of stress response genes after NMD attenuation outweigh the risks of the lack of RNA surveillance. It is also possible that cells retain residual NMD activity after stress, which is sufficient for RNA surveillance. During these intervals of low NMD activity, the activation of an autoregulatory circuit that leads to increased mRNA stability of NMD factors—which are normally targeted by NMD—rapidly restores NMD activity to appropriate levels once cellular conditions improve [74, 91, 141]. The discovery of alternative branches of the NMD pathway that are apparently independent of UPF2, UPF3, or the EJC introduces the possibility that when one branch of NMD is suppressed other branches still remain active and degrade aberrant transcripts [46, 142, 143]. In support of this idea, the activity of the UPF2-dependent branch of NMD is diminished during myogenesis but an alternative, UPF2-independent branch is stimulated, allowing both for increased expression of the NMD target myogenin and continued degradation of mutant mRNA transcripts [112]. An additional mechanism to cope with reduced NMD activity is autophagy, which purges cells of the mutated, misfolded, and aggregated proteins that accumulate in NMD-deficient cells [144].
NMD and human disease
Nonsense-mediated RNA decay and its regulation influence the development of human disease. While some disease phenotypes are exacerbated by the effects of NMD, others are suppressed by them, making NMD a “double-edged sword”. One example where NMD contributes to disease is β-thalassemia, which is often caused by mutations in the β-globin gene that generate a nonsense mRNA. Most recessive forms of β-thalassemia result from nonsense mutations in the first or second exon of the β-globin gene, with the corresponding mRNAs being targeted for degradation by NMD [145,146,147]. In these cases, the unaffected allele is still able to be expressed but the amount of protein produced is unable to compensate for loss of function of the mutant allele. Mutations that occur in the final exon of β-globin evade degradation and consequently are translated normally. However, the resulting truncated proteins have dominant negative activity that interferes with normal hemoglobin function [145].
Numerous other genetic diseases, including cystic fibrosis, polycystic kidney disease, and muscular dystrophy, are also caused by PTCs that trigger NMD of target mRNAs [145, 147]. Interestingly, different subtypes of muscular dystrophy can result from mis-expression of distinct genes that are associated with NMD. Duchenne’s muscular dystrophy results from loss of function of dystrophin, which can occur when mutations in the gene generate a PTC that targets the transcript for NMD. Facioscapulohumeral muscular dystrophy results from the misexpression of the DUX4 transcription factor in muscle. DUX4 is normally a substrate for NMD, but its misexpression in muscle leads to the inhibition of NMD, resulting in a regulatory feedback loop that further stabilizes the DUX4 transcript, leading to cellular toxicity [148].
Certain neurodevelopmental disorders are closely connected with dysregulation of NMD. Mutations in the NMD factor UPF3B have been found to cause syndromic and nonsyndromic intellectual disability (ID) [101, 102, 104, 106]. UPF3B mutations are also associated with a spectrum of disorders including attention-deficit hyperactivity disorder, autism and schizophrenia [102,103,104]. Dysregulation of other NMD factors such as UPF2 and SMG6, is also associated with various forms of ID [102].
Aberrant NMD also is associated with inflammation and cancer. Deletion of one allele of the NMD kinase SMG1 in a mouse model results in chronic inflammation as well as cancer predisposition [3]. Mutations in UPF1 have been identified in inflammatory myofibroblastic tumors (IMT) [149]. In IMT, decreased NMD function leads to increased expression of the transcript for the NIK protein kinase, which activates the NFkB pathway and promotes cytokine expression and inflammatory infiltrates [149]. Inhibition of NMD can cause chronic activation of the immune response, leading to autoimmunity [150]. Loss of function or overexpression of NMD factors have also been found to be associated with several other cancer types, including pancreatic cancer and neuroblastoma [151,152,153,154]. Deregulation of NMD contributes to tumorigenesis likely due to aberrant expression of oncogenes and tumor suppressor genes with PTCs [151, 155, 156].
Although decreased NMD efficiency can cause human disease and contribute to the severity of disease phenotypes, NMD inhibition can also be a strategy for disease treatment. Inhibiting NMD may alleviate the symptoms of certain genetic diseases caused by PTCs in a single gene—e.g. β-thalassemia, cystic fibrosis, Hurler’s syndrome, and Duchenne muscular dystrophy—by allowing expression of a mutant protein product that is partially functional [145]. However, this therapeutic strategy is limited by the ability of the truncated proteins to provide sufficient activity to compensate for the loss of function. A more promising solution may be to restore expression of full-length, functional proteins by combined treatment of NMD inhibitors (to stabilize nonsense transcripts) with drugs that allow stop-codon read-through. This strategy has been successfully used to restore full-length, functional proteins in a model of Hurler’s syndrome and in cancer cells with nonsense mutations in the p53 gene [157, 158]. A recent modification of this potential therapeutic strategy uses antisense oligonucleotides (ASOs), which are showing increasing promise in clinical trials, rather than small-molecule drugs to repress NMD activity, thereby expanding the repertoire of potential NMD-targeted therapeutic strategies [159]. Interestingly, increasing NMD activity, such as by overexpressing UPF1, can alleviate the phenotypes of amyotrophic lateral sclerosis (ALS) in both in vitro and in vivo models. A large fraction of ALS is caused by aberrant expression of TDP43, which deregulates splicing and generates many NMD targets [160, 161]. The observed effects of UPF1 overexpression suggest that NMD enhancers may be effective in treating certain forms of ALS and raise the possibility that a similar principle may apply to other disorders caused by aberrant RNA processing.
Due to the presence of a higher level of nonsense mRNAs caused by mutations and genomic instability in cancer cells, inhibition of NMD may cause accumulation of mutant proteins and activation of the unfolded protein response, leading to heightened cell death. Inhibition of NMD can also promote the expression of novel antigens on tumor cells, due to the translation of nonsense mRNAs generated by frameshift mutations or aberrant splicing [162, 163]. For these reasons, there has been a strong interest in developing small molecules to inhibit NMD activity (Table 2). Compounds such as cycloheximide and puromycin abrogate NMD by inhibiting translation, and other reagents that modify the specificity or efficacy of translation termination—suppressor tRNAs, aminoglycosides, PTC124, amlexanox—are also capable of stabilizing nonsense transcripts [95, 164,165,166,167,168,169,170]. Wortmannin and caffeine also inhibit NMD by decreasing SMG1 enzymatic activity, but these inhibitors are limited as tools because they also affect other PI3K family members such as ATM, ATR and DNA-PK [171, 172]. Inhibitors of SMG1 kinase activity with improved potency and selectivity, such as pyrimidine deriviatives, have been identified and shown to substantially diminish UPF1 phosphorylation in vitro and in cells [173]. Recently, other potent small molecule inhibitors selective for SMG1 kinase have been identified to inhibit UPF1 phosphorylation in cells and in mouse tumor xenograft models, where they promote anti-tumor efficacy (JMB, unpublished). Inhibitors to NMD factors other than SMG1 have also been reported. For example, patemine A was found to repress NMD activity by inhibiting the NMD function of eIF4AIII, whereas NMDI-1 blocks NMD by preventing the interaction between SMG5 and UPF1 [23, 174]. NMDI-14 was identified in a computational screen for molecules that physically prevent the interaction of SMG7 with UPF1 [158]. Promisingly, NMDI-1 and particularly NMDI-14 potently repress NMD at low concentrations with minimal cellular toxicity [158, 174]. In addition, the approved drugs 5-azacytidine and cardiac glycosides such as ouabain and digitoxin were recently found to inhibit NMD by upregulating Myc or by increasing intracellular calcium, respectively [137, 175]. These findings point to the potential of NMD-based therapeutic intervention by directly inhibiting NMD factors, or indirectly affecting the cellular microenvironment.
Perspectives
Nonsense-mediated RNA decay, initially discovered as a quality control mechanism that targeted mutant transcripts for degradation, is now widely appreciated as a key mechanism that regulates gene expression. NMD plays a crucial role in multiple cellular processes, including development, differentiation and disease physiology. While the main factors that drive NMD have been identified, many opportunities remain to fill in gaps in our understanding of NMD target selection and its impact on cell biology. A major area of ongoing NMD research will concern the complex regulatory networks that govern NMD activity in developmental and tissue-specific contexts. In addition to uncovering new pathways or processes where NMD is dynamically regulated, putative NMD transcripts must be validated and their effects on cell biology elucidated. Work discussed in this review has begun to address this challenge. The contribution of NMD to disease states, particularly neurological disorders and cancer will constitute another major direction of NMD research. The discovery of novel inhibitors—and potentially also enhancers—of the NMD pathway provide the possibility for therapeutic intervention with genetic diseases, neurological disorders, and cancer. NMD inhibition by chemical or genetic means has been demonstrated to restore expression of proteins in vitro, but the viability of these strategies in vivo—including in humans—remains to be tested. Furthering this promising work is paramount to applying our ever-expanding understanding of NMD to the treatment of human disease.
References
Mcllwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW. SMG1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA. 2010;107:12186–91.
Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet. 2001;10:99–105.
Roberts TL, Ho U, Luff J, Lee CS, Apte SH, MacDonald KP, Raggat LJ, Pettit AR, Morrow CA, Waters MJ, et al. SMG1 haploinsufficiency predisposes to tumor formation and inflammation. Proc Natl Acad Sci USA. 2013;110:E285–94.
Thoren LA, Nørgaard GA, Weischenfeldt J, Waage J, Jakobsen JS, Damgaard I, Bergström FC, Blom AM, Borup R, Bisgaard HC, et al. UPF2 is a critical regulator of liver development, function and regeneration. PLoS ONE. 2010;5(7):e11650.
Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008;22:1381–96.
Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, Chen YS, Groth M, Krueger A, Platzer M, et al. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 2015;34:1630–47.
Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, Lou CH, Espinoza JL, Song HW, Phan MH, et al. The antagonistic gene paralogs UPF3a and UPF3b govern nonsense-mediated RNA decay. Cell. 2016;165:382–95.
Anastasaki C, Longman D, Capper A, Patton EE, Caceres JF. Dhx34 and Nbas function in the NMD pathway and are required for embryonic development in zebrafish. Nucleic Acids Res. 2011;39:3686–94.
Wittkopp N, Huntzinger E, Weiler C, Saulière J, Schmidt S, Sonawane M, Izaurralde E. Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol Cell Biol. 2009;29:3517.
Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–77.
Weischenfeldt J, Waage J, Tian G, Zhao J, Damgaard I, Jakobsen JS, Kristiansen K, Krough A, Wang J, Porse BT. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol. 2012;13:R35.
Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017;50:175–85.
He F, Jacobson A. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet. 2015;49:339–66.
Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc Natl Acad Sci USA. 1979;76:5134–7.
Pulak R, Anderson P. mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 1993;7:1885–97.
Le Hir H, Izaurralde E, Maquat LE, Moore MJ. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. EMBO J. 2000;19:6860–9.
Le Hir H, Gatfield D, Izaurralde E, Moore MJ. The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–97.
Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE. Y14 and hUPF3b form an NMD-activating complex. Mol Cell. 2003;11:939–49.
Viegas MH, Gehring NH, Breit S, Hentze MW, Kulozik AE. The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the nonsense mediated decay pathway. Nucleic Acids Res. 2007;35:4542–51.
Singh KK, Wachsmuth L, Kulozik AE, Gehring NH. Two mammalian MAGOH genes contribute to exon junction complex composition and nonsense-mediated decay. RNA Biol. 2013;10:1291–8.
Steckelberg A, Boehm V, Gromadzka AM, Gehring NH. CWC22 connects pre-mRNA splicing and exon junction complex assembly. Cell Rep. 2012;2:454–61.
Kim VK, Yong J, Kataoka N, Abel L, Diem MD, Dreyfuss G. The Y14 protein communicates to the cytoplasm the position of exon–exon junctions. EMBO J. 2001;20:2062–8.
Dang Y, Low WK, Xu J, Gehring NH, Dietz HC, Romo D, Liu JO. Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J Biol Chem. 2009;284:23613–21.
Palacios IM, Gatfield D, St Johnston D, Izaurralde E. An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature. 2004;427:737–57.
Singh G, Jakob S, Kleedehn MG, Lykke-Andersen J. Communication with the exon-junction complex and activation of nonsense-mediated decay by human Upf proteins occur in the cytoplasm. Mol Cell. 2007;27:780–92.
Bono F, Ebert J, Unterholzner L, Güttler T, Izaurralde E, Conti E. Molecular insights into the interaction of PYM with the Mago-Y14 core of the exon junction complex. EMBO Rep. 2004;5:304–10.
Lykke-Andersen J, Mei-Di S, Steitz JA. Communication of the position of exon–exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science. 2001;293:1836–9.
Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–9.
Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111.
Ohnishi T, Yamashita A, Kashima I, Schell T, Anders KR, Grimson A, Hachiya T, Hentze MW, Anderson P, Ohno S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell. 2003;12:1187–200.
Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S. N- and C-terminal UPF1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012;40:1251–66.
Loh B, Jonas S, Izaurralde E. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013;27:2125–38.
Unterholzner L, Izaurralde E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell. 2004;16:587–96.
Cho H, Han S, Choe J, Park SG, Choi SS, Kim YK. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res. 2013;41:1319–28.
Lejeune F, Li X, Maquat LE. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylation, and exonucleolytic activities. Mol Cell. 2003;12:675–87.
Lykke-Andersen J. Identification of a human decapping complex associated with the hUpf proteins in nonsense-mediated decay. Mol Cell Biol. 2002;22:8114–21.
Chen CY, Shyu AB. Rapid deadenylation triggered by a nonsense codon precedes decay of the RNA body in a mammalian cytoplasmic nonsense-mediated decay pathway. Mol Cell Biol. 2003;23:4805–13.
Coullet P, Grange T. Premature termination codons enhance mRNA decapping in human cells. Nucleic Acids Res. 2004;32:488–94.
Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, Shyu AB. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol. 2005;12:1054–63.
Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoans. RNA. 2008;14:2609–17.
Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol. 2009;16:49–55.
Lykke-Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014;28:2498–517.
Schmidt SA, Foley PL, Jeong DH, Rymarquis LA, Doyle F, Tenenbaum SA, Belasco JG, Green PJ. Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res. 2015;43:309–23.
Hogg JR, Goff SP. UPF1 senses 3′ UTR length to potentiate mRNA decay. Cell. 2010;143:379–89.
Eberle AB, Stalder L, Mathys H, Orozco RZ, Muhlemann O. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol. 2008;6:e92.
Buhler M, Steiner S, Mohn F, Paillusson A, Muhlemann O. EJC-independent degradation of nonsense immunoglobulin-μ mRNA depends on 3′ UTR length. Nat Struct Mol Biol. 2006;13:462–4.
Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–8.
Karousis ED, Nasif S, Muhlemann O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip Rev RNA. 2016;7:661–82.
Hug N, Longman D, Caceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44:1483–95.
Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129:461–7.
Baboo S, Bhushan B, Jiang H, Grovenor CR, Pierre P, Davis BG, Cook PR. Most human proteins made in both nucleus and cytoplasm turn over within minutes. PLoS ONE. 2014;9:e99346.
Apcher S, Millot G, Daskalogianni C, Scherl A, Manoury B, Fahraeus R. Translation of pre-spliced RNAs in the nuclear compartment generates peptides for the MHC class I pathway. Proc Natl Acad Sci USA. 2013;110:17951–6.
Iborra FJ, Jackson DA, Cook PR. Coupled transcription and translation within nuclei of mammalian cells. Science. 2001;293:1139–42.
Cheng J, Maquat LE. Nonsense codons can reduce the abundance of nuclear mRNA without affecting the abundance of pre-mRNA or the half-life of cytoplamsic mRNA. Mol Cell Biol. 1993;13:1892–902.
Belgrader P, Cheng J, Zhou X, Stephenson LS, Maquat LE. Mammalian nonsense codons can be cis effectors of nuclear mRNA half-life. Mol Cell Biol. 1994;14:8219–28.
Belgrader P, Maquat LE. Nonsense but not missense mutations can decrease the abundance of nuclear mRNA for the mouse major urinary protein, while both types of mutations can facilitate exon skipping. Mol Cell Biol. 1994;14:6326–36.
Cheng J, Belgrader P, Zhou X, Maquat LE. Introns are cis effectors of the nonsense-codon-mediated reduction in nuclear mRNA abundance. Mol Cell Biol. 1994;14:6317–25.
Bhalla AD, Gudikote JP, Wang J, Chan WK, Chang YF, Olivas OR, Wilkinson MF. Nonsense codons trigger an RNA partitioning shift. J Biol Chem. 2009;284:4062–72.
Lozano F, Maertzdorf B, Pannell R, Milstein C. Low cytoplasmic mRNA levels of immunoglobulin kappa light chain genes containing nonsense codons correlate with inefficient splicing. EMBO J. 1994;13:4617–22.
Gersappe A, Pintel DJ. A premature termination codon interferes with the nuclear function of an exon splicing enhancer in an open reading frame-dependent manner. Mol Cell Biol. 1999;19:1640–50.
Muhlemann O, Mock-Casagrande CS, Wang J, Li S, Custodio N, Carmo-Fonseca M, Wilkinson MF, Moore MJ. Precursor RNAs harboring nonsense codons accumulate near the site of transcription. Mol Cell. 2001;8:33–43.
Buhler M, Wilkinson MF, Muhlemann O. Intranuclear degradation of nonsense codon-containing mRNA. EMBO Rep. 2002;3:646–51.
Kessler O, Chasin LA. Effects of nonsense mutations on nuclear and cytoplasmic adenine phosphoribosyltransferase RNA. Mol Cell Biol. 1996;16:4426–35.
Takeshita K, Forget BG, Scarpa A, Benz EJ. Intranuclear defect in beta- globin mRNA accumulation due to a premature translation termination codon. Blood. 1984;64:13–22.
Wang J, Chang YF, Hamilton JI, Wilkinson MF. Nonsense-associated altered splicing: a frame-dependent response distinct from nonsense-mediated decay. Mol Cell. 2002;10:951–7.
Trcek T, Sato H, Singer RH, Maquat LE. Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 2013;27:541–51.
Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–92.
Cao D, Parker R. Computational modeling and experimental analysis of nonsense-mediated decay in yeast. Cell. 2003;113:533–45.
He F, Li X, Spatrick P, Casillo R, Dong S, Jacobson A. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol Cell. 2003;12:1439–52.
Mendell JT, Sharifi NA, Meyers JL, Matinez-Murillo R, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–8.
Wittmann J, Hol EM, Hans-Martin J. hUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol Cell Biol. 2006;26:1272–87.
Tani H, Imamachi N, Salam KA, Mizutani R, Ijiri K, Irie T, Yada T, Suzuki Y, Akimitsu N. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol. 2012;9:1370–9.
Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol. 2011;31:3670–80.
Yepiskoposyan H, Aescimann F, Nilsson D, Okoniewski M, Mühlemann O. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA. 2011;17:2108–18.
Rehwinkel J, Letunic I, Raes J, Bork P, Izaurralde E. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA. 2005;11:1530–44.
Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100.
Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21:708–18.
Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with high conserved and ultraconserved DNA elements. Nature. 2007;446:926–9.
Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, Blencowe BJ. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol Cell Biol. 2008;28:4320–30.
Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001;20:1785–96.
Wilson GM, Sun Y, Sellers J, Lu H, Penkar N, Dillard G, Brewer G. Regulation of AUF1 expression via conserved alternatively spliced elements in the 3′ untranslated region. Mol Cell Biol. 1999;19:4056–64.
Malabat C, Feuerbach F, Ma L, Saveanu C, Jacquier A. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. eLife. 2015;4:e06722.
Belew AT, Hepler NL, Jacobs JL, Dinman JD. PRFdb: a database of computationally predicted eukaryotic programmed-1 ribosomal frameshift signals. BMC Genom. 2008;9:339.
Belew AT, Advani VM, Dinman JD. Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res. 2011;39:2799–808.
Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, Sulima SO, Kasprzak WK, Shapiro BA, Dinman JD. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature. 2014;512:265–9.
Advani VM, Belew AT, Dinman JD. Yeast telomere maintenance is globally controlled by programmed ribosomal frameshifting and the nonsense-mediated mRNA decay pathway. Translation. 2013;1:e24418.
Barbosa C, Peixeiro I, Romão L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9:e1003529.
Hurt JA, Robertson AD, Burge CB. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013;23:1636–50.
Toma KG, Rebbapragada I, Durand S, Lykke-Andersen J. Identification of elements in human long 3′ UTRs that inhibit nonsense-mediated decay. RNA. 2015;21:887–97.
Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, Song HW, Corbett MA, Gifford WD, Gecz J, et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell. 2011;42:500–10.
Huang L, Lou CH, Chan W, Shum EY, Shao A, Stone E, Karam R, Song HW, Wilkinson MF. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell. 2011;43:950–61.
Shetty SP, Copeland PR. Selenocysteine incorporation: a trump card in the game of mRNA decay. Biochimie. 2015;114:97–101.
Seyedali A, Berry MJ. Nonsense-mediated decay factors are involved in the regulation of selenoprotein mRNA levels during selenium deficiency. RNA. 2014;20:1248–56.
Usuki F, Yamashita A, Fujimura M. Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. J Biol Chem. 2011;286:6641–9.
Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reverse by protein synthesis inhibitors in vitro. J Biol Chem. 1995;270:28995–9003.
Gudikote JP, Wilkinson MF. T-cell receptor sequences that elicit strong downregulation of premature termination codon-bearing transcripts. EMBO J. 2002;21:125–34.
Frischmeyer-Guerrerio PA, Montgomery RA, Warren DS, Cooke SK, Lutz J, Sonnenday CJ, Guerrerio AL, Dietz HC. Perturbation of thymocyte development in nonsense-mediated decay (NMD)-deficient mice. Proc Natl Acad Sci USA. 2011;108:10638–43.
Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154:583–95.
Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, Wilkinson MF. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 2014;6:748–64.
Lou CH, Dumdie J, Goetz A, Shum EY, Brafman D, Liao X, Mora-Castilla S, Ramaiah M, Cook-Andersen H, Laurent L, et al. Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Rep. 2016;6:844–57.
Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoulbridge C, Edkins S, Stevens C, et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet. 2007;39:1127–33.
Nguyen LS, Kim HG, Rosenfeld JA, Shen Y, Gusella JF, Lacassie Y, Layman LC, Shaffer LG, Gécz J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet. 2013;22:1816–25.
Addington AM, Gauthier J, Piton A, Hamdan FF, Raymond A, Gogtay N, Miller R, Tossell J, Bakalar J, Inoff-Germain G, et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry. 2011;16:238–9.
Laumonnier F, Shoubridge C, Antar C, Nguyen LS, Van Esch H, Kleefstra T, Briault S, Fryns JP, Hamel B, Chelly J, et al. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol Psychiatry. 2010;15:767–76.
Jolly LA, Homan CC, Jacob R, Barry S, Gécz J. The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum Mol Genet. 2013;22:4673–87.
Xu X, Zhang L, Tong P, Xun G, Su W, Xiong Z, Zhu T, Zheng Y, Luo S, Pan Y, et al. Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin Genet. 2013;83:560–4.
Alrahbeni T, Sartor F, Anderson J, Miedzybrodzka Z, McCaig C, Muller B. Full UPF3B function is critical for neuronal differentiation of neural stem cells. Mol Brain. 2015;8:33.
Giorgi C, Yeo GW, Stone ME, Katz DB, Burge C, Turrigiano G, Moore MJ. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell. 2007;130:179–91.
Colak D, Ji SJ, Porse BT, Jaffrey SR. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell. 2013;153:1252–65.
Eom T, Zhang C, Wang H, Lay K, Fak J, Noebels JL, Darnell RB. NOVA-dependent regulation of cryptic NMD exons controls synaptic protein levels after seizure. elife. 2013;2:e00178.
Kim YK, Furic L, Desgroseillers L, Maquat LE. Mammalian staufen1 recruits UPF1 to specific mRNA 3′UTRs so as to elicit mRNA decay. Cell. 2005;120:195–208.
Gong C, Kim YK, Woeller CF, Tang Y, Maquat LE. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 2009;23:54–66.
Park E, Maquat LE. Staufen-mediated mRNA decay. Wiley Interdiscip Rev RNA. 2013. doi:10.1002/wrna.1168.
Balistreri G, Horvath P, Schweingruber C, Zünd D, Mclnerney G, Merits A, Mühlemann O, Azzalin C, Helenius A. The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host Microbe. 2014;16:403–11.
Garcia D, Garcia S, Voinnet O. Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe. 2014;16:391–402.
Mocquet V, Neusiedler J, Rende F, Cluet D, Robin JP, Terme JM, Duc Dodon M, Wittmann J, Morris C, Le Hir H, Ciminale V, Jalinot P. The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J Virol. 2012;86:7530–43.
Nakano K, Ando T, Yamagishi M, Yokoyama K, Ishida T, Ohsugi T, Tanaka Y, Brighty DW, Watanabe T. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: implications for retroviral replication. Microbes Infect. 2013;15:491–505.
Weil JE, Beemon KL. A 3′ UTR sequence stabilizes termination codons in the unspliced RNA of Rous sarcoma virus. RNA. 2006;12:102–10.
Quek BL, Beemon K. Retroviral strategy to stabilize viral RNA. Curr Opin Microbiol. 2014;18:78–82.
Ramage HR, Kumar GR, Verschueren E, Johnson JR, Von Dollen J, Johnson T, Newton B, Shah P, Horner J, Krogran NJ, et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell. 2015;57:329–40.
Taft RJ, Pheasant M, Mattick JS. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays. 2007;29:288–99.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8.
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802.
Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–7.
Chew GL, Pauli A, Rinn JL, Regev A, Schier AF, Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–34.
van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, Hao W, Macinnes AW, Cuppen E, Simonis M. Extensive localization of long noncoding RNAs to the cytosol and mono and polyribosomal complexes. Genome Biol. 2014;15:R6.
Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, Geisler S, Hu W, Coller J, Baker KE. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–66.
Wery M, Descrimes M, Vogt N, Dallongeville AS, Gautheret D, Morillon A. Nonsense-mediated decay restricts lncRNA levels in yeast unless blocked by double-stranded RNA structure. Mol Cell. 2016;61:379–92.
Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–41.
Martin L, Gardner LB. Stress-induced inhibition of nonsense-mediated RNA decay regulates intracellular cystine transport and intracellular glutathione through regulation of the cystine/glutamate exchanger SLC7A11. Oncogene. 2015;34:4211–8.
Karam R, Lou CH, Kroeger H, Huang L, Lin JH, Wilkinson MF. The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 2015;16:599–609.
Oren YS, McClure ML, Rowe SM, Sorcher EJ, Bester AC, Manor M, Kerem E, Rivlin J, Zahdeh F, Mann M, Geiger T, Kerem B, et al. The unfolded protein response affects readthrough of premature termination codons. EMBO Mol Med. 2014;6:685–701.
Gardner LB. Nonsense-mediated RNA decay regulation by cellular stress: implications for tumorigenesis. Mol Cancer Res. 2010;8:295–308.
Sieber J, Hauer C, Bhuvanagiri M, Leicht S, Krijgsveld J, Neu-Yilik G, Hentze MW, Kulozik AE. Proteomic analysis reveals branch-specific regulation of the unfolded protein response by nonsense-mediated mRNA decay. Mol Cell Proteom. 2016;15:1584–97.
Nickless A, Jackson E, Marasa J, Nugent P, Mercer RW, Piwnica-Worms D, You Z. Intracellular calcium regulates nonsense-mediated mRNA decay. Nat Med. 2014;20:961–6.
Popp MW, Maquat LE. Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat Commun. 2015;6:6632.
Tani H, Torimura M, Akimitsu N. The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS ONE. 2013;8:e55684.
Nelson JO, Moore KA, Chapin A, Hollien J, Metzstein MM. Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability. eLife. 2016;5:e12876.
Longman D, Hug N, Keith M, Anastasaki C, Patton EE, Grimes G, Cáceres JF. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013;41:8319–31.
Chan W, Huang L, Gudikote JP, Chang Y, Imam JS, MacLean JA II, Wilkinson MF. An alternative branch of the nonsense-mediated decay pathway. EMBO J. 2007;26:1820–30.
Gehring NH, Kunz JB, Neu-Yilik G, Breit S, Viegas MH, Hentze MW, Kulozik AE. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol Cell. 2005;20:65–75.
Wengrod J, Martin L, Wang D, Frischmeyer-Guerrerio P, Dietz HC, Gardner LB. Inhibition of nonsense-mediated RNA decay activates autophagy. Mol Cell Biol. 2013;33:2128–35.
Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;2004(36):801–8.
Maquat LE, Kinniburgh AJ, Rachmilewitz EA, Ross J. Unstable β-globin mRNA in mRNA-deficient β0 thalassemia. Cell. 1981;27:543–53.
Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–900.
Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, Bradley RK. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife. 2015;4:e04996. doi:10.7554/elife.04996.
Lu J, Plank TD, Su F, Shi X, Liu C, Ji Y, Li S, Huynh A, Shi C, Zhu B, et al. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J Clin Invest. 2016;126:3058–62.
Rigby RE, Rehwinkel J. RNA degradation in antiviral immunity and autoimmunity. Trends Immunol. 2015;36:179–88.
El-Bchiri J, Guilloux A, Dartigues P, Loire E, Mercier D, Buhard O, Sobhani I, de la Grange P, Auboeuf D, Praz F, et al. Nonsense-mediated mRNA decay impacts MSI-driven carcinogenesis and anti-tumor immunity in colorectal cancers. PLoS ONE. 2008;3:e2583.
Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–84.
Beheshti B, Braude I, Marrano P, Throner P, Zielenska M, Squire JA. Chromosomal localization of DNA amplifications in neuroblastoma tumors using cDNA microarray comparative genomic hybridization. Neoplasia. 2003;5:53–62.
Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamaous carcinoma. Nat Med. 2014;20:596–8.
Karam R, Carvalho J, Bruno I, Graziadio C, Senz J, Huntsman D, Carneiro F, Seruca R, Wilkinson MF, Oliveira C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27:4255–60.
De Rosa M, Morelli G, Cesaro E, Duraturo F, Turano M, Rossi GB, Delrio P, Izzo P. Alternative splicing and nonsense-mediated mRNA decay in the regulation of a new adenomatous polyposis coli transcript. Gene. 2007;395:8–14.
Keeling KM, Wang D, Dai Y, Murugesan S, Chenna B, Clark J, Belakhov V, Kandasamy J, Velu SE, Baasov T, et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE. 2013;8:e60478.
Martin L, Grigoryan A, Wang D, Wang J, Breda L, Rivella S, Cardozo T, Gardner LB. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014;74:3104–13.
Nomakuchi TT, Rigo F, Aznarez I, Krainer AR. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol. 2016;34:164–6.
Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Xingli L, Zhang Y, Tank EMH, Qiu H. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci USA. 2015;112:7821–6.
Jackson KL, Dayton RD, Orchard EA, Ju S, Ringe D, Petsko GA, Maquat LE, Klein RL. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene Ther. 2015;22:20–8.
Gilboa E. Expression of new antigens on tumor cells by inhibiting nonsense-mediated mRNA decay. Immunol Res. 2013;57:44–51.
Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature. 2010;465:227–30.
Temple GF, Dozy AM, Roy KL, Kan YW. Construction of a functional human suppressor tRNA gene: an approach to gene therapy for beta-thalassaemia. Nature. 1982;296:537–40.
Kiselev AV, Ostapenko OV, Rogozhkina EV, Kholod NS, Seit Nebi AS, Baranov AN, Lesina EA, Ivashchenko TE, SabetskiĬ VA, Shavlovskiĭ MM, et al. Suppression of nonsense mutations in the Dystrophin gene by a suppressor tRNA gene. Mol Biol (Mosk). 2002;36:43–7.
Buvoli M, Buvoli A, Leinwand LA. Suppression of nonsense mutations in cell culture and mice by multimerized suppressor tRNA genes. Mol Cell Biol. 2000;20:3116–24.
Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91.
Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and puromycin. Nucleic Acids Res. 1985;13:6265–72.
Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and p53 cDNAs in a mammalian translation system. J Mol Med (Berl). 2002;80:367–76.
Gonzalez-Hilarion S, Beghyn T, Jia J, Bebreuck N, Berte G, Mamchaoui K, Mouly V, Gruenert DC, Déprez B, Lejeune F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58.
Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated decay. Genes Dev. 2001;15:2215–28.
Denning G, Jamieson L, Maquat LE, Thompson EA, Fields AP. Cloning of a novel phosphatidylinositol kinase-related kinase: characterization of the human SMG-1 RNA surveillance protein. J Biol Chem. 2001;276:22709–14.
Gopalsamy A, Bennett EM, Shi M, Zhang WG, Bard J, Yu K. Identification of pyrimidine derivatives as hSMG-1 inhibitors. Bioorg Med Chem Lett. 2012;22:6636–41.
Durand S, Cougot N, Mahuteau-Betzer F, Nguyen CH, Grierson DS, Bertrand E, Tazi J, Lejeune F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol. 2007;178:1145–60.
Bhuvanagiri M, Lewis J, Putzker K, Becker JP, Leicht S, Krijgsveld J, Batra R, Turnwald B, Jovanovic B, Hauer C, et al. 5-azacytidine inhibits nonsense-mediated decay in a MYC-dependent fashion. EMBO Mol Med. 2014;6:1593–609.
Peccarelli M, Kebarra BW. Regulation of natural mRNAs by the nonsense-mediated mRNA decay pathway. Eukaryot Cell. 2014;13:1126–35.
Authors’ contributions
All authors read and approved the final manuscript.
Acknowledgements
We apologize to colleagues whose work is not cited due to space limitation. Z.Y. is supported by a NIH Grant (R01GM098535), an American Cancer Society Research Scholar Grant (RSG-13-212-01-DMC) and a Siteman Investment Program Grant (4036) from the Siteman Cancer Center of Washington University.
Competing interests
The authors declare that they have no competing interests. J.M.B. is an employee of Amgen, Inc.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nickless, A., Bailis, J.M. & You, Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci 7, 26 (2017). https://doi.org/10.1186/s13578-017-0153-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-017-0153-7