
Abstract
Phenotypic determination of antimicrobial resistance in bacteria is very important for diagnosis and treatment, but sometimes this procedure needs further genetic evaluation. Whole-genome sequencing plays a critical role in deciphering and advancing our understanding of bacterial evolution, transmission, and surveillance of antimicrobial resistance. In this study, whole-genome sequencing was performed on nineteen clinically extraintestinal Escherichia coli isolates from chicken, cows and swine and showing different antimicrobial susceptibility. A total of 44 different genes conferring resistance to 11 classes of antimicrobials were detected in 15 of 19 E. coli isolates (78.9%), and 22 types of plasmids were detected in 15/19 (78.9%) isolates. In addition, whole-genome sequencing of these 19 isolates identified 111 potential virulence factors, and 53 of these VFDB-annotated genes were carried by all these 19 isolates. Twelve different virulence genes were identified while the most frequent ones were gad (glutamate decarboxylase), iss (increased serum survival) and lpfA (long polar fimbriae). All isolates harbored at least one of the virulence genes. The findings from comparative genomic analyses of the 19 diverse E. coli isolates in this study provided insights into molecular basis of the rising multi-drug resistance in E. coli.
Similar content being viewed by others

Introduction
Antimicrobial resistance (AMR) in bacteria is an important issue related to the health of both human and animals (McDermott et al. 2016; Zawack et al. 2016). It has been reported that AMR bacteria are responsible for about 50,000 deaths in people each year in the USA and Europe, and 700,000 global death (Bottacini et al. 2017) (Zhang et al. 2017). Studies suggested that the agricultural use of antimicrobial agents increases the number of human infections caused by drug-resistant bacteria (O’Neill 2015). Thus, the AMR monitoring system should focus not only on humans, but also animal hosts and the associated environments (Zhang et al. 2017).
Escherichia coli is a well-known commensal of the gastrointestinal tract of numerous animals, and also involved in intestinal and extraintestinal pathologies (Tenaillon et al. 2010; Croxen and Finlay 2010). E. coli show a clonal population structure with the delineation of at least seven main phylogenetic groups (Desjardins et al. 1995; Clermont et al. 2011). The chromosomal elasticity of the strains helps E. coli to adapt to different environments (Touchon et al. 2009).
One of the key issues with E. coli is its role in the dissemination and emergence of bacterial antimicrobial resistance. Most of the resistance properties emerge from commensal bacteria in the gastrointestinal tract (Andremont 2003) where the bacteria grow to higher density, allowing horizontal transfers of resistance genes between strains from a single species, and even between species and genera. One of the mechanisms involved in the spread of AMR is the emergence of some specific clones that acquire resistance genes, mostly via mobile genetic elements including plasmids, gene cassettes, transposons, and other integrative genetic elements (Woodford et al. 2011).
Because of its high level of discriminatory power, microbial Whole Genome Sequencing (WGS) strategy plays an important role in the investigation and surveillance of foodborne disease outbreak (Kröger et al. 2012). Better understanding of bacterial evolution, outbreaks, and transmission events revealed with the advent of WGS approach has been shown in a number of recent studies as well as from the surveillance of antimicrobial resistance (Zankari et al. 2012). It has significant advantages compared to other commonly used drug resistant testing approaches. WGS as a diagnostic method to detect bacterial antimicrobial resistance is particularly important where congruence exists between phenotype and genotype, and where phenotypic testing is prohibitively slow for slow-growing bacteria. Epidemiology has benefited greatly from high-throughput WGS in the aspects of identifying and tracking drug-resistant organisms as well as of identifying their genetic diversity (Organization 2014).
The main aim of this study is to investigate the genetic diversity and relationship of E. coli clinical strains collected from chicken, cows and swine in China. The E. coli clinical strains were further characterized and assigned to the unique profiles of virulence factors and antimicrobial resistance genes.
Materials and methods
Bacterial strains
A total of 19 E. coli isolates (10 from chicken, 5 from swine, 4 from cows) were selected for WGS analysis in this study. The selection of these isolates was based on their significantly different phenotypes of antimicrobial resistance (Yassin et al. 2017a, b) (Table 1).
Whole genome sequencing and data analysis
Genomic DNA from all the 19 E. coli isolates were extracted, end repaired, ligated to specific adaptors and subject to paired-end sequencing using Illumina HiSeq 2500 by PE125 strategy at Beijing Novogene Bioinformatics Technology Company (Beijing, China). After filtering raw reads, the clean reads were de novo assembled into contigs using the CLC Genomics Workbench. The assembled genomes were analyzed by using online software tools provided by the Centre of Genomic Epidemiology (https://www.genomicepidemiology.org/). In addition, CGE ResFinder 2.1 was used to identify the antimicrobial resistance genes in the assembled genomes using (Zankari et al. 2012). The minimum percentage of the gene length detected and the identity threshold was set to be a 90.0% identity for a positive match between a target genome and the reference database.
The MLST server database v1.7 (Larsen et al. 2012) and the Virulence Finder server database v1.2 (Joensen et al. 2014) in the CGE website were used to identify virulence genes and housekeeping genes (adk, fumC, icd, gyrB, mdh, purA, recA). The scaffolds of each isolate were incorporated into these tools as described in CGE, with an identity threshold set to be 98%. The profile of replicons of bacterial plasmids was identified by the use of PlasmidFinder-1.3 (Carattoli et al. 2014).
Double index alignment of next-generation sequencing data (DIAMOND) (Buchfink et al. 2015) was applied to align the amino acid sequences against the VFDB database (Chen et al. 2015). The annotation of predicted gene with the description of the best fit was defined as amino acid sequences with alignment length > 90% of its own length and over 20% match identity.
Phylogenomic relationships among strains were assessed based on nucleotide alignments of the core genome gene content, including only the single-copy orthologues. An additional filter for paralogues was applied to the core genome in order to exclude families represented by more than a single member since they do not represent robust evolutionary markers (Gutiérrez and Maere 2014). Gene alignments were conducted using MUSCLE v.3.8.31 (Edgar 2004), followed by construction of a phylogenetic tree for each single-copy gene using the maximum-likelihood (≤5 samples) in PhyML v3.0 (Guindon and Gascuel 2003) and tree concatenation (Bottacini et al. 2017).
GenBank accession numbers
This Whole Genome Shotgun project 19 E. coli isolates investigated in this study has been deposited at DDBJ/ENA/GenBank under the following accession numbers: E. coli E565, QETO00000000; E. coli E535, QETQ00000000; E. coli E533, QETR00000000; E. coli E530, QETS00000000; E. coli E497, QETT00000000; E. coli E461, QETU00000000; E. coli E433, QETV00000000; E. coli E418, QETW00000000; E. coli E393, QETX00000000; E. coli E386, QETY00000000; E. coli E205, QETZ00000000; E. coli E175, QEUA00000000; E. coli E122, QEUB00000000; E. coli E100, QEUC00000000; E. coli E80, QEUD00000000; E. coli E34, QEUE00000000; E. coli E30, QEUF00000000; and E. coli E2, QEUG00000000.
Strain E497 from chicken, one of the multidrug-resistant E. coli strains investigated in this study, was deposited at China General Microbiological Culture Collection Center (CGMCC) with a deposit number of CGMCC-10601.
Results
Phenotypic analysis of antimicrobial resistance of 19 clinical E. coli strains
All 19 clinical E. coli isolates, except for E433, E535, E80 and E533, displayed MDR phenotype with resistance to 2–16 antimicrobials (Table 1). In total, we identified 14 resistance patterns, and none of the strains was resistant to ertapenem. Based on antibiogram results, the 19 clinical E. coli isolates were divided into five groups (Table 1).
Genotypic analysis of antibiotic resistance genes
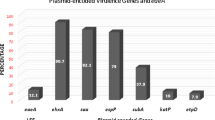
Four of 19 strains did not harbor any of the antimicrobial resistance genes analyzed, while the remaining 15 isolates had more than one resistance gene. A total of 44 different antimicrobial resistance genes were identified in these 15 isolates conferring resistance to 11 classes of antimicrobials (Fig. 1). One isolate (5.3%) possess mcr-1 gene in a full-length copy of a colistin resistance gene that showed 100% nucleotide similarity to the reference database sequence (Table 2).

Presence of antibiotic resistance gene in clinical WGS E. coli isolates. The X axis shows the antibiotic resistance gene, and the Y-axis is the number of resistance genes in clinical E. coli isolates. The colors of the bars denotes resistance to different classes of antimicrobials
Prevalence of plasmid replicons
Through WGS analysis, 22 plasmid replicons were identified in 15 of the 19 isolates. Fourteen isolates harbored multiple plasmid replicons. Eighteen types of Inc with different frequencies were found, including IncA/C2, IncFIA, IncFIB (AP001918), IncFIB(K), IncFIB(pLF82), IncFIC(FII), IncFII, IncFII(pCoo), IncFII(pHN7A8), IncHI2, IncHI2A, IncI1, IncI2, IncN,, IncQ1, IncR, IncX1 and IncY (Table 3).
Virulence genes
At least one virulence gene was detected in all 19 E. coli isolates evaluated. Twelve different virulence genes were identified while the most frequent ones were gad (glutamate decarboxylase), iss (increased serum survival) and lpfA (long polar fimbriae) which were identified in 14, 11 and 9 isolates, respectively (Table 4).
Mlst
The multilocus sequence typing of the isolates is shown in Table 5. Three isolates (E100, E386 and E433) belong to Sequencing Type (ST) 155 while two isolates (E461 and E565) belong to ST 23 and another two isolates (E80 and E533) belong to ST 297. The remaining 12 isolates belong to individual MLST types, ST2505, ST746, ST656, ST10, ST3345, ST4012, ST6856, ST602, ST2111, ST5019, ST548 and ST4753.
VFDB analysis
In this study, WGS of these 19 isolates and the analysis identified 111 potential virulence factors. Fifty-three of these VFDB-annotated genes were carried by all these 19 isolates. The VFDB-annotated genes are responsible to adherence, autotransporter, invasion, iron uptake, toxins, secretion system and secretion system related effectors. Different potential virulence factors with different abundance were observed, and the most abundant virulence factors was associated with adherence. In addition, some of the isolates contained pathogenic E. coli virulence factors, such as Chu (isolate E80) present in Enterohemorrhagic E. coli (EHEC) and Per (isolates E30, E205, E433, E533) present in Enteropathogenic E. coli (EPEC).
Phylogenomic analysis
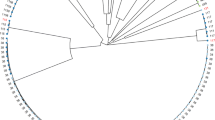
In this study, a total of 2858 core genes are present as single copies in isolates, and the resulting phylogenetic tree was computed using the 19 clinical E. coli isolates sequenced while E. coli CP009072 serves as an outgroup (Fig. 2). The phylogenetic analysis provided complete resolution of relationships among all isolates sampled, with maximum support (100) for all nodes. The E. coli isolates are clustered in seven phylogenetic groups, and E543 was derived firstly from the lineage of the remaining members of the subfamily (Fig. 2). The isolates of E100, E386, E433 and E80 appear to be monophyletic and more closely related to E418 and E497 than to E2, E461 and E565. E. coli comprised this clade plus another in which E122, E535 plus E393 and E30 plus E34 (both monophyletic) were together adjacent to E175, E533, E205 and E530 (Fig. 2).

Phylogenomic analysis of E. coli isolates. Phylogenetic Supertree computed on concatenated single-copy core genes and using the Neighbor-Joining method showing the existing relationship between 19 E. coli species. CP009072 was used as the outgroup. Evolutionary analyses were conducted in TreeBest. The numbers on the branch indicate branch credibility. The branch length shows the size of the evolutionary distance which calculates in the units of the number of base substitutions per site
Discussion
The genetic characteristics of clinical E. coli isolates can provide useful information about the potential for causing disease and resistance to treatment. In this study, we analyzed the WGS of E. coli isolates obtained from chickens, swine, and cows in Jiangsu province, China. These isolates exhibited a diverse range of genetic profiles.
These isolates have quite comprehensive profiles being resistant to beta-lactams, quinolones, aminoglycosides and colistin. Most of the strains displayed consistent antibiogram and antibiotic resistance gene profiles, but some of them showed inconsistency between the phenotype and the genotype. For instance, there is not any antibiotic resistance gene cluster profile displayed in the isolates E497 and E565 which however displayed resistance to a panel of antibiotics (Tables 1, 2). This is likely due to an unidentified regulatory mechanism in these isolates. In this study, one of the most important antimicrobial resistance genes identified was mcr-1, mediating resistance against colistin (Liu et al. 2016; Yassin et al. 2017a, 2017b). The plasmid-borne colistin resistance gene mcr-1 was found in bacterial strains of both humans and animals (Liu et al. 2016). This is of a great public health concern as these colistin is considered as “last-resort” drugs for human infections caused by multi-drug resistant Enterobacteriaceae (Shaheen et al. 2013). The identified mcr-1 gene by WGS in this study was verified by PCR and the mcr-1-positive E. coli was verified to be resistant to colistin (Yassin et al. 2017a, 2017b).
Plasmids as diverse and self-replicating extrachromosomal elements encode a variety of traits which include antimicrobial resistance, virulence, and environmental adaptability. Plasmids also plays a major role in bacterial adaptation to environmental (Smets and Barkay 2005). Inc plasmids target the replicons of the major plasmid families occurring in Enterobacteriaceae (HI2, HI1, I1-ã, X, L/M, N, FIA, FIB, FIC, W, Y, P, A/C, T, K, B/O) (Carattoli 2009). Currently, there are 27 known Inc groups occurring among the Enterobacteriaceae family (Frost et al. 2005; Carattoli 2009). Classification of plasmids into Inc groups is desirable because specific plasmid types have been associated with virulence and/or antimicrobial resistance (Gilmour et al., 2004; Hopkins et al. 2006; Carattoli et al. 2014). In this study, 18 types of Inc plasmids were detected. IncI 1 plasmid has been shown to contribute to adhesion and invasion of shiga-toxigenic E. coli due to presence of a cluster encoding IV pili (Kim and Komano 1997). While plasmids mediating antimicrobial resistance in Enterobacteriaceae is highly variable, some plasmid families are largely prevalent and also prevalently associated with specific resistance genes (Carattoli 2009).
As shown in Table 5, 19 clinical E. coli isolates possess 15 different ST types based on the MLST analysis. ST10 is one of the important multilocus sequence types possessed by one isolate (E122) in our findings, which confers resistant to colistin and is often reported as antibiotics against ESBL-producing E. coli (Chen et al. 2016). This ST type is also commonly found in chickens, other animals and humans (Chen et al. 2016) and this is consistent with our findings that this strain was isolated from chicken.
Among 12 different virulence genes possessed by all isolates, the highest frequencies appeared was gad, iss and lpfA which were frequently reported in pathogenic E. coli isolates (Bergholz et al. 2007; Solà-Ginés et al. 2015; Malik et al. 2017). Thirteen isolates which carried these gene harbored multiple antimicrobial resistance genes, and 9 of them carried genes for lpfA virulence and that has been described to be a potential virulence marker for pathogenic E. coli (Petty et al. 2014). However, the presence of a single or multiple virulence genes in an E. coli strain does not warrant that a strain is pathogenic unless that strain has the appropriate combination of the virulence genes to cause infections in the hosts (Boerlin et al. 1999). Pathogenic E. coli uses a complex multi-step mechanism of pathogenesis involving a number of virulence factors which consists of attachment, host cell surface modification, invasion, a variety of toxins and secretion systems, eventually leading to death of the target host cells (Kaper et al. 2004). Thus, virulence genes are ideal targets for determining the pathogenic potential of a given E. coli isolate (Kuhnert et al. 2000).
Virulence factors are important for microbial pathogenesis. A mutation of a virulence factor from a virulent pathogen will attenuate the pathogen strain (Volk et al. 1995). However, virulence factors may also exist in attenuated and even avirulent strains (Chen et al. 2015).Our study attempted to characterize the clinical isolates for the presence of virulence associated genes by comparison against a database collection of virulence factors, VFDB. In our study, we observed that 53 of the VFDB-annotated genes were shared within 19 clinical E. coli isolates. The most abundant adherence found in our isolates maybe related to the IncI 1 plasmids, which can encode the type IV pili. These virulence factors, along with their epidemic ability and resistance determinants, may have favored the dissemination of plasmids belonging to IncI 1 plasmid family (Carattoli 2009).
Based on nucleotide alignments of the core genome of individual strains, phylogenomic investigation allowed us to deduce the evolutionary relationships between strains while 16S rRNA sequence-based phylogeny does not provide sufficient resolution at the intra-species level (Ventura et al. 2006). The phylogenetic analysis of the 19 clinical E. coli isolates in our study showed no group correlations between the isolates from the same species or numbers of antibiotic resistance genes possessed by them. MLST analysis showed that strains E100, E386, E433 and strains E461, E565 belonged to ST155 and ST23, respectively, which are phylogenetically similar. Unlike comparative genomics-derived clustering which is based on the presence-absence of genes, phylogenomic analysis is based on sequence alignment of core genes and for this reason it is more suitable for an in depth investigation of phylogenetic relationships between closely related taxa (Bottacini et al. 2017).
This study confirmed that multiple drug resistance of most of the clinical E. coli isolates were probably due to the presence of different plasmids. Virulence genes carried by these isolates can increase potential risks on the health of human and animals. Virulence factors associated with adherence have the most abundance in Virulence Factors of Pathogenic Bacteria analysis. With the rapidly falling cost and turnaround time as well as availability of more user-friendly software, WGS promises to be transformative for rapid surveillance and genotypic antimicrobial susceptibility testing for microbes that are difficult to grow, and has great benefits in combination with phenotypic methods. The findings from comparative genomic analyses of the 19 diverse E. coli isolates provided insights into molecular basis of the rising multi-drug resistance in E. coli.
The WGS-based characterization of multidrug-resistant E. coli from extraintestinal infections in three animal species in this study revealed a diverse range of E. coli STs, and demonstrated the emergence and persistence of particular multidrug-resistant strains, which may have a competitive advantage in fitness under antimicrobial selection pressure compared with previous strains. Surveillance of the emergence and spread of dominant multidrug-resistant isolates with unique plasmids, resistance genes, virulence genes and STs may assist veterinarians in developing improved strategies for treatment and prevention of infections for which the choice of antimicrobials is limited.
Abbreviations
- WGS:
-
whole-genome sequencing
- AMR:
-
antimicrobial resistance
- MLST:
-
multi locus sequence typing
- CGE:
-
Centre of Genomic Epidemiology
- VFDB:
-
virulence factor database
- DIAMOND:
-
double index alignment of next-generation sequencing data
- EHEC:
-
Enterohemorrhagic E. coli
- EPEC:
-
Enteropathogenic E. coli
- HGT:
-
horizontal gene transfer
- ESBL:
-
extended-spectrum β-lactamase
- mcr :
-
mobile colistin resistance
- astA :
-
arginine succinyltransferase A
- capU :
-
Cappuccino, hexosyltransferase homolog
- celb :
-
Caldocellum saccharolyticum
- cma :
-
colicin-M
- gad :
-
glutamate decarboxylase alpha
- ireA :
-
iron-responsive element A
- iss :
-
increased serum survival
- lpfA :
-
long polar fimbriae A
- mchF :
-
microcin transporter protein F
- stb :
-
heat-stable enterotoxin B
- tsh :
-
temperature-sensitive hemagglutinin
References
Andremont A (2003) Commensal flora may play key role in spreading antibiotic resistance. ASM news 69:601–607
Bergholz TM, Tarr CL, Christensen LM, Betting DJ, Whittam TS (2007) Recent gene conversions between duplicated glutamate decarboxylase genes (gadA and gadB) in pathogenic Escherichia coli. Mol Biol Evol 24(10):2323–2333
Boerlin P, McEwen SA, Boerlin-Petzold F, Wilson JB, Johnson RP, Gyles CL (1999) Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J Clin Microbiol 37:497–503
Bottacini F, Morrissey R, Roberts RJ, James K, van Breen J, Egan M, Lambert J, van Limpt K, Knol J, Motherway MOC (2017) Comparative genome and methylome analysis reveals restriction/modification system diversity in the gut commensal Bifidobacterium breve. Nucleic Acids Res 48(4):1860–1877
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Carattoli A (2009) Resistance plasmid families in Enterobacteriaceae. Antimicrob Agents Chemother 53:2227–2238
Carattoli A, Zankari E, García-Fernández A, Larsen MV, Lund O, Villa L, Aarestrup FM, Hasman H (2014) In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903
Chen L, Zheng D, Liu B, Yang J, Jin Q (2015) VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res 44:D694–D697
Chen PA, Hung CH, Huang PC, Chen JR, Huang IF, Chen WL, Chiou Y, Hung WY, Wang JL, Cheng MF (2016) Characteristics of CTX-M extended-spectrum β-lactamase-producing Escherichia coli strains isolated from multiple rivers in Southern Taiwan. Appl Environ Microbiol 82:1889–1897
Clermont O, Olier M, Hoede C, Diancourt L, Brisse S, Keroudean M, Glodt J, Picard B, Oswald E, Denamur E (2011) Animal and human pathogenic Escherichia coli strains share common genetic backgrounds. Infect Genet Evol 11:654–662
Croxen MA, Finlay BB (2010) Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol 8:26
Desjardins P, Picard B, Kaltenböck B, Elion J, Denamurl E (1995) Sex in Escherichia coli does not disrupt the clonal structure of the population: evidence from random amplified polymorphic DNA and restriction-fragment-length polymorphism. J Mol Evol 41:440–448
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Frost LS, Leplae R, Summers AO, Toussaint A (2005) Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 3:722
Gilmour MW, Thomson NR, Sanders M, Parkhill J, Taylor DE (2004) The complete nucleotide sequence of the resistance plasmid R478: defining the backbone components of incompatibility group H conjugative plasmids through comparative genomics. Plasmid 52:182–202
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Gutiérrez J, Maere S (2014) Modeling the evolution of molecular systems from a mechanistic perspective. Trends Plant Sci 19:292–303
Hopkins KL, Liebana E, Villa L, Batchelor M, Threlfall EJ, Carattoli A (2006) Replicon typing of plasmids carrying CTX-M or CMY β-lactamases circulating among Salmonella and Escherichia coli isolates. Antimicrob Agents Chemother 50:3203–3206
Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM (2014) Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol 52:1501–1510
Kaper JB, Nataro JP, Mobley HL (2004) Pathogenic Escherichia coli. Nat Rev Microbiol 2:123
Kim SR, Komano T (1997) The plasmid R64 thin pilus identified as a type IV pilus. J Bacteriol 179:3594–3603
Kröger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hébrard M, Händler K (2012) The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci 109(20):7606–7607
Kuhnert P, Boerlin P, Frey J (2000) Target genes for virulence assessment of Escherichia coli isolates from water, food and the environment. FEMS Microbiol Rev 24:107–117
Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Pontén T, Ussery DW, Aarestrup FM (2012) Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361
Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, Doi Y, Tian G, Dong B, Huang X (2016) Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 16:161–168
Malik A, Nagy B, Kugler R, Szmolka A (2017) Pathogenic potential and virulence genotypes of intestinal and faecal isolates of porcine post-weaning enteropathogenic Escherichia coli. Res Vet Sci 115:102–108
McDermott PF, Tyson GH, Kabera C, Chen Y, Li C, Folster JP, Ayers SL, Lam C, Tate HP, Zhao S (2016) Whole-genome sequencing for detecting antimicrobial resistance in nontyphoidal Salmonella. Antimicrob Agents Chemother 60:5515–5520
O’Neill J (2015) Antimicrobials in agriculture and the environment: reducing unnecessary use and waste. Review on antimicrobial resistance. Available via DIALOG. https://amr-review.org/Publications.html. Accessed Dec 2015
Organization WH (2014) Antimicrobial resistance: global report on surveillance. World Health Organization. Available via DIALOG. http://www.who.int/drugresistance/documents/surveillancereport/en/ of subordinate document. Accessed April 2014
Petty NK, Zakour NLB, Stanton-Cook M, Skippington E, Totsika M, Forde BM, Phan MD, Moriel DG, Peters KM, Davies M, Rogers BA, Dougan G, Rodriguez-Baño J, Pascual A, Pitout JDD, Upton M, Paterson D, Walsh TR, Schembri MA, Beatson SA (2014) Global dissemination of a multidrug resistant Escherichia coli clone. Proc Natl Acad Sci USA 111(15):5694–5699
Shaheen BW, Nayak R, Boothe DM (2013) Emergence of a New Delhi metallo-β-lactamase (NDM-1)-encoding gene in clinical Escherichia coli isolates recovered from companion animals in the United States. Antimicrob Agents Chemother 57:2902–2903
Smets BF, Barkay T (2005) Horizontal gene transfer: perspectives at a crossroads of scientific disciplines. Nat Rev Microbiol 3:675
Solà-Ginés M, Cameron-Veas K, Badiola I, Dolz R, Majó N, Dahbi G, Viso S, Mora A, Blanco J, Piedra-Carrasco N, González-López JJ, Migura-Garcia L (2015) Diversity of multi-drug resistant avian pathogenic Escherichia coli (APEC) causing outbreaks of colibacillosis in broilers during 2012 in Spain. PLoS ONE 10(11):e0143191
Tenaillon O, Skurnik D, Picard B, Denamur E (2010) The population genetics of commensal Escherichia coli. Nat Rev Microbiol 8:207
Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O (2009) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5:e1000344
Ventura M, Canchaya C, Del Casale A, Dellaglio F, Neviani E, Fitzgerald GF, Van Sinderen D (2006) Analysis of bifidobacterial evolution using a multilocus approach. Int J Syst Evol Microbiol 56:2783–2792
Volk WA, Gebhardt B, Hammaskjold M, Kaomer R (1995) Essentials of medical microbiology. Lippincott Williams & Wilkins, Philadelphia
Woodford N, Turton JF, Livermore DM (2011) Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35:736–755
Yassin AK, Gong J, Kelly P, Lu G, Guardabassi L, Wei L, Han X, Qiu H, Price S, Cheng D (2017a) Antimicrobial resistance in clinical Escherichia coli isolates from poultry and livestock, China. PLoS One 12:e0185326
Yassin AK, Zhang J, Wang J, Chen L, Kelly P, Butaye P, Lu G, Gong J, Li M, Wei L, Wang Y, Qi K, Han X, Price S, Hathcock T, Wang C (2017b) Identification and characterization of mcr mediated colistin resistance in extraintestinal Escherichia coli from poultry and livestock in China. FEMS Microbiol Lett 364(24):fxn242
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV (2012) Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644
Zawack K, Li M, Booth JG, Love W, Lanzas C, Gröhn YT (2016) Monitoring antimicrobial resistance in the food supply chain and its implications for FDA policy initiatives. Antimicrob Agents Chemother 60:5302–5311
Zhang P, Shen Z, Zhang C, Song L, Wang B, Shang J, Yue X, Qu Z, Li X, Wu L (2017) Surveillance of antimicrobial resistance among Escherichia coli from chicken and swine, China, 2008–2015. Vet Microbiol 203:49–55
Authors’ contributions
CW, LC, XH and LW conceived of the study. AKY, JZ, JG, KQ, RRG, YZ and YY performed experiment and analyzed the data. CW, LW and XH wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article. All data are fully available without restriction.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported, in part, by Alabama Agricultural Experimental Station and the USDA National Institute of Food and Agriculture, Hatch project (ALA052-1-17026).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chen, L., Wang, L., Yassin, A.K. et al. Genetic characterization of extraintestinal Escherichia coli isolates from chicken, cow and swine. AMB Expr 8, 117 (2018). https://doi.org/10.1186/s13568-018-0646-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-018-0646-8