
Abstract
Mammary pathogenic Escherichia coli (MPEC) is an important causative agent of mastitis in dairy cows that results in reduced milk quality and production, and is responsible for severe economic losses in the dairy industry worldwide. Oxidative stress, as an imbalance between reactive oxygen species (ROS) and antioxidants, is a stress factor that is common in most bacterial habitats. The presence of ROS can damage cellular sites, including iron-sulfur clusters, cysteine and methionine protein residues, and DNA, and may cause bacterial cell death. Previous studies have reported that Autoinducer 2 (AI-2) can regulate E. coli antibiotic resistance and pathogenicity by mediating the intracellular receptor protein LsrR. This study explored the regulatory mechanism of LsrR on the H2O2 stress response in MPEC, showing that the transcript levels of lsrR significantly decreased under H2O2 stress conditions. The survival cell count of lsrR mutant XW10/pSTV28 was increased about 3080-fold when compared with that of the wild-type WT/pSTV28 in the presence of H2O2 and overexpression of lsrR (XW10/pUClsrR) resulted in a decrease in bacterial survival rates under these conditions. The β-galactosidase reporter assays showed that mutation of lsrR led to a remarkable increase in expression of the promoters of ahpCF, katG and oxyR, while lsrR-overexpressing significantly reduced the expression of ahpCF and katG. The electrophoretic mobility shift assays confirmed that LsrR could directly bind to the promoter regions of ahpCF and katG. These results revealed the important role played by LsrR in the oxidative stress response of MPEC.
Similar content being viewed by others


Introduction
Escherichia coli is one of the main pathogenic bacteria causing clinical mastitis [1, 2]. Mammary pathogenic E. coli (MPEC) is a class of extraintestinal pathogenic E. coli that usually adheres to and infects dairy cow mammary epithelial cells together with other pathogens, forming biofilms. The biofilm can help pathogens to evade the host immune system and continue to multiply in the mammary gland, leading to persistent intramammary infections [3,4,5]. As such, research on the role of biofilms in the pathogenesis of mastitis has become important.
Previous studies showed that bacteria express a series of stress proteins to protect themselves from environmental stimuli and to resist adverse environmental pressures such as antibiotics, oxidative stress, acid–base, and osmotic pressure [6,7,8,9,10]. It is generally understood that bacterial infection can trigger innate immune response [11, 12]. Once the pathogenic bacteria infect the host, they are phagocytosed by the host immune cells and macrophages and heterophile cells may produce oxidative bursts in response to pathogens [13, 14]. The rapid production of reactive oxygen species (ROS) including superoxide anions (O2−), hydrogen peroxide (H2O2) and the highly reactive hydroxyl radical (·OH) can damage many cellular sites, including iron-sulfur clusters, cysteine and methionine protein residues and DNA, and may eventually cause cell senescence and death [15,16,17]. H2O2 is a widely used bactericide for inactivating foodborne pathogens. Over time, a variety of E. coli regulatory pathways has evolved to adapt to H2O2 stress including synthesizing catalases (encoded by katG and katE), alkyl hydroperoxide reductase (Ahp, encoded by ahpCF), and stress protein YciF (encoded by yciF). A range of transcriptional regulators including the transcriptional regulator OxyR and the sigma factor RpoS could regulate the synthesis of these enzymes [6, 8, 18,19,20]. Previous studies showed that E. coli resistance to oxidative stress affects both survival and pathogenic potential in the host [21, 22]. However, the detailed molecular mechanism of bacterial adapting to H2O2 stress needs to be further explored.
Autoinducer 2 (AI-2) is produced by many Gram-negative and Gram-positive bacteria and is considered to be a quorum sensing (QS) signaling molecule involved in interspecies communication [23]. Previous studies have reported that the AI-2 QS system is involved in the bacterial response to oxidative stress [24,25,26]. The absence of luxS (involved in the synthesis of AI-2) in Streptococcus suis significantly enhanced its tolerance to H2O2, and the mutation of luxS in Deinococcus radiodurans resulted in a significantly higher susceptibility to H2O2 than wild-type strains [25, 26]. In E. coli, the process of AI-2 uptake involves an ATP-binding transporter complex encoding by the lsrACDB operon. The expression of the lsr operon was regulated by LsrR, a DNA-binding repressor and LsrK, a cognate signal kinase, whose genes (LsrRK) are located immediately upstream of the lsr operon and divergently transcribed. The LsrR represses the expression of the lsr operon and its own lsrRK operon by binding to the promoters, while phospho-AI-2 can release LsrR repression and then activating lsr operon expression [27,28,29]. The role of LsrR as a transcriptional regulator mediating QS signal AI-2-related cellular functions has been identified in previous studies and its significant influence on gene expression control has been revealed. According to a previous study, 146 genes were significantly affected by LsrR deletion [30], but whether there is a relationship between LsrR and the regulation of bacterial oxidative stress response has not yet been reported.
In the present study, we demonstrated that the survival ability of lsrR mutant under H2O2 stress was significantly increased compared to that of the wild type. The β-galactosidase reporter assays indicated that LsrR had an obvious inhibitory effect on the expression of ahpCF, katG, and oxyR. Electrophoretic mobility shift assays (EMSA) confirmed that LsrR inhibited the expression of ahpcF and katG by direct binding to their promoter regions. This study is therefore helpful to understand the bacterial response to host-derived ROS in MPEC, and might provide potential drug targets for the treatment and prevention of E. coli infection.
Materials and methods
Bacterial strains, plasmids, and culture conditions
All the strains and plasmids used in this study are listed in Table 1. The E. coli strain DCM5 was isolated from milk samples in dairy cows with mastitis and was identified by 16S rDNA sequencing. The E. coli cultures were routinely grown at 37 °C in Luria–Bertani (LB) broth (Oxoid, Basingstoke, UK) or on LB agar plates containing 1.5% agar (Oxoid). When needed, antibiotics (Sangon Biotech, Shanghai, China) were used at the following final concentrations: ampicillin (100 μg/mL), chloramphenicol (15 μg/mL), and kanamycin (50 μg/mL).
General DNA manipulation
Genomic DNA from E. coli DCM5 was prepared according to the instructions of TIANamp Bacteria DNA Kit (TianGen Biotech, Bei Jing, China). Plasmid DNA was extracted using a plasmid extraction kit (Sangon Biotech, Shanghai, China). PCR testing was carried out using Taq or PrimeSTAR® Max DNA Polymerase (Takara Bio Inc., Dalian, China). The PCR products and DNA restriction fragments were purified using a gel purification kit (Sangon Biotech, Shanghai, China). DNA restriction endonuclease (Thermo Fisher Scientific, Waltham, MA, USA) digestion and DNA ligation (Thermo Fisher Scientific) were performed by standard methods. Primer premier 5.0 software was used for sequence analysis and primer design. The primer sequences used are listed in Table 2.
Construction of the lsrR-deficient mutant
Homologous recombination was used for deletion of lsrR from the chromosome of DCM5 based on λ red recombinase system [31]. Briefly, using pKD3 plasmid as a template, the fragment containing the chloramphenicol-resistance cassette (cat) inserted within the lsrR gene was amplified with primers DCM5-lsrR-f and DCM5-lsrR-r and then transformed into DCM5 competent cells containing lambda Red recombinase expressed by pKD46 plasmid using electroporation. After electroporation, the cells were added immediately to 900 µL super optimal broth with catabolite repression broth (SOC) without antibiotics, incubated at 37 °C for 60 min, and then mutants were selected by plating cultures on LB plates with 15 μg/mL chloramphenicol. Following incubation for 24 h, the lsrR mutant was selected by PCR amplification using the primers check-lsrR-f and check-lsrR-r. Plasmid pCP20 was subsequently transformed into the mutant strain to delete the Cm cassette. The Cm-sensitive mutant strain was confirmed by PCR amplification using the primers check-lsrR-f and check-lsrR-r, and the PCR products were further confirmed by DNA sequencing, with the mutant strain was designated as DCM5ΔlsrR (XW10).
Complementation of the lsrR mutant
To construct the complemented plasmid pSTV28-lsrR, the lsrR gene and its promoter region were amplified from the DCM5 strain using the primers lsrR-KpnI-f and lsrR-BamHI-r, and then the PCR products were gel-purified and ligated into the BamHI and KpnI restriction sites of the plasmid pSTV28. The recombinant plasmid pSTV28-lsrR was extracted and further confirmed by PCR using primers M13-f and M13-r, and DNA sequencing. To obtain the complemented strain XW10/pClsrR, the plasmid pSTV28-lsrR was electroporated into the mutant strain XW10. The wild-type strain (WT) and the mutant strain XW10 were transformed with the empty vector pSTV28 to obtain WT/pSTV28 and XW10/pSTV28, respectively.
Construction of the lsrR overexpressing strain
The strain overexpressing lsrR was constructed as previously described [32]. The lsrR gene was amplified from the DCM5 strain using the primers lsrR-KpnI-f and lsrR-EcoRI-r, and then the PCR products were gel-purified and ligated into the KpnI and EcoRI restriction sites of the plasmid pUC19 to construct the overexpressing plasmid pUC19-lsrR. Subsequently, the recombinant plasmid pUC19-lsrR was extracted and further confirmed by PCR using primers M13-f and M13-r, and DNA sequencing. The purified recombinant plasmid pUC19-lsrR and the control empty vector pUC19 were electroporated into the wild-type strain WT to obtain strains WT/pUClsrR and WT/pUC19, respectively.
Bacterial growth curves
All strains were cultured overnight in fresh LB broth. Then each sample was diluted to an OD600 of approximately 0.03 in 100 mL of fresh LB broth with 15 μg/mL chloramphenicol or 100 μg/mL ampicillin and grown at 37 °C for 28 h with shaking. The OD600 was detected at 2 h intervals using a UV/Vis spectrophotometer (Thermo Scientific, Pittsburgh, PA, USA), and the growth curves of these strains were established. The experiment was repeated three times and all samples were measured in triplicate.
H 2 O 2 stress assays
The overnight cultures were diluted to an OD600 of approximately 0.03 in 3 mL of fresh LB broth with 15 μg/mL chloramphenicol or 100 μg/mL ampicillin, and incubated in culture tubes at 37 °C for 2 h with shaking. Then, 6 μL of 30% H2O2 was added in each experimental group cultures, and the cells continued to be incubated at 37 °C for 1 h with shaking. All cultures from the experimental group and control group (without addition of H2O2) were then diluted by serial ten-fold dilution with LB medium, and 3 μL of each dilution was dropped onto the LB agar plates and cultured at 37 °C overnight. At the same time, to perform the viable colony-forming units (CFUs) assays, 100 μL of each dilution was spread onto the LB agar plates and cultured at 37 °C overnight. The colonies on each plate were counted. The results were compared between the test groups and the control groups and all experiments were repeated three times.
β-galactosidase assays
The promoters of oxyR, katG, katE, rpoS, yciF, and ahpCF were amplified and cloned into the promoterless lacZ plasmid pRCL by restriction enzyme digestion and DNA ligation to construct the transcriptional lacZ fusion reporter plasmid. To avoid the lacZ gene on the chromosome of the strain from affecting the experimental results, the lacZ genes from the WT strain and the lsrR-deficient strain were mutated using homologous recombination methods based on λ red recombinase system, and then the constructed lacZ fusion reporter plasmids were introduced into strain WT/ΔlacZ and XW10/ΔlacZ, respectively. The E. coli cells containing the lacZ fusion reporter plasmid were cultured in 100 mL LB broth containing a final concentration of 15 μg/mL chloramphenicol for the specified time. After cultivation, cells were collected and diluted in Z-buffer containing 16.1 g/L Na2HPO4, 5.50 g/L NaH2PO4, 0.75 g/L KCl, and 0.246 g/L MgSO4 to 1 mL and assayed for β-galactosidase activity using ortho-Nitrophenyl-β-galactoside (ONPG) as a substrate. The following equation was used to calculate units of enzyme activity: Miller Units: [OD420 × 1000)/(OD600 × Volume (mL) × Time (min)]. The experiment was repeated three times independently.
Total RNA isolation, cDNA generation, and real-time reverse transcription-quantitative PCR processing
Real-time reverse transcription-quantitative PCR (RT-qPCR) assays were performed according to a previous study [32]. To detect the transcript levels of lsrR in the presence or absence of H2O2, the overnight cultures of DCM5(WT) cells were diluted at 1:100 and transferred into two parallel fresh 100 mL LB broth. When the cell density reached OD600 of 1, the cultures were cultured for another 40 min with or without 50 μL of 30% H2O2, and then collected for total RNA extraction. To verify the effect of lsrR overexpression on the expression of katG and ahpCF, the overnight cultures of WT/pUC19, and WT/pUClsrR were diluted to an OD600 of approximately 0.03 in fresh LB broth with 100 μg/mL ampicillin. The cultures were grown to the exponential phase at 37 °C with shaking. The cells were collected by centrifugation and resuspended in RNase-free water; subsequently, total RNA was extracted from the cells using Trizol reagent [TransGen Biotech (Beijing) Co. Ltd., Beijing, China]. Reverse transcription was carried out using the EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen) according to the manufacturer’s instructions. RT-qPCR was performed using the TransStart Tip Green qPCR SuperMix kit (TransGen) with the CFX96™ Real-Time System (Bio-Rad, USA). The 16S cDNA abundance was used to normalize the quantity of the target genes. All the real-time RT-qPCR assays were repeated at least three times.
Expression and purification of the LsrR protein
The C-terminal His6-tag LsrR protein was cloned and purified according to a previous study [27]. The open reading frame of the lsrR gene of E. coli DCM5 was amplified with primers lsrR-NcoI-f and lsrR-XhoI-r from DCM5 genomic DNA, and then cloned into the pET28a (+) vector. The recombinant plasmid pET-lsrR was confirmed by PCR using primers T7-f and T7-r, and further confirmed by DNA sequencing. The pET-lsrR vector was then transferred into E. coli BL21(DE3) by a chemical transformation method. The LsrR protein was induced to express in E. coli BL21(DE3) cells by adding isopropyl β-D-thiogalactopyranoside (IPTG) at a final concentration of 0.5 mM. The purification of His6-tagged LsrR fusion protein was performed with a HisTrap high-performance column as previously described [32]. The purified LsrR protein was stored in 10% glycerol at −80 °C until use. The LsrR protein purity was determined by SDS-PAGE, and the protein concentration was measured by an enhanced BCA protein assay kit (Beyotime, Shanghai, China).
Electrophoretic mobility shift assay (EMSA)
Four promoter DNA fragments, p-lsrR, p-oxyR, p-ahpCF and p-katG, were amplified from the WT genome by biotin-labeled probe primers (p-lsrR-biotin-f/p-lsrR-r, p-ahpCF-biotin-f/p-ahpCF-r, p-katG-biotin-f/p-katG-r, and p-oxyR-biotin-f/p-oxyR-r) respectively. The biotin-labeled promoter fragments were incubated at 25 °C for 30 min with various amounts of LsrR proteins in 4 µL 5 × binding buffer containing 50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 3 mM magnesium acetate, 0.1 mM EDTA, 0.1 mM dithiothreitol. After incubation, 2 µL 10 × loading buffer with bromophenol blue was added to the mixture and then electrophoresed in a 4% native polyacrylamide gel in a 0.5 × Tris–borate EDTA buffer. The band shifts were detected and analyzed according to the manufacturer’s EMSA kit instructions (Beyotime, Shanghai, China).
Mutation of ahpCF and katG promoter sites and enzyme activity detection
Mutation of binding sequences of the ahpCF and katG promoter was performed according to the previous method [27]. There were 6-bp nucleotide bases (AACAAT) in the katG promoter region, and 6-bp nucleotide bases (AAAACT) in the ahpCF promoter region, which may be necessary for LsrR binding. High ratios of A and T within these motifs also suggest that LsrR might preferentially bind to AT-rich sequences [27] so these 6-bp nucleotide bases in plasmids pRCL-pahpCF and pRCL-pkatG were replaced by the GCGCGC sequence to obtain plasmids pRCL-pahpCFM6 and pRCL-pkatGM6 (Table 1). The mutations were performed by using Mut Express II Fast Mutagenesis Kit V2 (Vazyme Biotech, Nanjing, China). A DNA fragment about 6.0 kb, containing the full length of the plasmid except the 6-bp nucleotide bases, was amplified by PCR using the plasmid pRCL-pahpCF or pRCL-pkatG as the template and M6-p-ahpCF-f/M6-p-ahpCF-r or M6-p-katG-f/M6-p-katG-r as the primers. The remaining steps were performed according to the Mut Express II Fast Mutagenesis Kit V2 instructions. The constructed mutant plasmid was identified by DNA sequencing then the mutant reporter plasmids were introduced into strain WT/ΔlacZ. The strains WTΔlacZ/pRCL-pahpCFM6 and WTΔlacZ/pRCL-pkatGM6 were obtained by screening and the expression activity of the mutant promoter was detected by β-galactosidase assays.
Statistical analyses
Statistical analyses were conducted using the GraphPad Primer 8.0 (GraphPad Software Inc., GraphPad Prism 8.0.1.244, San Diego, CA, USA, 2018) using a one-way ANOVA method. The test results are shown as the mean ± SD. A paired t test was used for statistical comparisons between groups. The level of statistical significance was set at a P value of ≤ 0.05.
Results
Identification of lsrR mutant and complementary strains of DCM5
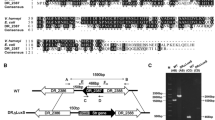
The lsrR gene was knocked out using homologous recombination to obtain the lsrR deletion mutant. The schematic diagram of the strategy for deleting the lsrR gene in DCM5 and the primers used for confirmation of the lsrR deletion are shown in Figure 1A. The mutant and complementary strains of lsrR were confirmed by PCR (Figure 1B). A 1660 bp product was amplified from the wild type strain WT/pSTV28, while 1330 bp products were respectively amplified from the mutant strain XW10/pSTV28 and the complementary strain XW10/pClsrR by using primers check-lsrR-f and check-lsrR-r (Lane 1–3). The plasmid pSTV28 and the complementary plasmid pClsrR were also confirmed by PCR (Figure 1B, Lane 4–6). The PCR products of 100 bp were amplified from strains WT/pSTV28 and XW10/pSTV28 using primers M13-f/M13-r, and a 1094 bp product was amplified from strain XW10/pClsrR.

Molecular determination and growth curve assays of WT, lsrR mutant, and complementary strains. A Schematic diagram of the strategy for constructing the lsrR deletion mutant. B Confirmation of the wild-type strain WT/pSTV28, mutant strain XW10/pSTV28 and the complementary strain XW10/pClsrR. M: 2000 bp DNA marker; Lane 1: a 1660-bp PCR product was amplified from the strain WT/pSTV28 with primers check-lsrR-f/check-lsrR-r; Lane 2: a 1330-bp PCR product was amplified from the mutant strain XW10/pSTV28 with primers check-lsrR-f/check-lsrR-r; Lane 3: a 1330-bp PCR product was amplified from the complementary strain XW10/pClsrR with primers check-lsrR-f/check-lsrR-r; Lane 4: a 150-bp PCR product was amplified from the strain WT/pSTV28 with primers M13-f/M13-r; Lane 5: a 150-bp PCR product was amplified from the mutant strain XW10/pSTV28 with primers M13-f/M13-r; Lane 6: a 1094-bp PCR product was amplified from the complementary strain XW10/pClsrR with primers M13-f/M13-r. C Growth curves of strains WT/pSTV28, XW10/pSTV28, and XW10/pClsrR in LB broth with 15 μg/mL chloramphenicol. D Growth curves of strains WT/pUC19 and WT/pUClsrR in LB broth with 100 μg/mL ampicillin. The data represent the means of three independent assays.
lsrR deletion and overexpression did not affect strains growth
To ensure that the growth conditions of the mutant XW10 strain and its parent strain were consistent with that of the complementary strain, the empty vector pSTV28 was transformed into the WT and XW10 strains. Growth curve assays were performed and results showed that the growth curves of XW10/pSTV28 and XW10/pClsrR were like that of WT/pSTV28 when cells were cultured in LB broth with 15 μg/mL chloramphenicol (Figure 1C). Likewise, to ensure that the growth condition of the WT strain was consistent with that of the lsrR overexpression strain, the empty vector pUC19 was transformed into the WT strain. The growth curve of strain WT/pUClsrR in LB broth with 100 μg/mL ampicillin was like that of strain WT/pUC19 as shown in Figure 1D.
Deletion of lsrR increased bacterial survival ability under the H2O2 stress
With the tolerance of pathogenic bacteria to H2O2 stress being increased, the infecting ability of pathogenic bacteria to the host is also increased. To evaluate the effect of lsrR on the survival of strain DCM5 in the presence of H2O2, the survival ability of the lsrR-deficient mutant strain XW10/pSTV28 was compared with that of the wild-type strain WT/pSTV28 and the complementary strain XW10/pClsrR. The results showed that there was no difference in the number of colonies grown on the plates of each group in absence of H2O2 (Figure 2A). However, the tolerance of strain XW10/pSTV28 to H2O2 stress was increased when compared with that of strain WT/pSTV28 after treating with H2O2 for 1 h, and the tolerance was partially restored in strain XW10/pClsrR (Figure 2B). The CFU assays were performed to further analyze the survival rates of strains WT/pSTV28, XW10/pSTV28, and XW10/pClsrR, and the colonies of hydrogen peroxide treatment group and control group on the plates were counted and compared after cultivating in LB broth with 15 μg/mL chloramphenicol overnight. In the absence of H2O2, there was no difference in the survival rate between the groups as shown in Figure 2C, D. However, with the addition of H2O2, the survival rate of strain XW10/PSTV28 was increased 3080-fold when compared with that of strain WT/PSTV28, and the survival rate was partially recovered in strain XW10/PClsrR. These results suggested that LsrR played a vital role in the bacterial oxidative stress response.

Analysis of the H2O2 stress response of strains WT/pSTV28, XW10/pSTV28, and XW10/pClsrR. The strains WT/pSTV28, XW10/pSTV28 and XW10/pClsrR with an initial OD600 of 0.03 were inoculated into LB broth containing 15 μg/mL chloramphenicol and incubated for 2 h. Then the cultures of the control group (A) and the experimental group (treated with 6 μL 30% hydrogen peroxide for 1 h) (B) were ten-fold serially diluted, and 3 μL of each dilution was dropped onto LB agar plates and cultured overnight. The above dilutions of the control group (C) and the experimental group (D) were spread onto the LB agar plates. After incubation overnight, the colony forming units were analyzed. (∗∗∗∗P < 0.0001).
Overexpression of lsrR decreased bacterial survival ability under the H2O2 stress
To further evaluate the effect of lsrR on the survival of strain DCM5 in the presence of H2O2, the survival ability of the lsrR overexpression strain WT/pUClsrR and its parental strain WT/pUC19 were compared under the H2O2 stress condition. The results showed that there was no difference in the number of colonies grown on the plates of each group in absence of H2O2 (Figure 3A), but strain WT/pUClsrR showed reduced tolerance to H2O2 stress compared with WT/pUC19 after being treated with H2O2 for 1 h (Figure 3B). To further analyze the survival rates of strains WT/pUC19 and WT/pUClsrR, CFU assays were performed. There was no difference in the survival rate between the groups in the absence of H2O2 as shown in Figures 3C, D. In the presence of H2O2, the survival rate of WT/pUClsrR was decreased approximately 21-fold when compared with that of WT/pUC19.This data further supported the important role of LsrR on the oxidative stress response of MPEC.

Analysis of the H2O2 stress response of strains WT/pUC19 and WT/pUClsrR. A The strains WT/pUC19 and WT/pUClsrR with an initial OD600 of 0.03 were inoculated into LB broth containing 100 μg/mL ampicillin and incubated for 2 h. Then the cultures of the control group (A) and the experimental group (treated with 6 μL 30% hydrogen peroxide for 1 h) (B) were ten-fold serially diluted, and 3 μL of each dilution was dropped onto LB agar plates and cultured overnight. The above dilutions of the control group (C) and the experimental group (D) were spread onto the LB agar plates. After incubation overnight, the colony forming units were analyzed. (∗∗∗P < 0.001).
Deletion of lsrR increased the expression of oxyR, ahpCF, and katG
To assess whether the regulatory effects of LsrR on oxyR, rpoS, ahpCF, katE, yciF, and katG were transcriptionally mediated, a system of β-galactosidase report plasmids was constructed to measure expression levels of the promoters of target genes. The data showed that the deletion of lsrR had no effect on the transcriptional activities of the katE, yciF, and rpoS promoters (Figures 4A–C). Compared with the WT strain, the deletion of lsrR significantly increased the transcription activity of the promoters of oxyR (1.95-, 1.71-, and 1.88-fold at 4, 8 and 16 h, respectively), katG (1.98-, 1.73-, and 1.58-fold at 4, 8, and 12 h, respectively), and ahpCF (7.0-, 3.94-, 2.66- and 2.76-fold at 4, 8, 12 and 16 h, respectively) seen in Figures 4D–F. Taken together, these results suggested that LsrR decreases the bacterial ability to respond to hydrogen peroxide stress by downregulating the expression of hydrogen peroxide stress-associated genes oxyR, katG and ahpCF.

Measurement of expression activities of the katE, yciF, rpoS, oxyR, katG, and ahpCF promoters. The β-galactosidase activities expressed in strains WT/ΔlacZ and XW10/ΔlacZ containing plasmid A pRCL-pkatE, B pRCL-pyciF, C pRCL-prpoS, D pRCL-poxyR, E pRCL-pkatG and, F pRCL-pahpCF were measured every 4 h. (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001).
H2O2 stress decreased the expression of lsrR, and overexpression of lsrR decreased the expression of ahpCF, and katG
To investigate whether the expression level of lsrR in ECM5 was affected by H2O2 stress, 0.5 μL 30% H2O2 was added and the transcript level of lsrR decreased by 7.5-fold as shown in Figure 5A. The RT-qPCR experiments were performed to determine whether overexpression of lsrR affected the transcript levels of the catalase encoding gene katG and alkyl hydroperoxide reductase encoding gene ahpCF. The data showed that the transcript level of ahpCF and katG of the lsrR-overexpressing strain WT/pUClsrR was decreased by 2.7- and 2.9-fold when compared with its parental strain WT/pUC19 as seen in Figures 5B, C. The expression level of lsrR was inhibited by H2O2 stress and lsrR reduced the expression of ahpCF and katG, so it can be inferred that MPEC can adapt to the H2O2 stress conditions by mediating LsrR expression.

Comparative measurement of the transcription (complementary DNA [cDNA] abundance) of genes (lsrR, ahpCF, and katG) by RT-qPCR. A The relative lsrR transcript level was determined in WT strain with or without addition 0.5 µL/mL 30% H2O2. B The relative ahpC transcript level was determined in strains WT/pUC19 and WT/pUClsrR cultured in LB broth with 100 µg/mL ampicillin. C The relative katG transcript level was determined in strains WT/pUC19 and WT/pUClsrR cultured in LB broth with 100 µg/mL ampicillin. (Error bars indicate SD; ∗∗P < 0.01, ∗∗∗P < 0.001).
LsrR binding to ahpCF and katG promoters
Our previous study showed that lsrR and lsrA promoter regions have two LsrR binding boxes, AAAACT and AAAACTGAA in the p-lsrA-box and AACAAT and AAGATTTAA in the p-lsrR-box, and the motifs contain high ratios of A and T [27]. On this basis, it was speculated that a putative binding site of LsrR existed in the promoter regions of ahpCF and katG genes (Figure 6A) and the results suggested that LsrR might repress the two genes by direct binding to their promoter regions. According to sequence alignment, the putative binding site of LsrR in the oxyR promoter region was not found.

The binding ability of LsrR to the p-ahpCF-box, p-katG-box, and p-oxyR-box was determined by gel shift assays. In each panel, from lanes (1) to (5), the LsrR concentration was 8, 0, 2, 4, and 8 mmol, respectively; the amounts of biotin-labeled probes in all lanes were 100 fmol. In lane 1, besides the labeled probes, 1 pmol unlabeled probes were added as the competitive control (Ctrl). A Site1 and site2 represent two known binding boxes of LsrR. The red boxes indicate two putative binding sites of LsrR in the promoter regions of ahpCF and katG, respectively. B The binding ability of LsrR to its promoter (positive control). C The binding ability of LsrR to the ahpCF promoter. D The binding ability of LsrR to the katG promoter. E The binding ability of LsrR to the oxyR promoter.
To verify the above hypothesis, electrophoretic mobility shift assays were performed to confirm whether LsrR binds to the oxyR, ahpCF and katG promoter regions. A positive control assay confirmed that LsrR could bind to its own promoter region (Figure 6B). As shown in Figure 6C, D, clearly shifted bands of protein-DNA complex were detected at LsrR concentrations of 4, 8 and 12 mM, and the intensity of the shifted band was enhanced as the amount of LsrR was increased. The results confirmed that LsrR could specifically bind to the promoter regions of ahpCF and katG, indicating that LsrR could directly regulate the transcription of ahpCF and katG. However, the shifted band of the protein-DNA complex was not detected when the promoter region of oxyR was used as a probe (Figure 6E), suggesting that LsrR cannot bind to the oxyR promoter regions. This suggested that LsrR might regulate the expression of oxyR through several indirect pathways and indicated that it had a complicated regulatory pattern in the regulation of bacterial oxidative stress response.
The β-galactosidase assays of the mutated promoters of ahpCF and katG
Based on sequence alignment, it was speculated that a putative binding site of LsrR existed in both promoter regions of ahpCF and katG genes. The EMSA results showed that LsrR could directly bind to the promoter regions of ahpCF and katG, which led to the hypothesis that these two putative sequences may be crucial for LsrR binding. To further verify this, mutant β-galactosidase reporter plasmids pRCL-pahpCFM6 and pRCL-pkatGM6 were constructed and the data in Figure 7A showed that compared with pRCL-pahpCF, pRCL-pahpCFM6 was associated with an apparent increase in the expression of lacZ throughout the growth cycle. The expression of lacZ in pRCL-pkatGM6 increased only before 4 h compared with pRCL-pkatG as shown in Figure 7B, so these results suggested that the sequence AAAACT in the promoter region of ahpCF was essential for the binding of LsrR in DCM5 strain. However, mutation of AACAAT in the promoter region of katG did not reduce the binding ability of LsrR throughout the growth cycle.

Expression activities of the ahpCF and katG promoters and their mutant promoters. β-galactosidase activities of the strains with plasmids A pRCL-pahpCF or pRCL-pahpCFM6, B pRCL-pkatG or pRCL-pkatGM6 were measured every 4 h. (∗∗P < 0.01, ∗∗∗P < 0.001).
Discussion
Bacteria can precisely regulate gene expression to promote their survival in various environments or hosts. Bacterial AI-2 quorum sensing is common and is involved in a variety of biological processes, including signal transduction, drug resistance, pathogenicity, motility, and interbacterial competition [23, 33,34,35,36]. In E. coli, AI-2 can regulate some physiological processes through the AI-2 receptor protein LsrR and the AI-2 metabolic pathway. The previous study showed that AI-2 relies on the intracellular receptor LsrR to increase the β-lactam antibiotic resistance of a broad-spectrum β-lactamase-positive E. coli isolated from cows with mastitis [37]. Another subsequent study indicated that exogenous AI-2 can down-regulate the transcription of folate biosynthesis-related genes folA, folC, luxS, metE, and metH through non-LsrR-dependent pathways, increasing avian pathogenic Escherichia coli (APEC) sensitivity to trimethoprime-sulfamethazole [38]. These studies supported the regulatory role of AI-2 or LsrR in bacterial adaption to environmental stimulus. However, whether there is a relationship between LsrR and the regulation of bacterial oxidative stress response has not been reported yet. This study explored the relationship between LsrR and bacterial oxidative stress regulation in MPEC and clarified the regulatory details of LsrR on the oxidative stress-related genes.
Oxidative stress can trigger multiple responses that have evolved as complex regulatory systems in bacterial pathogens, aiding bacteria to cause persistent infections [39,40,41]. A previous study indicated that the LsrR controls invasiveness of Salmonella Typhimurium by regulating Salmonella pathogenicity island-1(SPI-1) and flagella genes [42]. A subsequent study demonstrated that the lsr operon is involved in mediating the virulence of APEC but the regulatory mechanism remains unclear [43]. In Xanthomonas oryzae pv. oryzae, OxyR degrades hydrogen peroxide in rice by regulating the expression of catalase CatB, and then promotes bacterial infectivity [44]. The data from this study showed that the expression of LsrR was affected by H2O2 stress in the environment and that LsrR can directly regulate the expression of oxidative stress response associated genes ahpCF and katG suggesting that MPEC might respond to the H2O2 stress in an LsrR-dependent pathway. Because the ability to respond to oxidative stress of bacteria is very important for its infection process in the host, it was speculated that LsrR may also be associated with the regulation of bacterial pathogenicity in MPEC, so it was important to explore the regulatory role of LsrR on bacterial pathogenicity or other important physiological processes in future work.
The EMSA results showed that LsrR directly binds to the promoter regions of ahpCF and katG, which led to the hypothesis that pahpCF: AAAACT and pkatG: AACAAT may be crucial for LsrR binding. To verify this, mutant β-galactosidase enzyme activity reporter plasmids pRCL-pahpCFM6 and pRCL-pkatGM6 were constructed and their β-galactosidase activities were measured. The results suggested that the AAAACT sequence in the promoter region of ahpCF was essential for the binding of LsrR in DCM5 strain, which was consistent with our previous results [27], but the mutation of the AACAAT sequence in the promoter region of katG did not promote the transcriptional activity of katG promoter throughout the growth cycle. The previous study also has shown that LsrR binding sequences contained high ratios of A and T, but do not need to share a high level of sequence homology [27]. A subsequent study indicated that mutation of nucleotides in the vicinity of p-lsrR-box sequence AACAAT–AAGATTTAA also affected the transcriptional activity of the lsrR promoter, which might be associated with binding of CytR repressor [45], therefore it was speculated that the mutation of the binding site in the katG promoter region may cause the recognition and binding by other transcriptional repressors.
According to present and previous studies, a schematic diagram was made to illustrate the regulatory mode of LsrR on the H2O2 stress response of MPEC (Figure 8). These data showed that LsrR decreased H2O2 stress tolerance ability of MPEC by directly or indirectly inhibiting the transcriptions of H2O2 stress response associated genes including ahpCF, katG, and oxyR. The transcription of lsrR exhibited a significant decrease in response to the H2O2 stress. These results led to the conclusion that MPEC can adapt to H2O2 stress conditions by reducing the transcription of lsrR, which contributed to increased expression of H2O2 stress response associated genes. But how this bacterium regulates the lsrR expression under the H2O2 stress conditions remains obscure and needs to be further explored.

Schematic diagram of the LsrR-mediated regulation on bacterial H2O2 response in MPEC.
Abbreviations
- E. coli :
-
Escherichia coli
- MPEC:
-
mammary pathogenic E. coli
- APEC:
-
avian pathogenic Escherichia coli
- ROS:
-
reactive oxygen species
- H2O2 :
-
hydrogen peroxide
- QS:
-
quorum sensing
- IPTG:
-
isopropyl β-D-thiogalactopyranoside
- LB:
-
Luria–Bertani
- AI-2:
-
autoinducer 2
- ONPG:
-
ortho-Nitrophenyl-β-galactoside
- CFU:
-
colony-forming units
- PCR:
-
polymerase chain reaction
- RT-qPCR:
-
real-time reverse transcription-quantitative PCR
References
Boireau C, Cazeau G, Jarrige N, Calavas D, Madec JY, Leblond A, Haenni M, Gay E (2018) Antimicrobial resistance in bacteria isolated from mastitis in dairy cattle in France, 2006–2016. J Dairy Sci 101:9451–9462. https://doi.org/10.3168/jds.2018-14835
Blum SE, Leitner G (2013) Genotyping and virulence factors assessment of bovine mastitis Escherichia coli. Vet Microbiol 163:305–312. https://doi.org/10.1016/j.vetmic.2012.12.037
Dogan B, Klaessig S, Rishniw M, Almeida RA, Oliver SP, Simpson K, Schukken YH (2006) Adherent and invasive Escherichia coli are associated with persistent bovine mastitis. Vet Microbiol 116:270–282. https://doi.org/10.1016/j.vetmic.2006.04.023
Almeida RA, Dogan B, Klaessing S, Schukken YH, Oliver SP (2011) Intracellular fate of strains of Escherichia coli isolated from dairy cows with acute or chronic mastitis. Vet Res Commun 35:89–101. https://doi.org/10.1007/s11259-010-9455-5
Porcherie A, Cunha P, Trotereau A, Roussel P, Gilbert FB, Rainard P, Germon P (2012) Repertoire of Escherichia coli agonists sensed by innate immunity receptors of the bovine udder and mammary epithelial cells. Vet Res 43:14. https://doi.org/10.1186/1297-9716-43-14
Hindupur A, Liu D, Zhao Y, Bellamy HD, White MA, Fox RO (2006) The crystal structure of the E. coli stress protein YciF. Protein Sci 15:2605–2611. https://doi.org/10.1110/ps.062307706
Fernandez L, Hancock RE (2012) Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev 25:661–681. https://doi.org/10.1128/CMR.00043-12
Chiang SM, Schellhorn HE (2012) Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch Biochem Biophys 525:161–169. https://doi.org/10.1016/j.abb.2012.02.007
Xu Y, Zhao Z, Tong W, Ding Y, Liu B, Shi Y, Wang J, Sun S, Liu M, Wang Y, Qi Q, Xian M, Zhao G (2020) An acid-tolerance response system protecting exponentially growing Escherichia coli. Nat Commun 11:1496. https://doi.org/10.1038/s41467-020-15350-5
Bremer E, Kramer R (2019) Responses of microorganisms to osmotic stress. Annu Rev Microbiol 73:313–334. https://doi.org/10.1146/annurev-micro-020518-115504
Skelton RM, Shepardson KM, Hatton A, Wilson PT, Sreenivasan C, Yu J, Wang D, Huber VC, Rynda-Apple A (2019) Contribution of host immune responses against influenza D virus infection toward secondary bacterial infection in a mouse model. Viruses 11:994. https://doi.org/10.3390/v11110994
Zheng L, Xu Y, Lu J, Liu M, Bin D, Miao J, Yin Y (2016) Variant innate immune responses of mammary epithelial cells to challenge by Staphylococcus aureus, Escherichia coli and the regulating effect of taurine on these bioprocesses. Free Radic Biol Med 96:166–180. https://doi.org/10.1016/j.freeradbiomed.2016.04.022
El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, Dang PM (2016) Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev 273:180–193. https://doi.org/10.1111/imr.12447
Herb M, Schramm M (2021) Functions of ROS in macrophages and antimicrobial immunity. Antioxidants 10:313. https://doi.org/10.3390/antiox10020313
Hong Y, Zeng J, Wang X, Drlica K, Zhao X (2019) Post-stress bacterial cell death mediated by reactive oxygen species. Proc Natl Acad Sci USA 116:10064–10071. https://doi.org/10.1073/pnas.1901730116
Hong Y, Li L, Luan G, Drlica K, Zhao X (2017) Contribution of reactive oxygen species to thymineless death in Escherichia coli. Nat Microbiol 2:1667–1675. https://doi.org/10.1038/s41564-017-0037-y
Shivaprasad DP, Taneja NK, Lakra A, Sachdev D (2021) In vitro and in situ abrogation of biofilm formation in E coli by vitamin C through ROS generation, disruption of quorum sensing and exopolysaccharide production. Food Chem 341:128171. https://doi.org/10.1016/j.foodchem.2020.128171
Zhang L, Alfano JR, Becker DF (2015) Proline metabolism increases katG expression and oxidative stress resistance in Escherichia coli. J Bacteriol 197:431–440. https://doi.org/10.1128/JB.02282-14
Wan F, Yin J, Sun W, Gao H (2019) Oxidized OxyR up-regulates ahpCF expression to suppress plating defects of oxyR- and catalase-deficient strains. Front Microbiol 10:439. https://doi.org/10.3389/fmicb.2019.00439
Battesti A, Majdalani N, Gottesman S (2011) The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65:189–213. https://doi.org/10.1146/annurev-micro-090110-102946
Mei GY, Tang J, Carey C, Bach S, Kostrzynska M (2015) The effect of oxidative stress on gene expression of Shiga toxin-producing Escherichia coli (STEC) O157:H7 and non-O157 serotypes. Int J Food Microbiol 215:7–15. https://doi.org/10.1016/j.ijfoodmicro.2015.07.029
Nie W, Wang S, He R, Xu Q, Wang P, Wu Y, Tian F, Yuan J, Zhu B, Chen G (2020) A-to-I RNA editing in bacteria increases pathogenicity and tolerance to oxidative stress. PLoS Pathog 16:e1008740. https://doi.org/10.1371/journal.ppat.1008740
Pereira CS, Thompson JA, Xavier KB (2013) AI-2-mediated signalling in bacteria. FEMS Microbiol Rev 37:156–181. https://doi.org/10.1111/j.1574-6976.2012.00345.x
Sun F, Liang H, Kong X, Xie S, Cho H, Deng X, Ji Q, Zhang H, Alvarez S, Hicks LM, Bae T, Luo C, Jiang H, He C (2012) Quorum-sensing agr mediates bacterial oxidation response via an intramolecular disulfide redox switch in the response regulator AgrA. Proc Natl Acad Sci USA 109:9095–9100. https://doi.org/10.1073/pnas.1200603109
Lin L, Li T, Dai S, Yu J, Chen X, Wang L, Wang Y, Hua Y, Tian B (2016) Autoinducer-2 signaling is involved in regulation of stress-related genes of Deinococcus radiodurans. Arch Microbiol 198:43–51. https://doi.org/10.1007/s00203-015-1163-7
Cao M, Feng Y, Wang C, Zheng F, Li M, Liao H, Mao Y, Pan X, Wang J, Hu D, Hu F, Tang J (2011) Functional definition of LuxS, an autoinducer-2 (AI-2) synthase and its role in full virulence of Streptococcus suis serotype 2. J Microbiol 49:1000–1011. https://doi.org/10.1007/s12275-011-1523-1
Xue T, Zhao L, Sun H, Zhou X, Sun B (2009) LsrR-binding site recognition and regulatory characteristics in Escherichia coli AI-2 quorum sensing. Cell Res 19:1258–1268. https://doi.org/10.1038/cr.2009.91
Xavier KB, Miller ST, Lu W, Kim JH, Rabinowitz J, Pelczer I, Semmelhack MF, Bassler BL (2007) Phosphorylation and processing of the quorum-sensing molecule autoinducer-2 in enteric bacteria. ACS Chem Biol 2:128–136. https://doi.org/10.1021/cb600444h
Wu M, Tao Y, Liu X, Zang J (2013) Structural basis for phosphorylated autoinducer-2 modulation of the oligomerization state of the global transcription regulator LsrR from Escherichia coli. J Biol Chem 288:15878–15887. https://doi.org/10.1074/jbc.M112.417634
Li J, Attila C, Wang L, Wood TK, Valdes JJ, Bentley WE (2007) Quorum sensing in Escherichia coli is signaled by AI-2/LsrR: effects on small RNA and biofilm architecture. J Bacteriol 189:6011–6020. https://doi.org/10.1128/JB.00014-07
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. https://doi.org/10.1073/pnas.120163297
Yu L, Li W, Li Q, Chen X, Ni J, Shang F, Xue T (2020) Role of LsrR in the regulation of antibiotic sensitivity in avian pathogenic Escherichia coli. Poult Sci 99:3675–3687. https://doi.org/10.1016/j.psj.2020.03.064
Thompson JA, Oliveira RA, Djukovic A, Ubeda C, Xavier KB (2015) Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep 10:1861–1871. https://doi.org/10.1016/j.celrep.2015.02.049
Wu X, Lv X, Lu J, Yu S, Jin Y, Hu J, Zuo J, Mi R, Huang Y, Qi K, Chen Z, Han X (2017) The role of the ptsI gene on AI-2 internalization and pathogenesis of avian pathogenic Escherichia coli. Microb Pathog 113:321–329. https://doi.org/10.1016/j.micpath.2017.10.048
Daniels R, Vanderleyden J, Michiels J (2004) Quorum sensing and swarming migration in bacteria. FEMS Microbiol Rev 28:261–289. https://doi.org/10.1016/j.femsre.2003.09.004
Xue T, Zhao L, Sun B (2013) LuxS/AI-2 system is involved in antibiotic susceptibility and autolysis in Staphylococcus aureus NCTC 8325. Int J Antimicrob Agents 41:85–89. https://doi.org/10.1016/j.ijantimicag.2012.08.016
Xue T, Yu L, Shang F, Li W, Zhang M, Ni J, Chen X (2016) Short communication: The role of autoinducer 2 (AI-2) on antibiotic resistance regulation in an Escherichia coli strain isolated from a dairy cow with mastitis. J Dairy Sci 99:4693–4698. https://doi.org/10.3168/jds.2015-10543
Yu L, Li W, Zhang M, Cui Y, Chen X, Ni J, Yu L, Shang F, Xue T (2018) Autoinducer2 affects trimethoprim-sulfamethoxazole susceptibility in avian pathogenic Escherichia coli dependent on the folate synthesis-associate pathway. Microbiologyopen 7:e00582. https://doi.org/10.1002/mbo3.582
Zhang W, Zhao L, Zhou J, Yu H, Zhang C, Lv Y, Lin Z, Hu S, Zou Z, Sun J (2019) Enhancement of oxidative stress contributes to increased pathogenicity of the invasive pine wood nematode. Philos Trans R Soc Lond B Biol Sci 374:20180323. https://doi.org/10.1098/rstb.2018.0323
Pandey S, Sahukhal GS, Elasri MO (2019) The msaABCR operon regulates the response to oxidative stress in Staphylococcus aureus. J Bacteriol 201:e00417-e419. https://doi.org/10.1128/JB.00417-19
Liu NN, Uppuluri P, Broggi A, Besold A, Ryman K, Kambara H, Solis N, Lorenz V, Qi W, Acosta-Zaldivar M, Emami SN, Bao B, An D, Bonilla FA, Sola-Visner M, Filler SG, Luo HR, Engstrom Y, Ljungdahl PO, Culotta VC, Zanoni I, Lopez-Ribot JL, Kohler JR (2018) Intersection of phosphate transport, oxidative stress and TOR signalling in Candida albicans virulence. PLoS Pathog 14:e1007076. https://doi.org/10.1371/journal.ppat.1007076
Choi J, Shin D, Kim M, Park J, Lim S, Ryu S (2012) LsrR-mediated quorum sensing controls invasiveness of Salmonella typhimurium by regulating SPI-1 and flagella genes. PLoS One 7:e37059. https://doi.org/10.1371/journal.pone.0037059
Zuo J, Yin H, Hu J, Miao J, Chen Z, Qi K, Wang Z, Gong J, Phouthapane V, Jiang W, Mi R, Huang Y, Wang C, Han X (2019) Lsr operon is associated with AI-2 transfer and pathogenicity in avian pathogenic Escherichia coli. Vet Res 50:109. https://doi.org/10.1186/s13567-019-0725-0
Yu C, Wang N, Wu M, Tian F, Chen H, Yang F, Yuan X, Yang CH, He C (2016) OxyR-regulated catalase CatB promotes the virulence in rice via detoxifying hydrogen peroxide in Xanthomonas oryzae pv. oryzae. BMC Microbiol 16:269. https://doi.org/10.1186/s12866-016-0887-0
Hauk P, Stephens K, McKay R, Virgile CR, Ueda H, Ostermeier M, Ryu KS, Sintim HO, Bentley WE (2016) Insightful directed evolution of Escherichia coli quorum sensing promoter region of the lsrACDBFG operon: a tool for synthetic biology systems and protein expression. Nucleic Acids Res 44:10515–10525. https://doi.org/10.1093/nar/gkw981
Wang S, Yang D, Wu X, Yi Z, Wang Y, Xin S, Wang D, Tian M, Li T, Qi J, Ding C, Yu S (2019) The ferric uptake regulator represses Type VI secretion system function by binding directly to the clpV promoter in Salmonella enterica Serovar Typhimurium. Infect Immun 87:e00562-e619. https://doi.org/10.1128/IAI.00562-19
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Number 31672571) and the Doctoral Start-Up Foundation of Linyi University (Grant Number LYDX2020BS031). We thank International Science Editing for editing this manuscript.
Author information
Authors and Affiliations
Contributions
HW: investigation, methodology, experiments, writing—original draft, writing—review and editing, data curation. FS: methodology, data curation, writing—review and editing. JS and JX: methodology and experiments. XC: investigation, data curation. JN: formal analysis, writing—review and editing. LY: methodology, writing—review and editing. TX: conceptualization, writing—review and editing, supervision and funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, H., Shang, F., Shen, J. et al. LsrR, the effector of AI-2 quorum sensing, is vital for the H2O2 stress response in mammary pathogenic Escherichia coli. Vet Res 52, 127 (2021). https://doi.org/10.1186/s13567-021-00998-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-021-00998-8