
Abstract
This study reports the comparative analyses of four Flavobacterium columnare isolates that have different virulence and antimicrobial resistance patterns. The main research goal was to reveal new insights into possible virulence genes by comparing the genomes of bacterial isolates that could induce tissue damage and mortality versus the genome of a non-virulent isolate. The results indicated that only the genomes of the virulent isolates possessed unique genes encoding amongst others a methyl-accepting chemotaxis protein possibly involved in the initial colonization of tissue, and several VgrG proteins engaged in interbacterial competition. Furthermore, comparisons of genes unique for the genomes of the highly virulent (HV) carp and trout isolates versus the, respectively, low and non-virulent carp and trout isolates were performed. An important part of the identified unique virulence genes of the HV-trout isolate was located in one particular gene region identified as a genomic island. This region contained araC and nodT genes, both linked to pathogenic and multidrug-resistance, and a luxR-gene, functional in bacterial cell-to-cell communication. Furthermore, the genome of the HV-trout isolate possessed unique sugar-transferases possibly important in bacterial adhesion. The second research goal was to obtain insights into the genetic basis of acquired antimicrobial resistance. Several point-mutations were discovered in gyrase-genes of an isolate showing phenotypic resistance towards first and second-generation quinolones, which were absent in isolates susceptible to quinolones. Tetracycline-resistance gene tetA was found in an isolate displaying acquired phenotypic resistance towards oxytetracycline. Although not localized on a prophage, several flanking genes were indicative of the gene’s mobile character.
Similar content being viewed by others


Introduction
Flavobacterium columnare (F. columnare) is a Gram-negative, bacterial, freshwater fish pathogen that causes columnaris disease. The latter is characterized by gill, fin, and skin lesions [1, 2]. F. columnare belongs to the family Flavobacteriaceae [1], which is part of the phylum Bacteroidetes. The F. columnare cluster is subdivided into five genomovars: I, I/II, II, II-B and III [3].
Flavobacterium columnare is distributed globally and may infect many different cultured and wild freshwater fish species, including carp, catla, channel catfish, eel, goldfish, perch, salmonids, and tilapia [1, 2]. Tropical freshwater aquarium fish may also be affected [4]. In channel catfish (Ictalurus punctatus), F.columnare is in the top‐three diseases reported to cause morbidity and mortality resulting in yearly losses of millions of dollars [5]. The prevalence of columnaris disease in cyprinids is also noteworthy. Carp (Cyprinus carpio), for example, are amongst the world’s most-produced food fish [6, 7] and koi carp, in particular, have high economic value as ornamental fish species [8, 9].
Various reports have described differences in virulence among F. columnare strains [1, 10,11,12,13,14]. Colonization of the host tissue by F. columnare plays a crucial part in this. This process is initiated by the chemotaxis of the bacteria to the host tissues. Former studies on F. columnare isolates of channel catfish revealed that virulent isolates have an increased chemotactic response towards channel catfish mucus [13, 15], suggesting that chemotaxis could be associated with virulence. Bader et al. [16] reported that F. columnare cells were attached to the skin and gill mucus of channel catfish as early as 5 min after a bath immersion challenge. Similar results were previously obtained in carp and rainbow trout (Oncorhynchus mykiss) [13]. The latter revealed that virulent F. columnare carp and trout isolates attached faster and in higher numbers to the gill tissue, which eventually led to the mortality of the host. This mortality rate was not seen for fish exposed to a non-virulent isolate as in those, no tissue colonization by the bacterial cells was displayed [10, 11]. Sugars would also play an important role in the adhesion of F. columnare to the host tissue. Decostere et al. [17], for example, reported that treatment of either F. columnare bacterial cells or gill tissue with sodium metaperiodate (cleaves the C–C bond between vicinal hydroxyl groups of sugar) or with various sugars significantly reduced bacterial adhesion. This lead to the hypothesis that the interaction between a bacterial lectin and its host’s carbohydrate receptor explained the diminished adhesion [17].
Once adhered to the host tissue, bacterial cell-to-cell communication, a process known as quorum sensing, allows planktonic cells to switch to biofilm formation [18]. In this mechanism of gene regulation, bacteria coordinate the expression of certain genes in response to the concentration of small signal molecules. The exact mechanisms of bacterial cell-to-cell communication in F. columnare is currently unknown. However, in other Gram-negative bacteria, the involvement of two important proteins in the regulation of QS has been described [19]. An I-protein is the enzyme that synthesizes the signalling molecule [19, 20]. The signal molecules are detected by an R-protein, a transcriptional regulator. The signal molecules accumulate extracellularly and once a certain threshold concentration has been reached, they bind to a response regulator (LuxR homolog). Subsequently, the LuxR/signal molecule complex activates the transcription of QS-regulated genes [19, 21].
In the past years, the number of Flavobacterium genomes reported has increased. For F. columnare, some comparative genomic studies are available [22,23,24]. However, these studies focused on the genomes of different catfish and tilapia species, particularly on isolates that were retrieved from diseased fish only. In this study, we performed comparative analyses of four F. columnare strains, including two strains isolated from trout (Oncorhynchus mykiss), and two from carp (Cyprinus carpio). These isolates belong to genomovar I and possess different virulence patterns [10, 11]. These moreover show differences in antimicrobial susceptibility [25]. The comparative genomics approach conducted in this research is the first to focus on identifying virulence genes based on the comparison between F. columnare isolates with a different virulence and antimicrobial susceptibility pattern in carp and trout.
Materials and methods
Flavobacterium species, library preparation and whole-genome sequencing
Four F. columnare isolates belonging to genomovar I [25] were included in this study for genome sequencing. The virulence pattern and antimicrobial resistance profile of all isolates, as previously described [10, 11, 25], are shown in Table 1. Briefly, the two strains that could induce tissue colonization and cause 100% mortality within 24 h after the onset of an immersion challenge, were assigned highly virulent (HV), which is the case for, respectively, the carp and trout isolates 04017018 and JIP P11/91 [10, 11]. The carp isolate CDI-A was assigned low virulent (LV), as it could induce tissue colonization leading to mortality, but in up to ten percent of fish challenged [10, 11]. The trout isolate ATCC49512 was not able to adhere to the host tissue nor to induce mortality, and was hence labelled as non virulent (NV) [10, 11]. The genome sequence of isolate F. columnare ATCC 49512 is publicly available from the NCBI database [26]. The genomes of the other three strains were sequenced and assembled during this study. Briefly, the F. columnare strains were cultivated as described before [25] and whole DNA was extracted using the Qiagen (Venlo, The Netherlands) Blood & Tissue kit, following the manufacturer’s instructions. The qualities and quantities of the extracted DNA were evaluated using a NanoDrop spectrophotometer (NanoDrop™, Themo Fisher Scientific, Merelbeke, Belgium) and included for further library preparation and subsequent sequencing. F. columnare genomic DNA was then sheared on a Covaris S2 sonicator (Covaris, Woburn, Massachusetts, USA) aiming for 800 bp fragments. The fragmented DNA was used to build a sequencing library with the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, Massachusetts, USA). The sequencing library was size-selected on a 2% agarose E-gel. Fragments ranging from 600 to 900 bp were cut out of the gel and purified with the Zymoclean Gel Recovery Kit (Zymo Research, Irvine, California, USA). The isolated library fragments were submitted to 8 PCR cycles and recovered using AMPure XP magnetic beads (Beckman Coulter, Indianapolis, Indiana, USA). Library quality was checked on an Agilent Bioanalyzer using a High Sensitivity Chip (Agilent Technologies, Santa Clara, California, USA) and concentration was measured via qPCR according to the qPCR Quantification Protocol (Illumina, San Diego, California, USA). Sequencing was done as paired-end 300 on a MiSeq (Illumina).
Genome assembly and annotation
Removal of residual adapters and quality trimming of raw sequencing reads was done with Cutadapt [27], version 1.15, using a phred cut-off of 20 and removal of reads containing ambiguous nucleotides or having a final length shorter than 100 nucleotides. The quality of the reads was checked using FastQC [28], version 0.11.5. The trimmed paired-end reads were assembled into genome scaffolds with SPAdes [29], version 3.11.1, using options—careful and—k 21,33,55,77,99,127. Scaffolds smaller than 2000 nucleotides were discarded. The quality of the obtained genome assemblies was checked with QUAST [30], version 5.0.0, and BUSCO [31] version 3.0.2. Raw sequencing reads and the assembled genomes of the three isolates were submitted at NCBI (BioProject accession PRJNA431138) and are publicly available [32]. The GenBank accession numbers of the de novo assembled genomes are shown in Table 1. All new genomes, as well as the genome of strain ATCC 49512, were subjected to gene finding and automatic annotation using RAST (Rapid Annotations using Subsystems Technology) version 2.0 and SEED viewer [33].
Comparative analysis of F. columnare isolates
The pan-genome ortholog clustering tool (PanOCT) was applied for a pan-genomic analysis of the four isolates [34]. This tool looked for gene differences between these four isolates. Gene locations are formatted as NODEn:s-e where n is a unique scaffold number, s and e are the gene’s start and end position respectively. Scaffold names are based on their original names as generated by the de novo genome assembly software. The publicly available submitted genomes contain newly assigned scaffold names by NCBI, as well as the original name (formatted as NODE_n). To identify possible virulence factors encoded in F. columnare, the virulence factor database (VFDB) was downloaded [35, 36], and a local BLAST search was conducted using BLAST + version 2.2.31. To identify phage elements in the three newly sequenced F. columnare genomes, sequences were submitted to the PHAge Search Tool- Enhanced Release (PHASTER) server [37]. Additionally, the presence of genes involved in Flavobacterium secretion systems was investigated. MacSyFinder 1.0.5 and TXSScan were applied to detect protein secretion systems and their appendages in the genomes [38, 39].
Variant detection in the gyrase genes of isolates CDI-A and 04017018 was performed using CLC Genomics Workbench against those of F. columnare reference CP0003222.2. Jalview 2.11.1 [40] was used to verify whether these SNPs present in isolate CDI-A were associated with amino acid substitutions. Then, to investigate the impact of these amino acid substitutions on gyrase activity, the online PredictSNP software was used [41]. PredictSNP, MAPP, PhD-SNP, PolyPhen-1 and -2, SIFT, SNAP, nsSNPAna-lyzer, and PANTHER were run with default parameters. Using the online I-Mutant Suite 3.0 software [42], the impact of amino acid substitutions on gyrase stability was investigated. Finally, ConSurf Server [43] was run with default parameters to investigate if the amino acid substitutions were located in conserved regions.
Results
The four genomes were assembled at scaffold level and are therefore incomplete. However, their sizes (3.09 to 3.21 Mbp) are within the range of the complete genomes for this species as listed by NCBI (3.03 to 3.27 Mbp). The GC content (31.2 to 31.5%) and number of protein coding genes (2626 to 2770) was similar for the assembled genomes (Table 2). Amino acid composition (Additional file 1A) and predicted protein lengths (Additional file 1B) were similar between the four F. columnare genomes. Predicted subcellular localization of proteins was also similar (Additional file 1C).
The SEED viewer “sequence based comparison” tool allowed the virulent F. columnare isolates to be aligned to the reference genome and each other. The cut-off value for homologues was set at 70% protein sequence identity (i.e. BLAST e-cutoff value). This resulted in a list of genes being present in a reference genome in chromosomal order and displayed hits on the comparison organisms accordingly. This allowed determining 156 protein-encoding genes, of which 114 encoded for hypothetical proteins, that were present in the genomes of the three virulent isolates, while being absent in the NV F. columnare trout isolate. The protein-encoding genes with a known function as annotated by RAST are listed in Additional file 2. These genes were then compared to the VFDB, to look for those associated with virulence. The resulting protein encoding genes are marked in bold in Additional file 2 and include several transferases, membrane proteins, several VgrG proteins and a methyl-accepting chemotaxis protein (MCP).
Furthermore, the SEED viewer tool allowed to align the genomes of the HV F. columnare carp and trout isolates to those of the, respectively low (carp) and non-virulent (trout) isolates. For the comparison of the F. columnare carp genomes, 80 genes (of which 62 hypothetical) were present only in the genome of the HV carp isolate while absent in the LV one. Regarding virulence, the homologous virulence-associated proteins encoded for the HV carp isolate genome include a transcriptional regulator araC [NODE42:complement (3102–3539)] and a glycosyltransferase involved in lipopolysaccharide biosynthesis (LPS) [NODE6:complement (123808–124008)]. An overview of the unique protein-encoding genes (excluding the hypothetical ones) in the HV carp isolate genome, including the ones predicted to be involved in virulence when identifying them via the VFDB, are presented in Additional file 3. The HV carp isolate also showed acquired resistance against oxytetracycline as determined before [25]. The gene encoding the AraC family protein in the HV carp isolate genome is flanked by a transposase, a putative prophage element, and a gene coding for TetA antimicrobial resistance protein, as shown in Figure 1.

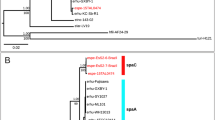
Schematic representation of unique genes on NODE42 in carp isolate 04017018. This region identified in this highly virulent carp isolate 04017018 possesses unique genes (black) when compared to the genome of low virulent trout isolate CDI-A. Genes marked in bold are also predicted to be involved in virulence when identifying them via Virulence Factor Database. Hypothetical ones are not labelled.
For trout, 374 genes (of which 263 hypothetical ones) were present only in the HV trout and absent in NV trout isolate. Next to the genes reported in Additional file 2, genes involved in transposable elements, transcription, antimicrobial resistance, universal stress and ulcer associated adenine specific DNA methyltransferase were identified. Regarding virulence, the homologous virulence-associated proteins encoded for the HV trout isolate genome include a gene encoding for a membrane protein (NODE11:22223–23335), nodT [NODE17:complement (54957–56384), araC (NODE17:36752–37645), luxR (NODE17:complement (8305–8706)], and a unique vgrG [NODE47:complement(5031–5630)]. An overview of the unique protein-encoding genes (excluding the hypothetical ones) in the HV trout isolate genome, including the ones predicted to be involved in virulence when identifying them via the VFDB, are presented in Additional file 4. For the HV F. columnare strain isolated from trout, some very interesting virulence genes are grouped on NODE17, which is recognized as a genomic island by the IslandPaht-DIMOB and SIGI-HMM method. The latter is represented in Figure 2.

Schematic representations of unique genes grouped on NODE17 (genomic island) in trout isolate JIPP11/91. When comparing the genome of this highly virulent trout isolate with the one from the non-virulent trout isolate ATCC 49512, several unique genes, present only in the highly virulent isolate are grouped on NODE17, which is recognized as a genomic island by the IslandPaht-DIMOB and SIGI-HMM method. Several of these unique genes are also predicted to be involved in virulence when identifying them via Virulence Factor Database. These are marked in bold. The hypothetical ones are not labelled.
MacSyFinder and TXSScan could detect multiple genes encoding for protein members of several secretion systems. Most of the systems are positioned on different scaffolds but are grouped within a scaffold. The four genomes carry a complete T1SS, consisting of three essential components: ABC-transporter (represented by gene abc), porin (omf), and the inner membrane-anchored adaptor protein (mfp). Furthermore, the T9SS carries genes involved in gliding (gldJ, gldK, gldL, gldM, gldN), spreading (sprA, sprE, and sprT) and porphyrin accumulation on the cell surface (porO, porU, porV). When observing the T4P, Pil B, C, M, and Q were identified in all sequenced F. columnare isolates. All four genomes seem to possess sctN in the T3SS, tssH in the T6SSi and a complete T6SSiii. In T4SS type B, several differences were identified. Apart from t4cp1 in the HV carp isolate, no genes were identified in the T4SS typeB for isolates 04017018 and ATCC49512. No other differences could be encountered. Table 3 gives a comparative overview of the secretion systems.
When searching for known point mutations associated with fluoroquinolone resistance in the LV carp isolate CDI-A, 11 SNPs were detected in several gyrase genes upon comparing those with the ones from the reference genome of F. columnare, the latter not displaying resistance towards fluoroquinolones [25]. These SNPs were present in gyrA (T to G, position 244), two single point mutations in Topoisomerase IV subunit A (G to T, position 1221; T to G position 1221), three-point mutations in Topoisomerase IV subunit B (G to A, position 154; T to G, position 1779; A to G, position 1782), and five-point mutations in DNA topoisomerase I (A to G, position 273; G to A position 405; G to A, position 468; G to A, position 617; A to G, position 2151) (Additional file 5). However, seven of these point mutations were also present in the genome of the HV carp isolate, which also did not reveal resistance towards any of the quinolone antimicrobial agents. The four unique SNPs which were present in isolate CDI-A, were the one in gyrA (T to G, position 244), and three in DNA topoisomerase I (A to G, position 273; G to A position 405; G to A, position 468) (Additional file 5). Using Jalview, only the SNP present in gyrA resulted in an amino acid substitution from serine to alanine. The ConSurf sever gave a conservation score of 5 for this amino acid position, indicating semi-conservation. Two of the PredictSNP tools, namely SIFT and PANTHER, indicated that this amino acid substitution might affect gyrA activity, with an average accuracy of 73.4%. Using I-Mutant 3.0, this amino acid substitution was also predicted to decrease protein stability [DDG: − 0.8, reliability index (RI): 7]. There were no point mutations revealed in the DNA gyrase subunit B. Results are presented in Additional file 6. Furthermore, a gene encoding tetracycline resistance tetA was found in the genome of the HV carp isolate 04017018, displaying phenotypic resistance towards oxytetracycline.
To check for the presence of phages, the PHASTER tool identified 18 prophage sequences that were similar in the three newly assembled F. columnare strains (Additional file 7, Additional file 8A). These were all located on the same scaffold in a region of 12,451 bp. Some genes could be identified as subparts of the Bacillus phage G genome (PHAGE_Bacill_G_NC_023719). In the reference genome ATCC 49512, the latter prophage sequences were not identified. Instead, the PHASTER tool identified 37 prophage sequences identified as subparts of a Flavobacterium sp. (PHAGE_Flavob_23T_NC_041859) (Additional file 7, Additional file 8B). These were allocated on the same scaffold in a region of 50431 bp. However, as the scores of the prophages in the three newly assembled isolates and the reference strain were 30, respectively 40, and hence less than 70, the prophages seemed to be incomplete based on PHAST's completeness score calculation [44].
Discussion
In the past years, more information has become available on the ability of, and stimulating factors for F. columnare to colonize surfaces and form biofilms [11, 15, 45, 46]. Bacterial motility is an important factor for the rapid colonization of a surface [46, 47]. Our former research conducted with the HV, LV and NV F. columnare isolates—also applied in this study—revealed that the HV F. columnare carp and trout isolates attached faster and in higher numbers to the gill tissue compared to the LV carp isolate [10, 11]. However, the latter also showed the capacity to rapidly colonize host tissue and cause tissue damage, followed by mortality of the fish. This was not the case for the NV trout isolate, which was not able to colonize tissues nor cause mortality [10, 11]. Therefore, the hypothesis arose that chemotaxis of the bacterial cells might play a crucial role in the colonization process. This hypothesis was reinforced based on the outcome of the SEED sequence based comparison analysis that was performed in this study. The software was used to differentiate protein-encoding genes present in the virulent isolates only and by comparing that generated output with the VFDB. A protein encoded gene annotated in RAST as a methyl-accepting chemotaxis protein (MCP) was present (Additional file 2). Extra analyses in the search for chemotaxis proteins via the VFDB in the four genomes revealed the presence of chemotaxis-involved genes cheA, cheB, cheY, and also cheW-like proteins (results not shown). However, extra analyses with InterProScan in the search for signature sequences for chemotaxis such as pfam00015, or pfam00672 did not deliver further supportive evidence. This makes it difficult to state that chemotaxis is active in F. columnare based on currently identified genes and proteins. The importance of the mcp-gene is worth further investigation as chemotaxis has been described in the literature for F. columnare [13, 15] and this phenomenon must have a driving set of genes. If we could learn to understand and control chemotaxis processes, this might be an important step in the combat against columnaris disease. Comparing the effect of chemotaxis and colonization in wild type versus deletion mutants of this mcp-gene in the F. columnare isolates might be the first step in this puzzle.
The gliding motility in F. columnare is well-known [1]. The rate of motility has been linked to biofilm formation and the production of virulence factors in different pathogenic Gram-negative bacteria [47,48,49], including F. columnare [45, 46]. Prior to attachment, the bacterial cells are highly motile and glide over the surface. However, in this research, no differences were encountered between the four genomes in the secretion systems distribution involved in gliding. The few differences found in the other SS were not just observed only in the virulent or only in the non-virulent isolates. Hence these cannot explain possible differences in virulence. The mere presence or absence of these systems can hence probably not be attributed to virulence. This does not exclude that, based on environmental influence, a different expression pattern of the secretion systems and proteins involved in secretion can influence virulence or biofilm formation, as has been proven before for the isolates included in this study [45, 50].
As shown in Additional file 2, the identified virulence genes present in genomes of the virulent isolates but absent in the genome of the NV isolate, are related to asparagine synthetase and sugar transferases. In the human bacterial pathogen Streptococcus pyogenes, the expression of asparagine synthetase is important in the expression of streptolysin toxins. Sugar transferases, on the other hand, are important enzymes in establishing glycosidic linkages. As for F. columnare, sugars are known to play an important role in the adhesion of the bacteria to the host tissue [17]. It is no surprise that the genomes of the isolates that displayed virulence contain several unique sugar transferases compared to the genome of the NV trout isolate. The exact role of the asparagine synthetase and sugar transferases in the genome of the virulent isolates needs further exploration.
Further comparison of the genomes shows that the genome of the HV trout isolate possesses a unique gene encoding VgrG protein when compared to the NV trout isolate. Moreover, when comparing the genomes of the three virulent isolates versus the non-virulent one, the genomes of the virulent isolates possess two more unique genes encoding for the VgrG proteins (Additional file 2). VgrG is one of the key markers of the secretion apparatus of T6SS [51]. The T6SS is a versatile secretion system and might be involved in virulence, antagonism, nutrient acquisition, and horizontal gene transfer [52]. Contact‐dependent interbacterial competition appears to be the major function of T6SS, a function that can influence the composition of microbial communities [53]. While many bacterial pathogens, including Pseudomonas aeruginosa, Vibrio cholerae, and Serratia marcescens, are known to use their T6SSs under laboratory conditions [54,55,56,57], the mechanisms of how these T6SSs contribute to survival in the environment (and in host infection) have not been determined [54].
SEED Viewer allowed to identify another virulence-related gene encoding protein, a transcriptional regulator of the AraC family, in the genome of the HV carp isolate while absent in the one from the LV carp isolate (Additional file 3). Members of the AraC family of transcriptional regulators are thought to counteract histone-like nucleoid-structuring (H-NS)-induced silencing in select environments [58]. Under non-permissive metabolic and environmental conditions, H-NS represses gene expression by coating and/or condensing (bridging) DNA strands [59]. Depending on the bacterial growth phase, and in response to changes in environmental factors (e.g., temperature, osmolarity, and pH), H-NS will release DNA to enable gene expression [60,61,62]. In Enterobacteriaceae, the HNS protein contributes crucially to the fitness, including pathogenic and multidrug-resistant strains [60]. The latter makes sense for the HV carp isolate, resistant to oxytetracycline as determined before [25]. The AraC family protein in the HV carp isolate genome is flanked by a transposase, a putative prophage element, and a gene coding for TetA antimicrobial resistance protein (Figure 1).
A unique AraC-protein was also identified in the genome of the HV trout isolate as part of a genomic island NODE17 (Figure 2). Orthology analysis revealed that all genes in the latter region seem to be unique for the genome of the HV trout isolate when compared to the NV one, and encompasses at least three virulence gene-encoded proteins, namely AraC, NodT, and a LuxR family protein. The gene encoding AraC is flanked by genes encoding a putative mobilization protein and DNA topoisomerase III (Figure 2). As does AraC, the NodT protein could also play a role in antimicrobial resistance. NodT family proteins comprise a group of outer membrane lipoproteins of the Resistance-Nodulation-cell Division (RND) type efflux systems. These play a major role in both intrinsic and acquired multidrug resistance in Gram-negative bacteria [63]. Although no antimicrobial resistance had been found in the HV trout isolate in former research [25], the genome of this isolate has a series of genetic elements on board involving antimicrobial resistance mechanisms. However, these could be inactivated or silenced until reactivation. Apart from that, RND systems would also be important for the pathogenicity of several bacteria, e.g., Salmonella enterica [63]. The importance of these RND systems in both multidrug resistance and pathogenicity has made them a target of new drugs aimed at inhibiting their function [63] and might form a new interesting research field in Flavobacterium species as well. The third virulence-associated gene-encoded protein identified on NODE17 is the LuxR family protein, involved in quorum sensing (Figure 2). Currently, not much is known on how F. columnare cells communicate. Possibly indole, a sigal molecule identified in many pathogenic bacteria such as E. coli, several Shigella strains, Enterococcus faecalis, and V. cholera [64], might be involved. F. columnare is known to produce indole [1] and a gene encoded protein involved in indole-3-glycerol phosphate synthase was identified in the four F. columnare isolates studied in this manuscript (results not shown). Whether the latter has a signalling function, and what the role of the luxR-gene plays in QS of F. columnare remains unclear. Therefore, the way by which biofilm is regulated and formed in F. columnare, definitely deserves further research. Another remarkable feature in this part of the genome of the HV trout isolate is that the unique luxR family protein gene is flanked by tra-genes, the latter playing a crucial role in the transfer of genetic material, provided they are functional. NODE17 has been recognized as a genomic island by the IslandPaht-DIMOB and SIGI-HMM method. Losing (or acquiring) a genomic island may play a crucial role in the evolution of many bacteria, as it is involved in disseminating variable genes, including antibiotic resistance and virulence genes, and catabolic genes leading to the formation of new metabolic pathways [65]. The exact role in the genome of the HV F. columnare trout isolate remains to be investigated.
The four F. columnare isolates showed differences in the antimicrobial resistance profile based on minimum inhibitory concentration (MIC) values studied before [25]. This made it interesting to compare the four genomes in the search for underlying antimicrobial resistance mechanisms. Compared to the reference genome, not displaying any kind of phenotypic antimicrobial resistance, several single point mutations were discovered in four out of five gyrase genes present in F. columnare isolate CDI-A, the koi carp isolate displaying acquired resistance towards both first- and second-generation quinolones. Quinolone resistance would arise stepwise through selective amplification of mutants when drug concentrations are above the MIC-values of the susceptible population and below the MIC-values of the least susceptible mutant subpopulation [66]. Resistance towards quinolones due to mutations is frequently found in the quinolone-resistance-determining-region of the DNA gyrase subunit A (gyrA) [67]. Mutations in gyrA have been described to generate resistance to the first generation quinolones and reduced susceptibility to other quinolones [68]. Here, a unique SNP in the gyrase gene gyrA was found for isolate CDI-A, leading to an amino acid substitution. Software tools predicted that this substitution might affect gyrase activity and stability. This may indicate that this SNP is involved in decreased susceptibility of this isolate for fluoroquinolones. Nevertheless, further investigation is necessary, as these results do not necessarily imply a causal relationship between the presence of a SNP and antimicrobial resistance.
Furthermore, for the HV carp isolate 04017018, which displayed acquired phenotypic resistance towards oxytetracycline, a unique protein-encoding gene of interest was the tetracycline resistance gene tetA. Oxytetracycline is one of the most commonly used tetracyclines worldwide for the treatment of bacterial fish diseases [69]. Class A tet determinants can confer high-level tetracycline resistance [70], and have been found on transferable plasmids and in the chromosome of bacteria. In the genome of the HV carp isolate, tetA is flanked by a mobile element and a gene coding for chromosome (plasmid) partitioning protein parB (Figure 1), which is indicative of its mobile character. The PHASTER tool could identify several prophage sequences located on the same scaffold, but the scores of the prophage regions were less than 70, excluding the presence of a complete prophage in any of the four F. columnare isolates (Additional file 7). Hence, if tetA is positioned on a mobile element, it is not a prophage, according to these results. Further analyses are needed to investigate the mobile character of this gene.
Overall, a global analysis of the F. columnare genomes demonstrates that several unique genes involved in virulence are present in the genomes of the virulent F. columnare carp and trout isolates when compared to the non- or less virulent ones. The genomic analyses furthermore provide insights into possible acquired antimicrobial resistance mechanisms adopted by the bacterial cells. This study is the first to focus on identifying virulence genes based on the comparison between F. columnare isolates with different virulence and antimicrobial susceptibility patterns in carp and trout. Although much remains to be investigated with regard to mechanisms of e.g. chemotaxis, biofilm formation and quorum sensing, the findings in this study might form a basis for new genomic, virulence, and antimicrobial resistance studies.
Availability of data and materials
The genome sequence of isolate Flavobacterium columnare ATCC 49512 is publicly available from the NCBI database [26]. The sequencing reads and genomes of the 3 strains that were sequenced and assembled during this study are available in the NCBI-database under project PRJNA431138 [32] with the accession numbers as presented in Table 1 of this manuscript.
Abbreviations
- F. columnare :
-
Flavobacterium columnare
- gld :
-
Gliding-gene
- gyrA:
-
DNA gyrase gene subunit A
- HV:
-
Highly virulent
- LV:
-
Low virulent
- mcp :
-
Methyl-accepting chemotaxis protein gene
- MIC:
-
Minimum Inhibitory Concentration
- NV:
-
Non-virulent
- PanOCT:
-
Pan-genome ortholog clustering tool
- PHASTER:
-
PHAge Search Tool-Enhanced Release
- por :
-
Porphyrin accumulation on the cell surface-gene
- QS:
-
Quorum sensing
- RND:
-
Resistance Nodulation Cell Division
- SNP:
-
Single nucleotide polymorphism
- spr :
-
Spreading-gene
- T4SS:
-
Type IV Secretion system
- T6SS:
-
Type VI Secretion system
- tetA:
-
Tetracycline A gene
- VFDB:
-
Virulence factor database
References
Bernardet JF, Bowman JP (2006) The genus Flavobacterium. In: Dworkin M, Falkow S (eds) The prokaryotes: a handbook on the biology of bacteria: vol 7: proteobacteria: delta and epsilon subclasses. Deeply rooting bacteria. Springer Science+Business Media, LLC, New York
Declercq AM, Haesebrouck F, Van den Broeck W, Bossier P, Decostere A (2013) Columnaris disease in fish: a review with emphasis on bacterium-host interactions. Vet Res 44:27
LaFrentz BR, Waldbieser GC, Welch TJ, Shoemaker CA (2014) Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J Fish Dis 37:657–669
Decostere A, Haesebrouck F, Devriese LA (1998) Characterization of four Flavobacterium columnare (Flexibacter columnaris) strains isolated from tropical fish. Vet Microbiol 62:35–45
Peterman MA, Posadas BC (2019) Direct economic impact of fish diseases on the East Mississippi catfish industry. N Am J Aquac 81:222–229
FAO (2018) The State of World Fisheries and Aquaculture 2018—meeting the sustainable development goals. Rome. Licence: CC BY-NC-SA 3.0 IGO. http://www.fao.org/3/i9540en/i9540en.pdf. Accessed 15 Sept 2020
Singh T (1997) Common culture practices for cyprinids in Asia. Southeast Asian J Trop Med Public Health 28(Suppl 1):73–76
Smith KF, Schmidt V, Rosen GE, Amaral-Zettler L (2012) Microbial diversity and potential pathogens in ornamental fish aquarium water. PLoS ONE 7:e39971
Chapman FA (2000) Ornamental fish culture, freshwater. In: Stickney RRJ (ed) Encyclopedia of aquaculture. Hoboken, Wiley and sons
Declercq AM, Chiers K, Haesebrouck F, Van den Broeck W, Dewulf J, Cornelissen M, Decostere A (2015) Gill infection model for columnaris disease in common carp and rainbow trout. J Aquat Anim Health 27:1–11
Declercq AM, Chiers K, Van den Broeck W, Dewulf J, Eeckhaut V, Cornelissen M, Bossier P, Haesebrouck F, Decostere A (2015) Interactions of highly and low virulent Flavobacterium columnare isolates with gill tissue in carp and rainbow trout. Vet Res 46:25
Decostere A, Haesebrouck F, Charlier G, Ducatelle R (1999) The association of Flavobacterium columnare strains of high and low virulence with gill tissue of black mollies (Poecilia sphenops). Vet Microbiol 67:287–298
Klesius PH, Shoemaker CA, Evans JJ (2008) Flavobacterium columnare chemotaxis to channel catfish mucus. FEMS Microbiol Lett 288:216–220
Kunttu H (2010) Characterizing the bacterial fish pathogen Flavobacterium columnare and some factors affecting its pathogenicity. Jyväskyla Studies Biol Environ Sci 206:1–69
LaFrentz BR, Klesius PH (2009) Development of a culture independent method to characterize the chemotactic response of Flavobacterium columnare to fish mucus. J Microbiol Methods 77:37–40
Bader JA, Nusbaum KE, Shoemaker CA (2003) Comparative challenge model of Flavobacterium columnare using abraded and unabraded channel catfish, Ictalurus punctatus (Rafinesque). J Fish Dis 26:461–467
Decostere A, Haesebrouck F, Van Driessche E, Charlier G, Ducatelle R (1999) Characterization of the adhesion of Flavobacterium columnare (Flexibacter columnaris) to gill tissue. J Fish Dis 22:465–474
Papenfort K, Bassler B (2016) Quorum-sensing signal-response systems in Gram-negative bacteria. Nat Rev Microbiol 14:576–588
Lazdunski AM, Ventre I, Sturgis JN (2004) Regulatory circuits and communication in Gram-negative bacteria. Nat Rev Microb 2:581–592
Camilli A, Bassler BL (2006) Bacterial small-molecule signaling pathways. Science 311:1113–1116
Defoirdt T, Brackman G, Coenye T (2013) Quorum sensing inhibitors: how strong is the evidence? Trends Microbiol 21:619–624
Tekedar HC, Karsi A, Reddy JS, Nho SW, Kalindamar S, Lawrence ML (2017) Comparative genomics and transcriptional analysis of Flavobacterium columnare strain ATCC 49512. Front Microbiol 8:588
Kumru S, Tekedar HC, Blom J, Lawrence ML, Karsi A (2020) Genomic diversity in flavobacterial pathogens of aquatic origin. Microb Pathog 142:104053
Kayansamruaj P, Dong HT, Hirono I, Kondo H, Senapin S, Rodkhum C (2017) Comparative genome analysis of fish pathogen Flavobacterium columnare reveals extensive sequence diversity within the species. Infect Genet Evol 54:7–17
Declercq AM, Boyen F, Van den Broeck W, Bossier P, Karsi A, Haesebrouck F, Decostere A (2013) Antimicrobial susceptibility pattern of Flavobacterium columnare isolates collected worldwide from 17 fish species. J Fish Dis 36:45–55
The National Center for Biotechnology Information database. https://www.ncbi.nlm.nih.gov/. Accessed 14 Jan 2021
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10
Babraham Bioinformatics Fast QC tool—a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 15 Sept 2020
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A (2018) Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
NCBI-database project PRJNA431138 Flavobacterium columnare. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA431138. Accessed 14 Jan 2021
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R (2014) The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42(1):D206–D214
Fouts DE, Brinkac L, Beck E, Inman J, Sutton G (2012) PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res 40:e172
Virulence Factors of Pathogenic Bacteria. http://www.mgc.ac.cn/VFs/main.htm. Accessed 15 Sept 2020
Chen L, Zheng D, Liu B, Yang J, Jin Q (2016) VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res 44:D694–D697
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21
Abby SS, Néron B, Ménager H, Touchon M, Rocha EPC (2014) MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas Systems. PLoS ONE 9:e110726
Abby SS, Cury J, Guglielmini J, Néron B, Touchon M, Rocha EPC (2016) Identification of protein secretion systems in bacterial genomes. Sci Rep 6:23080
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLOS Comput Biol 10:e1003440
Capriotti E, Calabrese R, Casadio R (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22:2729–2734
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucl Acids Res 44:W344–W350
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS (2011) PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352
Declercq AM, Cai W, Naranjo E, Thongda W, Eeckhaut V, Bauwens E, Arias C, De La Fuente L, Beck BH, Lange MD, Peatman E, Haesebrouck F, Aerts J, Decostere A (2019) Evidence that the stress hormone cortisol regulates biofilm formation differently among Flavobacterium columnare isolates. Vet Res 50:24
Cai W, De La Fuente L, Arias CR (2013) Biofilm formation by the fish pathogen Flavobacterium columnare: development and parameters affecting surface attachment. Appl Environ Microbiol 79:5633–5642
Álvarez B, Secades P, Prieto M, McBride MJ, Guijarro JA (2006) A mutation in Flavobacterium psychrophilum tlpB inhibits gliding motility and induces biofilm formation. Appl Environ Microbiol 72:4044–4053
Gardel CL, Mekalanos JJ (1996) Alteration in Vibrio cholera motility phenotypes correlate with changes in virulence factor expression. Infect Immun 64:2246–2255
Lee J-H, Rho JB, Park K-J, Kim CB, Han Y-S, Choi SH, Lee K-H, Park S-J (2004) Role of flagellum and motility in pathogenesis of Vibrio vulnificus. Infect Immun 72:4905–4910
Declercq AM, Aerts J, Ampe B, Haesebrouck F, De Saeger S, Decostere A (2016) Cortisol directly impacts Flavobacterium columnare in vitro growth characteristics. Vet Res 47:84
Wu C-F, Lien Y-W, Bondage D, Lin J-S, Pilhofer M, Shih Y-L, Chang J-H, Lai E-M (2020) Effector loading onto the VgrG carrier activates type VI secretion system assembly. EMBO Rep 21:e47961
Ringel PD, Hu D, Basler M (2017) The role of type VI secretion system effectors in target cell lysis and subsequent horizontal gene transfer. Cell Rep 21:3927–3940
Verster AJ, Ross BD, Radey MC, Bao Y, Goodman AL, Mougous JD, Borenstein E (2017) The landscape of type VI secretion across human gut microbiomes reveals its role in community composition. Cell Host Microbe 22:411–419
Green ER, Mecsas J (2016) Bacterial secretion systems: an overview. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.VMBF-0012-2015
Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD (2011) Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475:343–347
Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ (2007) Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci USA 104:15508–15513
English G, Trunk K, Rao VA, Srikannathasan V, Hunter WN, Coulthurst SJ (2012) New secreted toxins and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol Microbiol 86:921–936
Shahul Hameed UF, Liao C, Radhakrishnan AK, Huser F, Aljedani SS, Zhao X, Momin AA, Melo FA, Guo X, Brooks C, Li Y, Cui X, Gao X, Ladbury JE, Jaremko Ł, Jaremko M, Li J, Arold ST (2019) H-NS uses an autoinhibitory conformational switch for environment-controlled gene silencing. Nucleic Acids Res 47:2666–2680
Winardhi RS, Yan J, Kenney LJ (2015) H-NS regulates gene expression and compacts the nucleoid: insights from single-molecule experiments. Biophys J 109:1321–1329
Ono S, Goldberg MD, Olsson T, Esposito D, Hinton JC, Ladbury JE (2005) H-NS is a part of a thermally controlled mechanism for bacterial gene regulation. Biochem J 391:203–213
Stella S, Falconi M, Lammi M, Gualerzi CO, Pon CL (2006) Environmental control of the in vivo oligomerization of nucleoid protein H-NS. J Mol Biol 355:169–174
Wang H, Ayala JC, Benitez JA, Silva AJ (2015) RNA-seq analysis identifies new genes regulated by the histone-like nucleoid structuring protein (H-NS) affecting Vibrio cholerae virulence, stress response and chemotaxis. PLoS ONE 10:e0118295
Blair JMA, Piddock LJV (2009) Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr Opin Microbiol 12:512–519
Lee J-H, Lee J (2010) Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev 34:426–444
Juhas M, Roelof van der Meer J, Gaillard M, Harding RM, Hood DW, Crook DW (2009) Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33:376–393
Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X (2009) Quinolones: action and resistance updated. Curr Top Med Chem 9:981–998
Shah SQ, Nilsen H, Bottolfsen K, Colquhoun DJ, Sørum H (2012) DNA gyrase and topoisomerase IV mutations in quinolone-resistant Flavobacterium psychrophilum isolated from diseased salmonids in Norway. Microb Drug Resist 18:207–214
Marien M, Decostere A, Duchateau L, Chiers K, Froyman R, Nauwynck H (2007) Efficacy of enrofloxacin, florfenicol and amoxicillin against Ornithobacterium rhinotracheale and Escherichia coli O2:K1 dual infection in turkeys following APV priming. Vet Microbiol 121:94–104
Rigos G, Troisi GM (2005) Antibacterial agents in Mediterranean finfish farming: a synopsis of drug pharmacokinetics in important euryhaline fish species and possible environmental implications. Rev Fish Biol Fisher 15:53–73
Wang W, Guo Q, Xu X, Sheng Z, Ye X, Wang M (2014) High-level tetracycline resistance mediated by efflux pumps Tet(A) and Tet(A)-1 with two start codons. J Med Microbiol 63:1454–1459
Acknowledgements
The isolates were kindly provided by Dr Jean-François Bernardet (Unité de Virologie et Immunologie Moléculaires, INRAE/UVSQ Centre Île-de-France—Jouy-en-Josas—Antony, France) and Dr ir. Olga L.M. Haenen (Fish and Shellfish Diseases Laboratory, Central Veterinary Research Institute (CVI), Wageningen, the Netherlands). Prof. Dr Cova Arias (Ɨ) and Dr Haitham Mohammed of Auburn University, USA, Alabama are thanked for the genomovar determination of the F. columnare isolates of which a full genome sequencing was performed in this study. The Special Research Grant (Bijzonder Onderzoeksfonds, BOF, Grant Number 01Z06210 and Grant Number BOF.PDO.2015.0020.01) of Ghent University, Belgium is gratefully acknowledged for financial support.
Funding
The Special Research Grant (Bijzonder Onderzoeksfonds): BOF, Grant Number 01Z06210 was used as PhD-grant and Grant Number BOF.PDO.2015.0020.01 was applied as postdoc grant for author AM Declercq.
Author information
Authors and Affiliations
Contributions
AMD: substantial contributions to the conception, design, acquisition, analysis, interpretation of the data, and drafting of the entire manuscript. LT: substantial contributions to the acquisition and analysis of the generated genomic data and extra software analyses and drafting the manuscript with emphasis on materials and methods and results sections. YG: substantial contributions to the acquisition and analysis of the generated genomic data and extra software analyses. CDW: substantial contributions to the acquisition and analysis of the generated genomic data and extra software analyses and drafting the manuscript with emphasis on materials and methods and results sections. FH: substantial contributions to the conception, design, interpretation of the data, and drafting of the entire manuscript. FVN: substantial contributions to the acquisition and analysis of the generated genomic data and extra software analyses, and SNP analyses in the search for antimicrobial resistance mechanisms. AS: substantial contributions to the conception, design, acquisition, analysis, interpretation of the data, and drafting of the entire manuscript. AD: substantial contributions to the conception and design of the research. All authors have approved the submitted version and have agreed both to be personally accountable for the author's own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even parts in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Additional file 1.
Comparison of Flavobacterium columnare strains. (A) Amino acid composition for F. columnare strains. Letter abbreviations. A, Alanine; C, Cysteine; D, Aspartic Acid; E, Glutamic Acid; F, Phenylalanine; G, Glycine; H, Histidine; I, Isoleucine; K, Lysine; L, Leucine; M, Methionine; N, Asparagine; P, Proline; Q, Glutamine; R, Arginine; S, Serine; T, Threonine; V, Valine; W, Tryptophan; Y, Tyrosine. (B) Predicted protein lengths for F. columnare strains. (C) Predicted subcellular localization of proteins encoded in F. columnare strains using PSORTb. ATCC 49512: low virulent (LV) F. columnare trout isolate, JIP P11/91: highly virulent (HV) F. columnare trout isolate, CDI-A: LV F. columnare carp isolate, 04017018 : HV F. columnare carp isolate.
Additional file 2.
Protein-encoding genes present only in the virulent F. columnare genomes. The latter 42 genes are only present in genomes of the highly virulent carp (04017018) and trout (JIPP11/91) and low virulent carp (CDI-A) F. columnare isolates that could cause tissue damage and mortality. These genes are not present in the genome of the non-virulent F. columnare trout isolate ATCC49512. Hypothetical genes (114) were not included here. Genes marked in bold are also predicted to be involved in virulence when identifying them via Virulence Factor Database.
Additional file 3.
Unique protein-encoding genes in the highly virulent carp isolate genome. 80 genes were identified to be present in the genome of the highly virulent carp isolate 04017018 while absent in that of the low virulent carp isolate CDI-A. Of these 80 genes, the 62 hypothetical genes were left out in the representation above. Genes marked in bold are also predicted to be involved in virulence when identifying them via Virulence Factor Database. *Gene locations are formatted as NODEn:s-e where n is a unique scaffold number, s and e are the gene’s start and end position respectively.
Additional file 4.
Unique protein-encoding genes in the highly virulent trout isolate genome. 374 genes were identified in the genome of the highly virulent F. columnare JIP P11/91 isolate while being absent in that of the non-virulent F. columnare ATCC 49512. Of these 374 genes, the 263 hypothetical genes were left out in the representation above. Genes marked in bold are also predicted to be involved in virulence when identifying them via Virulence Factor Database. *Gene locations are formatted as NODEn:s-e where n is a unique scaffold number, s and e are the gene’s start and end position respectively.
Additional file 5.
Single nucleotide variants (SNVs) in gyrase genes of Flavobacterium columnare isolates. This table shows the SNVs present in the gyrase genes of F. columnare isolates CDI-A and 04017018 when compared to gyrase genes of the reference genome ATCC 49512. However, as only isolate CDI-A displayed phenotypic antimicrobial resistance towards both first- and second-generation quinolones, the unique SNVs in the gyrase genes of the latter isolate are highlighted in grey background. No SNVs were encountered in DNA gyrase subunit B (EC 5.99.1.3). Average read quality score of the bases supporting a variant was higher than 35 (which is considered very good quality).
Additional file 6.
Unique single nucleotide variants (SNVs) present in gyrA of isolate CDI-A. The impact of the SNP (single nucleotide polymorphism) on protein activity/stability was predicted by comparison of amino acid characteristics, by using PredictSNP and I-Mutant Suite 3.0 tools, and by investigating which amino acids were present in the reference strain of F. columnare and of HV carp isolate 04017018, both of the latter not displaying acquired resistance towards fluoroquinolones, using BLAST and ConSurf.
Additional file 7.
Characteristics of prophage regions in the four Flavobacterium columnare isolates. The prophage regions were identified in the genome of the reference isolate (ATCC49512) and the three newly assembled F. columnare isolates 04017018, CDI-A and JIP P11/91 via PHASTER. Prophage regions with scores less than 70 are incomplete.

Additional file 8.
Schematic representation of genes present in the prophages. A: For the three newly sequenced bacterial genomes, the most probable prophage (Bacill_G_NC_023719) retrieved was similar for all three genomes. B: For the reference genome ATCC 49512, the latter prophage (Bacill_G_NC_023719) was not retrieved, however another most probable prophage (Flavob_23T_NC_041859) was identified. The prophages identified for all 4 isolates, were considered incomplete. Legend: Att: Attachment site, Coa: Coat protein (head protein), Hyp: Hypothetical proteins, PLP: Phage-like Protein, Tra: Transposase, Sha: Tail shaft.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Declercq, A.M., Tilleman, L., Gansemans, Y. et al. Comparative genomics of Flavobacterium columnare unveils novel insights in virulence and antimicrobial resistance mechanisms. Vet Res 52, 18 (2021). https://doi.org/10.1186/s13567-021-00899-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-021-00899-w