
Abstract
M. bovis is one of the leading causes of respiratory disease and antimicrobial use in cattle. The pathogen is widespread in different cattle industries worldwide, but highest prevalence is found in the veal industry. Knowledge on M. bovis strain distribution over the dairy, beef and veal industries is crucial for the design of effective control and prevention programs, but currently undocumented. Therefore, the present study evaluated the molecular epidemiology and genetic relatedness of M. bovis isolates obtained from Belgian beef, dairy and veal farms, and how these relate to M. bovis strains obtained worldwide. Full genomes of one hundred Belgian M. bovis isolates collected over a 5-year period (2014–2019), obtained from 27 dairy, 38 beef and 29 veal farms, were sequenced by long-read nanopore sequencing. Consensus sequences were used to generate a phylogenetic tree in order to associate genetic clusters with cattle sector, geographical area and year of isolation. The phylogenetic analysis of the Belgian M. bovis isolates resulted in 5 major clusters and 1 outlier. No sector-specific M. bovis clustering was identified. On a world scale, Belgian isolates clustered with Israeli, European and American strains. Different M. bovis clusters circulated for at least 1.5 consecutive years throughout the country, affecting all observed industries. Therefore, the high prevalence in the veal industry is more likely the consequence of frequent purchase from the dairy and beef industry, than that a reservoir of veal specific strains on farm would exist. These results emphasize the importance of biosecurity in M. bovis control and prevention.
Similar content being viewed by others

Introduction
Mycoplasma bovis (M. bovis) causes mostly pneumonia, arthritis, otitis in calves and mastitis in adult cattle [1, 2] resulting in high antimicrobial use (AMU) and enormous economic losses in cattle farming sectors worldwide [2,3,4]. In Belgium, 100% of the veal farms are seropositive for M. bovis [5, 6], whereas M. bovis is involved in 33% of acute pneumonia outbreaks in beef and dairy farms [7]. Treatment of M. bovis is frequently unsatisfactory, probably due to a combination of intrinsic and acquired antimicrobial resistance, immuno-evasive properties of the pathogen and failure of the animal to generate an effective immune response [8, 9]. Together with the absence of an effective commercially available vaccine, the control of M. bovis is particularly challenging.
A contemporary fear is that the veal sector, currently combining a high AMU and a farm level M. bovis prevalence of 100%, is a reservoir for multi-resistant sector-specific M. bovis strains [6, 10, 11]. Currently, there is insufficient knowledge about the epidemiology of circulating M. bovis strains to answer this question. Several epidemiological studies observed clonal emergence and identified dominant lineages of the M. bovis bacterium, based on antimicrobial resistance patterns and different strain typing methods [12,13,14,15]. In the past, different approaches were used to subtype M. bovis strains, including random amplification of polymorphic DNA (RAPD), arbitrarily primed PCR (AP-PCR), amplification fragment length polymorphism (AFLP), pulsed-field gel electrophoresis (PFGE), multiple-locus variable-number tandem repeat (MLVA), and multi-locus sequence typing (MLST) [13, 14, 16,17,18]. Unfortunately, results from these typing methods are difficult to compare and only focus on a small fraction of the genomic information, resulting in a limited insight in the genetics and incongruence among studies [17, 19, 20]. Therefore, whole genome sequencing (WGS) could be a great opportunity, considering its highly discriminative capacity and reproducibility compared to older typing methods [21, 22].
Several studies already investigated whether specific M. bovis strains were associated with affected organs, such as udder, respiratory tract or joints [14, 23, 24], geographical location [23, 24] or health status [23, 25]. Only one study determined epidemiology based on AP-PCR in three farms from three different husbandry conditions (dairy calf ranch, feedlot and closed beef herd), presuming that management factors could influence the distribution of M. bovis [16]. These husbandry conditions are not comparable with the three main sectors in Europe. In Europe, a lot of short-distance movements of cattle between farms is seen, and the veal industry is an important side market of the dairy and beef industry [26, 27]. It is currently not clear whether sector-specific M. bovis strains are present and what their genetic relation is to previously sequenced M. bovis isolates. Therefore, the present study first evaluated the molecular epidemiology and genetic relatedness of M. bovis isolates obtained from Belgian beef, dairy and veal farms. Furthermore, it studied the relationship of these isolates to M. bovis strains from other countries.
Materials and methods
Mycoplasma bovis collection and identification
One hundred M. bovis isolates were obtained from 94 Belgian farms (27 dairy, 38 beef and 29 veal) over a 5-year period (2014–2019). All isolates were obtained from diagnostic samples collected by field veterinarians from clinical cases, in compliance with EU legislation on ethics in animal experimentation [2010/63/EU]. Isolates were collected in 2014 (n = 1), 2016 (11), 2017 (63), 2018 (19) and 2019 (6), originating from the provinces East-Flanders (n = 10), West-Flanders (25), Antwerp (38), Limburg (6), Flemish Brabant (10), Heynowes (6), Namen (2) and Liege (1). The origin of two isolates was unclear. The samples were retrieved from the respiratory tract (89), middle ear (3), milk (4), joint (2) and other fluids (3) of calves and adult cattle, as shown in Additional file 1. The samples were cultured on selective indicative agar [28], and identification was verified with MALDI-TOF MS (score value ≥ 1.7), as described earlier [29] and Kraken2 analysis. Isolates had a passage history of maximum 3–5 times and all isolates were stored at maximum − 20 °C until further analysis.
Preparation and DNA extraction
In ten separate runs, all M. bovis isolates were thawed and cultured in 10 mL modified PPLO broth (pH 7.8) (Difco™, BD Diagnostic Systems, Sparks, Md.), supplemented with 25% inactivated horse serum (Gibco™), 0.7% technical yeast extract, 0.5% sodium pyruvate (ReagentPlus, Sigma-Aldrich®), 0.5% D-( +)-glucose monohydrate (Sigma-Aldrich) and 0.005% phenol red. After 4 days of incubation (37ºC, 5% CO2) a bacterial suspension of approximately 108 CFU/mL was obtained. Bacterial DNA was obtained using the ZymoBIOMICS DNA Miniprep kit (Zymo Research) according to the manufacturer’s instructions. Quantity and quality were verified using NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Low quality samples were further cleaned using CleanNGS (CleanNa) beads. All runs included the M. bovis PG45 type strain (ATCC 25523) and modified PPLO broth as the positive and negative control, respectively.
Library preparation and MinION long-read sequencing
Quality-checked native M. bovis DNA was immediately used for library preparation using the Rapid Barcoding Sequencing Kit (SQK-RBK004; Oxford Nanopore Technologies (ONT)), following manufacturers’ instructions. For each run, ten field strains, one positive control (PG45) and one negative control (sterile broth) were multiplexed (400 ng DNA per sample). A new R9.4.1 Flow cell (ONT) was used for a 48 h sequencing run on MinION device (ONT). Raw fast5 read files were collected using MinKnow v.3.6.5.
Bioinformatics pipeline
All data were analyzed on an Ubuntu 18.04.3 LTS system. In order to speed up bioinformatics analyses, GPU resources (GeForce RTX 2080 Ti/PCIe/SSE2) were exploited where possible. Raw fast5 files were basecalled using Guppy basecaller (GPU v.3.3.0; ONT), followed by demultiplexing, adapter trimming, and quality filtering (Q-score ≥ 7) of fastq files with qcat (v.1.1.0; ONT) and NanoFilt (v. 2.5.0; [30]), respectively. Reference-based assemblies were generated using the M. bovis PG45 type strain sequence (NC_014760.1) by mapping filtered reads onto the reference using GraphMap (v.0.5.2; [31]). Final consensus sequences were generated using Medaka (GPU v.0.10.0; ONT). All strains were identified as M. bovis using Kraken2 (v2.0.8; [32]) by aligning the reads against the minikraken_v1_8GB database with standard settings. Overall consensus assembly accuracies were verified by comparing total Single Nucleotide Polymorphisms (SNPs) using the CSI phylogeny package (v1.4, Center for Genomic Epidemiology, Denmark; [33]) as compared to the M. bovis PG45 type strain (NC_014760.1) reference sequences. To validate the use of long-read sequencing, SNPs of ten independent M. bovis PG45 assemblies were compared to those in a single MiSeq experimental dataset. All M. bovis consensus genomes are available for download on the NCBI GenBank database under the BioProject PRJNA639688 and accession numbers (SAMN15246515-SAMN1524662). Sequencing summaries can be found in Additional file 1.
Phylogenetic analysis
Phylogenetic analysis was performed on all newly generated consensus sequences alone or in combination with 250 previously published M. bovis sequences using the FastTree-based CSI Phylogeny v1.4 (see Additional file 2). All analyses included the M. bovis PG45 type strain (NC_014760.1) as reference and M. agalactiae PG2 (NC_009497.1) as outgroup. Resulting Newick files were visualized with MEGA-X software [34].
Cluster and strain determination
Due to the lack of relatedness criteria for SNP typing schemes of M. bovis and the need to establish these per organism and experimental design [35], clusters were defined by visual inspection of the phylogenetic tree and by taking into account bootstrap support. In addition, the matrix of pairwise SNP counts was extracted from CSI Phylogeny for further inspection. Mean SNP differences were calculated between within-cluster isolates, and outliers were defined with the 1.5xIQR rule, using the Outlier Calculator (https://miniwebtool.com/outlier-calculator/).
Geographical distribution
Esri®ArcMap™ (version 10.7.1) software was used to visualize the geographical distribution and density of M. bovis isolates over Belgium. Herd size was based on the national Identification and Registration system, containing, on the first of January 2017, a total of 23 995 cattle herds in Belgium (23 733 conventional herds; 262 veal), and a total of 2 517 850 cattle. The spatial distribution of the Belgian cattle (both cattle and veal calves) was displayed using kernel smoothing. Coordinates of Belgian cattle herds were converted into a continuous raster using the kernel density estimation function weighted by number of cattle (Spatial Analyst, ArcMAP X, ESRI, Redlands, CA, USA).
Results
Phylogenetic analysis of Belgian isolates
A median sequencing depth of 618X (range: 32X-2689X) was obtained from long reads with an average N50 read length of 5706 ± 1514 bp for all Belgian M. bovis isolates. First, implementation of long-read sequencing of M. bovis genomes was validated by comparing total SNPs from ten independently sequenced M. bovis PG45 sequences and a single M. bovis PG45 MiSeq dataset, showing 53 ± 3 SNPs and 27 SNPs difference, respectively, compared with the 1 003 404 bp of the M. bovis PG45 reference genome (NC_014760.1). The observed average SNP difference of 0.005% for the long-read sequencing approach was considered acceptable to allow meaningful phylogenetic analyses. In addition, control strain M. bovis PG45 results were mutually compared over all ten runs, showing a mean SNP difference of 20 (range 8–30, standard deviation 4.6), which was also demonstrated an acceptable inter-experimental variation.
Taking into account all Belgian M. bovis strains, and also including the outgroup M. agalactiae, 51.4% of the M. bovis genome or 515 324 nucleotide positions were used for phylogeny. The minimum and maximum SNP differences among Belgian M. bovis isolates were 33 and 3775, respectively.
Visual inspection of the phylogenetic analysis of 100 M. bovis isolates resulted in 5 clusters (I-V): 3 large clusters (n ≥ 10 isolates), 2 smaller clusters (n < 10), and 1 distinct strain (VK30) as shown in Figure 1. Cluster I to V contained 3, 7, 16, 33, and 40 M. bovis isolates, respectively. Inspection of pairwise SNP differences per cluster, showed more homogeny within clusters II and III (mean ΔSNPs of 87 and 316, respectively), compared with cluster I, IV and V (mean ΔSNPs of 834, 1027, and 1435) (Table 1). Mean SNP differences among within-cluster isolates and outlier calculations showed outliers within cluster III (Mb222, Mb231), IV (Mb201, Mb240, Mb175, TOVK) and V (Mb116, Mb166, VK11, VK23).

SNP-based phylogenetic tree of 100 M. bovis isolates from Belgian dairy, beef and veal farms. The figure was created using MEGA-X software with M. bovis isolates obtained over 2014–2019. The tree was rerooted to M. agalactiae PG2, which was included as an outgroup. Clusters (I-V), and VK30 are represented by different colors. The designation of the isolates features the sector (■ dairy; ● beef; ♦ veal), year of isolation (2014–2019), affected organ (R: respiratory tract; M: milk; E: ear, J: joint and O: other) and sequence identification (see Additional file 1). The scale bar indicates the number of substitutions per site, and bootstrap values are represented on nodes.
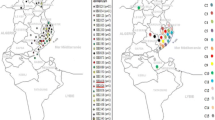
Between and within clusters, no association could be observed for the different cattle sectors or year of isolation (Figure 1). Two different isolates from the same herd (veal) and same sampling period (Mb49 and Mb50) did not cluster together (II and V). All clusters persisted in Belgium for at least 1.5 consecutive years throughout the country. M. bovis strains isolated from the middle ear (n = 3) were clustered within cluster IV, while those obtained from milk, joint and other samples were scattered over different clusters (Figure 1). Finally, no clear association between geographic location of sampled farms and M. bovis clusters was observed, as shown in Figure 2.

Geographical distribution of different M. bovis clusters over 2014–2019 and cattle density in Belgium (2017). The map was created using Esri®ArcMap™ (version 10.7.1) software. Clusters (I-V) are represented by different colors and the radius of the circle represents the number of isolates from one village. Mixed colors within one circle represent the presence of different clusters within one village.
Phylogenetic analysis of M. bovis worldwide
All 100 Belgian isolates were added to the worldwide phylogenetic tree. The percentage of the reference genome covered by all isolates, including the PG45 standard strain and the M. agalactiae outgroup strain PG2 was 39.3%, therefore 394 303 positions were found in all analyzed genomes. The minimum and maximum SNP differences among all M. bovis isolates including the reference strain, were 0 and 4871, respectively. Belgian clusters are situated in different parts of the phylogenetic tree worldwide (Figure 3). Cluster I is related to strains isolated in the USA (2007; mean ΔSNPs of 636) and Israel (2016; mean ΔSNPs of 1369). Cluster II is closely related to one recent French strain (2016; mean ΔSNPs of 171) and is situated in a larger cluster related to older strains from Israel and Eastern Europe (2001–2009; ΔSNPs < 200), and other more recently isolated strains from Israel and Eastern Europe (2011–2017; ΔSNPs < 500). Belgian cluster III and V do not cluster together with non-Belgian isolates, while cluster IV is closely related (ΔSNPs < 300 without cluster IV outliers) to M. bovis strains obtained in Israel (2012–2017) and Eastern Europe (2013–2016). VK11 remains an outlier that does not collate with the rest of cluster V. Consistent with Figure 1, VK30 is well separated from the other Belgian isolates and is very closely related (mean ΔSNPs of 171) to strains obtained from milk in the USA (2017).

SNP-based topology of 350 M. bovis isolates in MEGA-X. The tree was rerooted to M. agalactiae PG2 (EPS 1952 PG2), which was included as outgroup. Belgian clusters (I-V) were collapsed as far as possible and represented by different colors (I: yellow; II: purple; III: brown; IV: blue, V: red; VK30: green). The designation of the isolates contains the coded name of country of origin (ISO 3166-1; Alpha-3 code), continent of origin (● Europe; o Non-Europe), year of isolation (1952–2019), affected organ (R: respiratory tract; M: milk; E: ear, J: joint and O: other or unknown) and sequence identification (see Additional file 2) or the number of collapsed Belgian isolates between brackets. Bootstrap values are represented on nodes.
Discussion
In this study, one-hundred M. bovis isolates from different Belgian cattle sectors (beef, dairy or veal) were phylogenetically compared to investigate whether sector-specific strains exist and whether such strains are related to M. bovis strains previously isolated and sequenced worldwide.
In this study, we chose to apply the ONT long-read secquencing approach [36], because no default WGS approaches are defined for Mycoplasma spp. and short-read sequencing biases have been described for genomes with highly repetitive regions.
WGS approaches have become more attractive over the last years, as the cost for next-generation sequencing has significantly reduced. Single Nucleotide Polymorphism (SNP) analysis using Illumina short read data from M. bovis isolates already showed to be an effective way for M. bovis genotyping [24, 25]. Long-read nanopore sequencing (Oxford Nanopore Technologies) is known to create much faster results, and was recently applied in veterinary medicine as well [37, 38]. Yet, lower single read accuracies are currently obtained with ONT in comparison to Illumina [39]. Therefore, the implementation of long-read sequencing to generate M. bovis genome assemblies was verified, showing only on average 53 SNPs difference of the long-read approach with the publically available M. bovis PG45 genome, representing an acceptable error rate of 0.005%. As a result, the authors believe that nanopore sequencing is a highly accessible tool, allowing practical use outside academia in routine diagnostics and real time surveillance.
From the study results, several interesting observations were made. First of all, the obtained M. bovis isolates belonged to at least five different M. bovis clusters, of which three dominant clusters were identified. This is in agreement with an Israeli study based on WGS-SNP, where six clusters were observed of which three were dominant. Remarkably, one cluster contained more than 50% of the isolates in that study [25]. Several other studies also showed one or two dominant lineages, although different typing methods were used [13, 15, 18, 24, 40]. In contrast to Aebi et al. [41], where mostly herd-specific M. bovis isolates were seen in Switzerland, we observed close relatedness of M. bovis isolates over the different herds. This might be a result of more frequent purchasing cattle from different origins and transportation in Belgium, because 40% of cattle is transported at least once (and up to eight times) over a 5-year lifespan in Belgium [7, 16, 17, 26, 42]. As such, the higher heterogeneity observed in cluster I, IV and V compared with clusters II and III, may be caused by different rates of genetic drift between clonal lines [17].
Secondly, no sector-specific strains or clusters were identified in the present study, which does not entirely come as a surprise. In Belgium, veal calves are purchased from both dairy and beef farms, and fattened and slaughtered in specialized veal farms and slaughterhouses, respectively [27]. Also, approximately 15–20% of the farms in Belgium are mixed farms. Therefore, contact among different cattle sectors is intense. In addition, herd visitors and artificial insemination might play a role in spreading of M. bovis or introducing new strains on farms [43,44,45].
Thirdly, when we take a closer look at how the different clusters have spread over Belgium, no clear association with location was observed. This was also concluded in studies performed in the UK and USA [17, 23, 42]. Nevertheless, in the provinces of Antwerp and Western Flanders seem to be hotspots for M. bovis outbreaks. Besides a high number of local transports, Antwerp and the Flanders area are also the main gates for cattle import, which makes these areas predisposed for the introduction of new M. bovis strains [26]. Unfortunately, M. bovis genomes were not available for isolates obtained from the top import countries for Belgium, which are Germany and the Netherlands.
Although this study was not designed to draw definitive conclusions about year of isolation and affected organs, some preliminary observations can be made. For example, no association between M. bovis strain and year of isolation was observed. On the other hand, we saw that representatives of all clusters persisted for at least 1.5 consecutive years on Belgian territory. The persistence of strains within a country or herd has been described before [16, 24, 41]. Furthermore, shifts between dominant lineages from older to new strains have been reported before as well [13, 15, 46, 47]. Also, we did not observe an association between cluster and affected organ, which is in line with previous studies [14, 23, 24]. Yet, it was remarkable that all isolates obtained from the middle ear were clustered, which could suggest the middle ear as possible predilection site for certain M. bovis strains. However no definitive conclusions can be drawn as there were only few isolates obtained from this isolation site in the present study. In addition, we isolated different M. bovis strains (Mb49, Mb50) from two veal calves on the same farm at the same time. The observation of two different strains in one herd or even one animal has been described before [14, 16, 42, 48], in contrast to Arcangioli et al. [46], who isolated only one identical dominant profile in the same feedlot.
Finally, it was evident that Belgian isolates were mostly related to European and Israeli M. bovis isolates, even though only a few genomes of European M. bovis isolates have been published in the NCBI database. This seems plausible, as Belgian farmers mostly purchase cattle from European farms, while Israel also partly imports cattle from Eastern-Europe. The fact that Israeli isolates are often related to Chinese and Australian strains, is also due to import of cattle, as outlined in detail elsewhere [25, 40]. Some of the Belgian outlier strains were related to American isolates, which might be explained by the fact that M. bovis was first isolated in the USA and outbreaks in Europe were only seen years later. So, we can only speculate whether these outlier strains could have been imported or evolved geographically distinct from each other. Clusters of the Belgian isolates were not clustered exactly in the same way in the Belgian vs. the worldwide phylogenetic tree. A possible explanation could be the loss of overall coverage between the construction of both phylogenetic trees (51% for the Belgian to 40% worldwide). This might be due to (1) heterogeneity among isolates worldwide and/or (2) the use of genomes obtained by different laboratories, using different sequencing protocols, as the quality can be influenced by strain maintenance, DNA extraction, library preparation, sequencing, and the bioinformatics analysis [49].
In conclusion, multiple M. bovis clusters were circulating in Belgium in 2014–2019, and were persisting for several years. Neither the veal industry, nor any other cattle industry could be identified as source of strain persistence. Connections between dairy, beef and veal industry are intense and M. bovis appears to easily spread among these sectors. The M. bovis issues in the veal industry seem more likely the consequences of strain import from dairy and beef, rather than persistence of a limited number of veal specific strains. This information can contribute to better control and prevention of M. bovis infections by improved biosecurity.
Availability of data and materials
All M. bovis consensus genomes are available for download on the NCBI GenBank database under the BioProject PRJNA639688 and accession numbers (SAMN15246515-SAMN1524662). Sequencing summaries can be found in Additional file 1.
Abbreviations
- AFLP:
-
amplification fragment length polymorphism
- AMU:
-
antimicrobial use
- AP-PCR:
-
arbitrarily primed PCR
- MLST:
-
multi-locus sequence typing
- MLVA:
-
multiple-locus variable-number tandem repeat
- ONT:
-
Oxford Nanopore Technologies
- PFGE:
-
pulsed-field gel electrophoresis
- RAPD:
-
random amplification of polymorphic DNA
- SNPs:
-
single Nucleotide Polymorphisms
- WGS:
-
whole genome sequencing
References
Maunsell FP, Woolums AR, Francoz D et al (2011) Mycoplasma bovis infections in cattle. J Vet Intern Med 25:772–783. https://doi.org/10.1111/j.1939-1676.2011.0750.x
Maunsell FP, Donovan GA (2009) Mycoplasma bovis Infections in Young Calves. Vet Clin North Am Food Anim Pract 25:139–177. https://doi.org/10.1016/j.cvfa.2008.10.011
Nicholas RAJ, Ayling RD (2003) Mycoplasma bovis: Disease, diagnosis, and control. Res Vet Sci 74:105–112. https://doi.org/10.1016/s0034-5288(02)00155-8
Fox LK, Kirk JH, Britten A (2005) Mycoplasma mastitis: a review of transmission and control. J Vet Med Ser B Infect Dis Vet Public Heal 52:153–160. https://doi.org/10.1111/j.1439-0450.2005.00845.x
Pardon B, De Bleecker K, Dewulf J et al (2011) Prevalence of respiratory pathogens in diseased, non-vaccinated, routinely medicated veal calves. Vet Rec. https://doi.org/10.1136/vr.d4406
Pardon B (2012) Morbidity, mortality and drug use in white veal calves emphasis on respiratory disease. PhD Thesis, Ghent University, Faculty of Veterinary Medicine, Department of Large Animal Internal Medicine. http://hdl.handle.net/1854/LU-3007344
Pardon B, Callens J, Maris J et al (2020) Pathogen-specific risk factors in acute outbreaks of respiratory disease in calves. J Dairy Sci 103:2556–2566. https://doi.org/10.3168/jds.2019-17486
Gautier-Bouchardon AV (2018) Antimicrobial resistance in Mycoplasma spp. Microbiol Spectr 6:425–446. https://doi.org/10.1128/microbiolspec.arba-0030-2018
Maunsell FP, Chase C (2019) Mycoplasma bovis: Interactions with the Immune System and Failure to Generate an Effective Immune Response. Vet Clin North Am Food Anim Pract 35:471–483. https://doi.org/10.1016/j.cvfa.2019.08.003
Arcangioli MA, Duet A, Meyer G et al (2008) The role of Mycoplasma bovis in bovine respiratory disease outbreaks in veal calf feedlots. Vet J 177:89–93. https://doi.org/10.1016/j.tvjl.2007.03.008
Bokma J, Boone R, Deprez P, Pardon B (2019) Risk factors for antimicrobial use in veal calves and the association with mortality. J Dairy Sci 102:607–618. https://doi.org/10.3168/jds.2018-15211
Gautier-Bouchardon AV, Ferré S, Le Grand D et al (2014) Overall decrease in the susceptibility of Mycoplasma bovis to antimicrobials over the past 30 years in France. PLoS One 2:e87672. https://doi.org/10.1371/journal.pone.0087672
Becker CAM, Thibault FM, Arcangioli MA, Tardy F (2015) Loss of diversity within Mycoplasma bovis isolates collected in France from bovines with respiratory diseases over the last 35 years. Infect Genet Evol 33:118–126. https://doi.org/10.1016/j.meegid.2015.04.019
Rosales RS, Churchward CP, Schnee C et al (2015) Global multilocus sequence typing analysis of Mycoplasma bovis isolates reveals two main population clusters. J Clin Microbiol 53:789–794. https://doi.org/10.1128/JCM.01910-14
Bürki S, Spergser J, Bodmer M, Pilo P (2016) A dominant lineage of Mycoplasma bovis is associated with an increased number of severe mastitis cases in cattle. Vet Microbiol 196:63–66. https://doi.org/10.1016/j.vetmic.2016.10.016
Butler JA, Pinnow CC, Thomson JU et al (2001) Use of arbitrarily primed polymerase chain reaction to investigate Mycoplasma bovis outbreaks. Vet Microbiol 78:175–181. https://doi.org/10.1016/S0378-1135(00)00286-8
McAuliffe L, Kokotovic B, Ayling RD, Nicholas RAJ (2004) Molecular epidemiological analysis of Mycoplasma bovis isolates from the United Kingdom shows two genetically distinct clusters. J Clin Microbiol 42:4556–4565. https://doi.org/10.1128/JCM.42.10.4556-4565.2004
Spergser J, Macher K, Kargl M et al (2013) Emergence, re-emergence, spread and host species crossing of Mycoplasma bovis in the Austrian Alps caused by a single endemic strain. Vet Microbiol 164:299–306. https://doi.org/10.1016/j.vetmic.2013.02.007
Maiden MCJ, Bygraves JA, Feil E et al (1998) Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145. https://doi.org/10.1073/pnas.95.6.3140
Register KB, Lysnyansky I, Jelinski MD et al (2020) Comparison of two multilocus sequence typing schemes for Mycoplasma bovis and revision of the PubMLST reference method. J Clin Microbiol 58:e00283–e320. https://doi.org/10.1128/jcm.00283-20
Deng X, den Bakker HC, Hendriksen RS (2016) Genomic epidemiology: whole-genome-sequencing–powered surveillance and outbreak investigation of foodborne bacterial pathogens. Annu Rev Food Sci Technol 7:353–374. https://doi.org/10.1146/annurev-food-041715-033259
Leekitcharoenphon P, Nielsen EM, Kaas RS et al (2014) Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS One 9:e87991. https://doi.org/10.1371/journal.pone.0087991
Register KB, Thole L, Rosenbush RF, Minion FC (2015) Multilocus sequence typing of Mycoplasma bovis reveals host-specific genotypes in cattle versus bison. Vet Microbiol 175:92–98. https://doi.org/10.1016/j.vetmic.2014.11.002
Parker AM, Shukla A, House JK et al (2016) Genetic characterization of Australian Mycoplasma bovis isolates through whole genome sequencing analysis. Vet Microbiol 196:118–125. https://doi.org/10.1016/j.vetmic.2016.10.010
Yair Y, Borovok I, Mikula I et al (2020) Genomics-based epidemiology of bovine Mycoplasma bovis strains in Israel. BMC Genomics 21:70. https://doi.org/10.1186/s12864-020-6460-0
Ensoy C, Faes C, Welby S et al (2014) Exploring cattle movements in Belgium. Prev Vet Med 116:89–101. https://doi.org/10.1016/j.prevetmed.2014.05.003
Pardon B, Catry B, Boone R et al (2014) Characteristics and challenges of the modern Belgian veal industry. Vlaams Diergeneeskd Tijdschr 83:155–163
Bokma J, Van Driessche L, Deprez P et al (2020) Rapid identification of Mycoplasma bovis from bovine bronchoalveolar lavage fluid with MALDI-TOF MS after enrichment procedure. J Clin Microbiol 58:e00004–20. https://doi.org/10.1128/jcm.00004-20
Bokma J, Pardon B, Van Driessche L et al (2019) Optimizing identification of Mycoplasma bovis by MALDI-TOF MS. Res Vet Sci 125:185–188. https://doi.org/10.1016/j.rvsc.2019.06.010
De Coster W, D’Hert S, Schultz DT et al (2018) NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 34:2666–2669. https://doi.org/10.1093/bioinformatics/bty149
Sović I, Šikić M, Wilm A et al (2016) Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat Commun 7:11307. https://doi.org/10.1038/ncomms11307
Wood DE, Lu J, Langmead B (2019) Improved metagenomic analysis with Kraken 2. Genome Biol 20:257. https://doi.org/10.1186/s13059-019-1891-0
Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O (2014) Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. https://doi.org/10.1371/journal.pone.0104984
Kumar S, Stecher G, Li M et al (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Schürch AC, Arredondo-Alonso S, Willems RJL, Goering RV (2018) Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin Microbiol Infect 24:350–354
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: Ten years of next-generation sequencing technologies. Nat Rev Genet 17:333–351. https://doi.org/10.1038/nrg.2016.49
Quick J, Ashton P, Calus S et al (2015) Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol 16:114. https://doi.org/10.1186/s13059-015-0677-2
Theuns S, Vanmechelen B, Bernaert Q et al (2018) Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Sci Rep 8:9830. https://doi.org/10.1038/s41598-018-28180-9
Rang FJ, Kloosterman WP, de Ridder J (2018) From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol 19:90. https://doi.org/10.1186/s13059-018-1462-9
Menghwar H, He C, Zhang H et al (2017) Genotype distribution of Chinese Mycoplasma bovis isolates and their evolutionary relationship to strains from other countries. Microb Pathog 111:108–117. https://doi.org/10.1016/j.micpath.2017.08.029
Aebi M, Bodmer M, Frey J, Pilo P (2012) Herd-specific strains of Mycoplasma bovis in outbreaks of mycoplasmal mastitis and pneumonia. Vet Microbiol 157:363–368. https://doi.org/10.1016/j.vetmic.2012.01.006
Soehnlen MK, Kariyawasam S, Lumadue JA et al (2011) Molecular epidemiological analysis of Mycoplasma bovis isolates from the Pennsylvania Animal Diagnostic Laboratory showing genetic diversity. J Dairy Sci 94:1893–1899. https://doi.org/10.3168/jds.2010-3967
Gonzalez RN, Sears PM, Merrill RA, Hayes GL (1992) Mastitis due to Mycoplasma in the state of New York during the period 1972–1990. Cornell Vet 82:29–40
Gille L, Pilo P, Valgaeren BR et al (2016) A new predilection site of Mycoplasma bovis: Postsurgical seromas in beef cattle. Vet Microbiol 186:67–70. https://doi.org/10.1016/j.vetmic.2016.02.011
Haapala V, Pohjanvirta T, Vähänikkilä N et al (2018) Semen as a source of Mycoplasma bovis mastitis in dairy herds. Vet Microbiol 216:60–66. https://doi.org/10.1016/j.vetmic.2018.02.005
Arcangioli MA, Aslan H, Tardy F et al (2012) The use of pulsed-field gel electrophoresis to investigate the epidemiology of Mycoplasma bovis in French calf feedlots. Vet J. https://doi.org/10.1016/j.tvjl.2011.05.004
Hata E, Harada T, Itoh M (2019) Relationship between antimicrobial susceptibility and multilocus sequence type of Mycoplasma bovis isolates and development of a method for rapid detection of point mutations involved in decreased susceptibility to macrolides, lincosamides, tetracyclines. Appl Environ Microbiol 85:e00575–e619. https://doi.org/10.1128/AEM.00575-19
Sulyok KM, Kreizinger Z, Fekete L et al (2014) Phylogeny of Mycoplasma bovis isolates from Hungary based on multi locus sequence typing and multiple-locus variable-number tandem repeat analysis. BMC Vet Res 10:108. https://doi.org/10.1186/1746-6148-10-108
Portmann AC, Fournier C, Gimonet J et al (2018) A validation approach of an end-to-end whole genome sequencing workflow for source tracking of Listeria monocytogenes and Salmonella enterica. Front Microbiol 9:446. https://doi.org/10.3389/fmicb.2018.00446
Acknowledgements
We thank everyone involved in the collection of the isolates (DGZ (Animal Health Service Flanders), ARSIA, Clinic of Large Animal Internal Medicine (Ghent University), and practicing veterinarians) and the support of the technical staff at the involved laboratories, with special thanks to Arlette Van de Kerckhove, Serge Verbanck, and Marthe Pauwels.
Funding
Jade Bokma is supported by a grant of the Belgian Federal Public Service, Health, Food Chain Safety and Environment (FOD)[RF 17/6313, MALDIRESP/MA]. The use of MinION was also supported by FOD [RF17/6312, PigMinION], and the Industrial Research Fund of Ghent University [F2018/IOF-Advanced/487]. The MALDI-TOF MS was financed by the Research Foundation Flanders (FWO-Vlaanderen) as Hercules project [G0H2516N, AUGE/15/05]. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
JB, NV, ST, FB and BP conceptualized the experiments and were part of the validation, formal analysis, interpretation and visualization of the presented work. SR assisted in the visualization of the work. JB, NV, KB and JC performed the data collection. JB and NV executed the experiments. Resources were provided by HN, FH, ST, FB and BP. JB, NV, FB and BP wrote the manuscript draft, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. All isolates were obtained from diagnostic samples collected by field veterinarians from clinical cases, this is in compliance with EU legislation on ethics in animal experimentation [2010/63/EU].
Consent for publication
Not applicable.
Competing interests
B.P. has received honoraria for acting as speaker or consultant for pharmaceutical (Zoetis, MSD, Vetoquinol, Dopharma, Boehringer Ingelheim, Dechra, Hipra, Ceva, Merial and Elanco) and agricultural (Boerenbond, Algoet nutrition) companies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bokma, J., Vereecke, N., De Bleecker, K. et al. Phylogenomic analysis of Mycoplasma bovis from Belgian veal, dairy and beef herds. Vet Res 51, 121 (2020). https://doi.org/10.1186/s13567-020-00848-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-020-00848-z