
Abstract
The G1-H9N2 avian influenza virus (AIV) has caused significant economic losses in the commercial poultry industry due to reduced egg production and increased mortality. The field observations have shown that H9N2 viruses circulate and naturally mix with other pathogens and these simultaneous infections can exacerbate disease. To avoid an incorrect virus characterization, due to co-infection, isolates were purified by in vitro plaque assays. Two plaque purified G1-H9N2 clones, selected on different cell types, named MDCK-and CEF-clone in regards to the cell culture used, were studied in vivo, revealing two different virulence phenotypes. Subsequently, the underlying mechanisms were studied. Specifically, the phenotypical outcome of SPF bird infection by the two clones resulted in completely different clinical outcomes. These differences in clinical outcome were used to study the factors behind this output in more detail. Further studies demonstrated that the more severe disease outcome associated with the MDCK-clone involves a strong induction of pro-inflammatory cytokines and a lack of type I interferon production, whereas the mild disease outcome associated with the CEF-clone is related to a greater antiviral cytokine response. The immunosuppressive effect of the MDCK-clone on splenocytes was further demonstrated via ChIFN-γ lack production after ex vivo mitogenic stimulation. Genome sequencing of the two clones identified only four amino acid differences including three in the HA sequence (HA-E198A, HA-R234L, HA-E502D-H9 numbering) and one in the NA sequence (NA-V33M). In the present study, valuable insights on the mechanisms responsible for AI pathogenicity and molecular mechanisms of H9N2 infections in chicken were obtained while highlighting the impact of the cells viruses are grown on their virulence.
Similar content being viewed by others


Introduction
Avian influenza, commonly known as avian flu or bird flu, is caused by influenza A viruses. Avian influenza viruses (AIVs) are members of the Orthomyxoviridae family and they have segmented, negative-sense, single-stranded RNA genomes. Avian influenza infections are some of the most contagious and devastating diseases that currently affect poultry populations. Infections from AIVs can lead to clinical pathologies that range from asymptomatic and mild clinical symptoms to sudden and complete mortality. Influenza A viruses are divided into subtypes based on the expression profile of two glycoproteins on their surface—the hemagglutinin (HA) and the neuraminidase (NA). To date, 18 subtypes of hemagglutinin (H1 to H18) and 11 subtypes of neuraminidase (N1 to N11) have been identified [1]. As a result, many different combinations of HA and NA proteins are possible. The natural reservoir of type A influenza viruses are wild aquatic birds, Anseriformes and Charadriiformes, such as wild ducks, gulls, and shorebirds [2, 3]. Occasionally, other species such as poultry and mammalian species become infected [4]. AIVs can be classified as low pathogenic (LPAI) or highly pathogenic (HPAI) based on the genetic features of a virus and the severity of disease it causes in chickens. The HPAI has only been associated with some strains of the H5 or H7 subtype [5].
The LPAI H9N2 was first detected in turkeys in the United States in 1966 [6] and distinct lineages have been described in the meantime. A recent study on H9 phylogeny revealed that the panoramic view of the global distribution of H9N2 is more complicated than the separation in North American and Eurasian lineages as thought in the past [7]. To date, four primary lineages, h9.1–h9.4, have been designated to represent its distribution. Lineages h9.1 and h9.2 correspond to viruses which have been isolated in North America in 1966 and in the 1990s, respectively; lineage h9.3 is widely distributed among regions of Asia, Europe, Africa, the Pacific, and North America; and lineage h9.4 has circulated exclusively in Asia with h9.4.1 and h9.4.2 sublineages, comprising the G1-like (h9.4.1.1) and Y280-like (h9.4.2.4) viruses.
The H9 G1-like viruses are the most widespread, with G1-H9N2 infections reported in Asia, the Middle East and North Africa [8, 9]. In the field, G1-H9N2 viruses induce moderate to severe clinical signs in chickens with highly variable rates of mortality (10–60%) and reduced laying rates (14–75%) that lead to economic losses [10, 11]. Numerous in vivo and in vitro studies have been conducted to investigate G1-H9N2 pathogenesis in chickens [12,13,14,15,16,17]. However, significant contradictions have been observed among the results of these studies and field data. In some experimental studies, H9N2 infections did not induce obvious clinical signs or death and replication of the virus was limited to the upper respiratory tract in specific-pathogen-free (SPF) chickens under laboratory conditions [12,13,14]. In contrast, other studies reported that H9N2 causes moderate to severe respiratory infections and low mortality in commercial broiler chickens, in parallel with the replication of the virus in multiple tissues [15,16,17]. Many factors can affect disease outcome. These contradictory results may be explained by pathogenic variability among H9N2 strains, including molecular determinants which have been found to govern the pathogenicity of AIV in chickens [18] or via secondary infections in the field. Indeed, field observations have shown that H9N2 viruses circulate and naturally mix with other pathogens such as Escherichia coli, Mycoplasma gallisepticum, Staphylococcus aureus, Haemophilus paragallinarum, Chlamydia psittaci, Ornithobacterium rhinotracheale, live bronchitis virus vaccine [19]. And these co-infections can exacerbate disease outcome, causing considerable economic losses due to poor weight gain, dramatic drop in egg production and high mortality. In Israel, the common co-circulation of H9N2 and Newcastle disease virus (NDV) since the year 2000 [20] creates complicated scenarios with the reciprocal interference between the two viruses depending on many factors such as intervals of infection, type of virus, dose, virulence and biological properties of viruses, birds species [21]. In fact, H9N2 has been associated with breakthroughs in the NDV mandatory vaccination control program [22]. Ellakany et al. observed that H9N2 infection reduced ND vaccine efficacy [23]. The presence of H9N2 in the host can induce a negative impact on the production of anti-Newcastle disease antibodies (i) either by the viral interference in which the growth of lentogenic Lasota strain—a commonly used as live vaccines against virulent form of NDV—could be suppressed or delayed [24] (ii) or by the immunosuppressive effect in which the immune organs could be damaged or destroyed following H9N2 infection [25, 26]. Furthermore, even in the presence of good herd immunity against NDV, H9N2 is also able to cause a vaccinal break as the simultaneous infection of H9N2 made birds more susceptible to velogenic NDV by lowering the minimum dose required to establish an infection and exacerbating clinical signs [27]. While it is hypothesized that the H9N2 virus acts as an immunosuppressive agent [28], the immunosuppressive effect of Israeli H9N2 and the mechanism behind remain to be fully characterized. To study the mechanisms underlying enhanced pathogenicity of H9N2 AIV and its immunosuppression, two H9N2 clones purified from the same isolate but exhibiting different pathogenicity in chickens were used as appropriate models. The objectives of this study include: (i) assessing the pathogenesis of the two H9N2 clones in SPF chickens (ii) investigating the potential immunosuppressive effects of these H9N2 viruses (iii) exploring the host’s innate immune responses by measuring the expression of different innate immune-related genes (iv) and analysing the genome sequences of both H9N2 clones.
Materials and methods
Virus strains
The low pathogenic isolate used in this study was the virus stock A/chicken/Israel/1163/2011 H9N2 LPAI (GenBank accession number: JQ973660.1). This strain was originally isolated from tracheal swabs collected from a 5-week-old broiler flock. The virus culture was amplified in embryonated SPF chicken eggs [29], allantoic fluid stored at −80 °C, and subsequently used as a virus stock. The median 50% egg infectious dose (EID50) per mL of this virus was determined by titration on SPF eggs and Reed and Muench calculations [30].
Virus purification by plaque assay
Cell culture
Chicken embryo fibroblast (CEF) cells were prepared from 9-day-old SPF embryonated eggs (Lohmann Valo, Germany) as previously described [31] and were maintained in L15/McCoy medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 2% fetal bovine serum (FBS), 50 µg/mL gentamicin and 1 mM glutamine (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in 5% CO2.
Infection
Confluent monolayers (80–90%) of CEF cells were infected with H9N2 virus at varying multiplicity of infection (MOI) (1, 0.1, 0.01, and 0.001) with supplemental tolylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma-Aldrich, St. Louis, MO, USA) at 0.05 µg/mL, in the absence of FBS. After 1 h at 37 °C and 5% CO2, the inoculum was removed and cell medium supplemented with antibiotics (50 µg/mL gentamicin, 1 mM glutamine) and 0.05 µg/mL TPCK-trypsin, in the absence of FBS, containing 2% low melting point SeaPlaque™ agarose (Lonza, Basel, Switzerland) replaced the infecting medium. The plates were subsequently incubated upside down at 37 °C in 5% CO2 for 3 days.
Virus isolation
Virus plaque was formed when a virus particle infected the host cell and inducing cytopathic effects in this and the surrounding cells. Virus plaque was subsequently collected as previously described [32]. Each plaque represented a single pure H9N2 clone. Purified virus was subsequently amplified in SPF embryonated chicken eggs. Egg fluids were collected and titrated (10−4 to 10−9 dilution) before being aliquoted and stored at −80 °C.
The similar process of plaque purification was performed on MDCK cell culture at Instituto Zooprofilattico Sperimentale delle Venezie, Italy. Two virus clones were purified from two different cell cultures and named CEF-clone and MDCK-clone.
Experimental design
In vivo experiments were performed in SPF White Leghorn chickens (Lohmann Valo, Germany) under biosecurity level 3 (BSL-3) conditions. The chickens had access to feed and water ad libitum throughout the experiments. Animal experiments were conducted with the authorization and supervision of the Biosafety and Bioethics Committees at Sciensano-Avian Virology and Immunology unit (Brussels, Belgium) according to National and European regulations.
In the first experiment, 4 week-old chickens were randomly divided into two groups, one group (n = 43) was infected with the H9N2 CEF-clone via the oculo-nasal route (50 µL intranasal and 50 µL as an eye-drop) at a viral dose of 106 EID50 in 100 µL phosphate-buffered saline (PBS) and the other group (n = 15) was left untreated and kept separately as non-infected negative controls.
In the second experiment, 4 week-old chickens were randomly divided into two groups, one group (n = 37) was infected with the H9N2 MDCK-clone as described above and the other group (n = 15) was left untreated and kept separately as non-infected negative controls.
In both experiments, the infected chickens were monitored daily for clinical signs (including conjunctivitis, excessive lacrimation and ruffled feathers) and mortality. The degree of severity of clinical signs was scored as follows: 0 = no clinical signs, 1 = mild to moderate clinical signs, 2 = moderate to severe clinical signs, 3 = dead. Daily clinical score was the average clinical score of the remaining chickens which was calculated from the sum of the individual clinical scores from remaining chickens divided by the number of animals. At 3 and 5 days post-infection (dpi), spleens from infected and non-infected chickens (n = 5) were collected for splenocyte isolation and ex vivo mitogenic activation (as described below).
Swabs (tracheal and cloacal) were collected from the infected chickens at 2, 3, 5, 7, 10, 15, and 21 dpi (n = 5 in Experiment 1; n = 3 in Experiment 2) and then were stored in brain-heart infusion (BHI) broth supplemented with antibiotics (107 U/L penicillin, 2 g/L streptomycin, 1 g/L gentamicin, 0.65 g/L kanamycin) at −80 °C until further analysed.
Tissues (brain, trachea, lung, liver, duodenum, spleen, kidney, and bursa of Fabricius) were resected from infected chickens at 1, 3, 5, 7, 10, 15, 21 dpi and non-infected chickens at 1, 3, 5 dpi. The collected tissues were stored in RNAlater® solution (Ambion, Applied Biosystems, Carlsbad, CA, USA) and quickly frozen at −80 °C until subjected to RNA extraction for studies of viral presence (n = 3) and cytokine expression (n = 5).
Blood was collected immediately after the sacrifice of the birds at the end of the experimental period (21 dpi) (n = 5 in Experiment 1; n = 3 in Experiment 2). Sera were separated by centrifugation at 5000 rpm for about 10 min and were kept at −20 °C until used for the study of seroconversion by hemagglutination inhibition (HI) test.
RNA extraction and cDNA synthesis
RNA extraction from swabs and tissues (approximately 30 mg) was performed as previously described [33] by using the MagMax AI/ND 96 Viral RNA Isolation kit and the MagMAX-96 Total RNA Isolation kit (Ambion, Applied Biosystems, Carlsbad, CA, USA), respectively, following the manufacturer’s instructions. The purified RNA (100 ng) from the trachea, duodenum and spleen were then reverse-transcribed to cDNA with the GoScript™ Reverse Transcription System (Promega, Madison, WI, USA) for the study of cytokine expression. The cDNA products were stored at −20 °C until they were further analysed.
Measurement of virus shedding via swabs and tissues
Quantitative real-time reverse transcription (RRT)-PCR targeting of the type A influenza matrix (M) gene was performed using the AgPath-ID One-Step RT-PCR Kit (Ambion, Applied Biosystems, Carlsbad, CA, USA). Primers specific for universal M For/Rev and probe used were previously described by Spackman et al. [34] with a final concentration of 900 nM of each primer (M + 25F and M − 124R) and 400 nM of the TAQMAN®-probe in a total reaction volume of 25 µL, containing 2 µL of purified RNA. Amplification and fluorescence detection were performed on LightCycler®480 Real-Time PCR system (Roche, Mannheim, Germany) with cycling condition: 50 °C for 30 min and 95 °C for 15 min, followed by 40 cycles of 95 °C 15 s–54 °C 35 s–72 °C 10 s. A crossing-point (Cp) value of 40 was chosen as cut-off and samples with higher Cp were considered as negative. Absolute quantification was done relative to a standard curve based on tenfold dilutions of an in vitro transcribed RNA template. Data are expressed as the number of viral RNA copies (log10) per mL for swabs and per 30 mg for tissues. The detection limit was 2log10 RNA copies/mL swabs and 2.5log10 RNA copies/30 mg tissue.
Hemagglutinin inhibition (HI) test
HI tests were performed according to the Manual of Diagnostic Tests and Vaccines for Terrestrial Animals [35]. The homologous virus was used as antigen in the HI test. The geometric mean of HI titers was expressed as reciprocal log2.
Measurements of immunosuppression
Splenocytes (n = 5) were isolated and then either cultured in RPMI 1640 medium (Invitrogen) in the presence or absence of pokeweed mitogen (PWM) (Sigma-Aldrich, St. Louis, MO, USA), as previously described [36]. PWM is a stimulator of chicken T- and B-cell proliferation and was showed to induce IFN-γ production by splenocytes [36]. T-cell stimulation was measured in supernatants after 48 h at 39 °C using ChIFN-γ-capture specific enzyme-linked immunosorbent assays (ELISAs), as previously described [36]. Cellular responses were expressed as optical density (OD) values. Negative chickens with an OD value < 0.1 for mitogen activation were excluded from further antigen activation analysis. Supernatant of mitogenic activated splenocytes from non-infected chickens as an in-house positive control were used in each plate.
Measurement of innate immune response-related gene expression
Expression of innate immune response-related genes was obtained by using the Power SYBR® Green RNA-to-CT™ 1-Step kit (Applied Biosystems, Carlsbad, CA, USA) with primers specific for target genes (Table 1) on the LightCycler®480 Real-Time PCR system (Roche, Mannheim, Germany), as previously described [37].
Data were normalized to levels of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and then were quantified according to the 2−∆∆Ct method [38]. Changes in gene expression that were detected in the CEF-infected group and in the MDCK-infected group are presented as fold-increases relative to the levels detected in the non-infected group. Samples in which target genes were not detected were excluded from these calculations.
Whole virus genome Sanger sequencing
RNA was extracted with a HIGH PURE® Viral Nucleic Acid kit (Roche, Mannheim, Germany), according to the manufacturer’s instructions. Reverse transcription (RT) was performed using the Uni12 primer and SuperScript III™ reverse transcriptase (Invitrogen) following the manufacturer’s instructions. RT-PCR was conducted using AccuPrime Taq DNA Polymerase, High Fidelity (Thermo Fisher Scientific, Waltham, MA, USA). All segments were amplified with Hoffman’s primers [20]. The PCR products were purified using the High Pure PCR Product Purification Kit (Roche, Mannheim, Germany) after separation on 1.5% agarose gel. The fragments were sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA), following the manufacturer’s instructions. Sequenced fragments were analysed and aligned by using DNAsis Max v 2.05 (Hitachi Software) and a consensus sequence was determined from all replicate sequences.
Statistical analysis
Analysis of data was performed with Minitab 13 (Minitab Ltd, Coventry, UK) and STATA 10 (Stata Corp LP, TX, USA) statistical programs for Windows 2000. Analyses of innate immune response-related gene expression data were performed with Student’s t-test. If normality and homogeneity of variance were not validated, then the non-parametric Wilcoxon-Mann–Whitney’s test was applied. Differences with a P-value ≤ 0.05 were considered statistically significant.
Results
Pathogenicity in SPF chickens
Clinical manifestations
Chickens infected with the CEF-clone exhibited no clinical signs of infection. In contrast, the chickens infected with the MDCK-clone exhibited obvious clinical signs which included conjunctivitis, excessive lacrimation and ruffled feathers. These signs were visible from 2 dpi, most remarkable at 4 dpi, and then reduced by 6 dpi (Figure 1). No mortality was recorded in any of the infected groups over the course of the experimental period.

Course of clinical signs for chickens infected with either CEF-H9N2 or MDCK-H9N2. Clinical signs were scored as: 0 = no sign, 1 = mild to moderate signs, 2 = moderate to severe signs, 3 = dead. Daily clinical score was calculated from the sum of the individual clinical scores from remaining chickens divided by the number of animals.
Viral dissemination
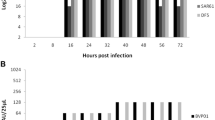
Excretion of the CEF-clone was detected in one out of five (1/5) tracheal swabs at 2 dpi (Figure 2). Excretion continued at 3 dpi (3/5) and lasted until 7 dpi (2/5). No viral RNA was detected in any of the cloacal swabs at any sampling point. In none of the examined tissues viral RNA copies of the CEF-clone were detected (data not shown). In contrast, the MDCK-clone exhibited enhanced replication in the chickens with excretions that were detected in both the tracheal and cloacal swabs. High number of viral RNA copies were first detected at 2 dpi (3/3) in the tracheal swabs in this MDCK group (Figure 2). The number of RNA copies in tracheal swabs subsequently decreased although replication of the MDCK-clone was still detected at the end of the experimental period (21 dpi). When compared to the CEF-clone, the MDCK-clone induced significantly higher excretion in tracheal swabs at 2 and 5 dpi. Viral excretion of the MDCK-clone via the cloacal route was also detected at 2 dpi (3/3), at 5 dpi (3/3), and 7 dpi (3/3) (Figure 2). However, from 10 dpi no more viral RNA was detected in cloacal swabs. In addition, the viral RNA was detected in various examined organs from the chickens that were inoculated with the MDCK-clone (Figure 3): in brain, trachea, lung, duodenum, kidney, and bursa of Fabricius tissues, viral RNA copies were detected at 2 dpi, 5 dpi (3/3) and 7 dpi (3/3) while in liver and spleen the viral RNA was detected as early as 1 dpi, 3 dpi and 5 dpi. The viral RNA was undetectable in these organs at 10 dpi.

Viral RNA copy number in swabs collected from chickens infected with either CEF-H9N2 or MDCK-H9N2. Chickens were infected with two different H9N2 clones and viral excretion was detected in swabs collected at 2, 3, 5, 7, 10, 15, and 21 dpi. The data were expressed as mean ± standard deviation. The limit of detection is 2log10 copies/mL. An asterisk indicates statistically significant differences between the two clones (P < 0.05).

Viral RNA copy number in various tissues of chickens infected with MDCK-H9N2. Chickens were infected with MDCK-H9N2 clone and viral excretion in various tissues was subsequently examined at 1, 3, 5, 7, 10, 15, and 21 dpi. The data were expressed as mean ± standard deviation. The limit of detection is 2.5log10 copies/30 mg.
Serology
At 21 dpi, the mean log2 antibody titers for the CEF- and MDCK-clone-infected groups were 7.2 ± 0.84 (5/5) and 10 ± 0 (3/3), respectively (data not shown).
Immunosuppression measured by ex vivo mitogenic activation of splenocytes
In the present study, the immunosuppressive effect of the two H9N2 clones was evaluated based on the ability of splenocytes to produce ChIFNγ following ex vivo mitogenic stimulation. PWM was used to activate splenocytes obtained from infected and non-infected chickens at 3 dpi and 5 dpi. ChIFN-γ production measured by ELISA by splenocytes obtained from both CEF- and MDCK-clone infected chickens was downregulated at 3 dpi but only significantly lower from negative group (P < 0.05) for MDCK-clone infected chickens (Table 2). At 5 dpi, mitogenic activation was fully restored in CEF-clone infected chickens, while a downregulation was still observed in the MDCK-clone infected birds, although not statistically significant (Table 2).
Innate immune response
Expression of toll-like receptor (TLR) 7 and 3
The expression of TLR-7 and TLR-3 was induced in trachea and duodenum by both clones. The chickens infected with CEF-clone expressed significantly higher levels of TLR-7 mRNA in the trachea and duodenum at 3 dpi and 5 dpi (Figure 4A) and higher levels of TLR-3 mRNA in the duodenum at 1 dpi and 3 dpi (Figure 4B). In the spleen, no changes in TLR-7 or TLR-3 expression were measured in either of the infected groups.

Innate immune response-related gene expression profiles in chickens infected with either CEF-H9N2 or MDCK-H9N2. At 1, 3, and 5 dpi, trachea, duodenum, and spleen tissues were resected from chickens infected with CEF-H9N2 or MDCK-H9N2 and mRNA levels of TLR-7 (A), TLR-3 (B), IFN-α (C), IFN-β (D), IFN-γ (E), IL-6 (F), and IL-1β (G) were detected in each by RRT-PCR. Fold-changes in gene expression were calculated relative to the corresponding levels detected in non-challenged chickens. Data were normalized to GAPDH expression, calculated according to the 2−∆∆Ct method [38], and expressed as mean ± standard deviation. An asterisk indicates statistically significant differences between the two clones (P < 0.05).
Expression of antiviral cytokines
In the present study, IFN-α mRNA was induced rapidly after infection and increased in the trachea and duodenum at 1 dpi and 3 dpi in the chickens infected by the CEF-clone. In contrast, the MDCK-clone induced slightly elevated levels of IFN-α mRNA expression in the trachea alone. The difference in IFN-α mRNA expression was statistically significant in the trachea at 1 dpi and in the duodenum at 3 dpi (Figure 4C). Similarly, the level of IFN-β mRNA was comparatively up-regulated in the chickens infected with CEF-clone in the trachea and duodenum (Figure 4D). However, the difference in the expression levels of IFN-β was only significant in the duodenum at all of the tested time points. In contrast to type I IFNs, significantly higher levels of IFN-γ mRNA was induced by MDCK-clone in the trachea at 3 dpi and in the spleen at 1 dpi and 3 dpi (Figure 4E).
Expression of pro-inflammatory cytokines
IL-6 mRNA was induced to a greater extent by the MDCK-clone in the trachea and duodenum at all of the tested time points, while a significant difference was detected in the trachea at 5 dpi (Figure 4F). Similarly, IL-1β mRNA was induced to a significantly greater level by the MDCK-clone compared to the CEF-clone in all of the tissues examined at 3 dpi and 5 dpi (Figure 4G).
Genome sequencing
A total of four amino acid substitutions were detected in the full genome consensus sequence between the CEF- and MDCK-clone. Three of these substitutions involved the HA gene: E198A, R234L, and E502D in the CEF-compared to the MDCK-clone. In the NA sequence, a single substitution was detected at residue 33, with a valine to methionine shift between the CEF- and MDCK-clone (V33M).
Discussion
In this study, two clones from a single G1-H9N2 field isolate, obtained via plaque purification on two different cell cultures, were evaluated for their difference in pathogenicity. These two purified clones, the CEF and MDCK-clones demonstrated significant differences in pathogenicity in SPF chickens, allowing studying the underlying mechanism(s) mediating the H9N2 virulence in chicken. Under experimental conditions, the CEF-clone was avirulent in SPF chicken with a local respiratory replication detected in tracheal swabs. In contrast, the MDCK-clone exhibited systemic replication which has been described for other H9N2 strains, such as A/Chicken/Saudi Arabia/SP02525/3AAV/2000 and A/Chicken/HS/K5/01 (H9N2) with viral RNA detection in multiple tissues including brain, trachea, lung, ileum, liver, kidney and spleen [39, 40]. However, the information on the potential transmissibility of two H9N2 clones was lacking as sentinel animals were not included in this study. In addition, the work presented here demonstrated that the MDCK-clone could play a role as a stronger immunosuppressive agent. Indeed, in splenocytes from chickens infected with MDCK-clone, an absence of IFN-γ production after ex vivo mitogenic activation confirmed the presence of unresponsive T lymphocytes. Influenza A viruses can cause immunosuppression in chickens by lysing or functionally impairing lymphocytes [41]. Some H9N2 strains have been demonstrated to be lymphotropic and cause severe immunosuppression in chickens by this mechanism [25]. Here, a causal link between the systemic replication of MDCK-clone and its immunosuppressive effect is suggested. However, further investigations are needed to confirm the direct effect of MDCK-clone on lymphocytes by a set of in vitro experiments and flow cytometry.
As pathogenesis and immunosuppression do not only rely on the replicative ability of influenza viruses alone but also on the host’s immune response, early immune responses following either H9N2 clone infection were characterized. Host innate immunity mediates the first line of defense towards influenza infection and TLR-7 and TLR-3 are the main sensors for influenza viruses, responsible for triggering the host’s innate immune responses in chickens [42]. We observed that an avirulent CEF-clone triggered the expression of these two TLRs, whereas the MDCK-clone did not mediate a TLR-7 and TLR-3 response. Interestingly, these results are similar with previous findings that levels of TLR-7 and TLR-3 mRNA are barely up-regulated in response to highly pathogenic H7 infections, while rapid upregulation occurs following a lower pathogenic H7 infection [43]. After influenza virus components being sensed, type I IFNs are produced by different innate immune cells and paramount in the defense against influenza infection with evidence that a greater and longer response of IFN-α was shown to be a crucial factor in host’s strategy against avian influenza [44]. In the present study, the elevated response of type I IFNs in chickens infected with CEF-clone may reflect the effort of the host’s immune system to achieve viral clearance, conversely, the absence of type I IFNs following the infection with MDCK-clone could be associated with the presence of viral mRNA in different organs and prolonged viral shedding in the trachea. The latter case may be a reflection of NS1 interference—a known IFN antagonist [45]. Interestingly, F103L mutation which is related to increased interferon antagonism for avian influenza virus [46] was found in NS1 segment of both clones. The deficient expression of type I IFN following MDCK-clone infection coincided with a high viral RNA load, speculating that the assumed high levels of NS1 viral protein could explain this deficiency by the immunomodulatory capacity described for this protein. The principle of NS1 antagonism in type I IFN induction cascade includes (i) sequestering dsRNA away from host sensors (ii) targeting TRIM25 to inhibit RIG-I activation (iii) interfering the processing and nuclear export of cellular mRNAs, thus limiting type I IFNs expression post-transcriptionally [47]. In addition, the CEF- and MDCK-clone also strikingly differed in Il-6 and Il-1β inductions. These pro-inflammatory cytokines are considered to be the main mediators of inflammation and their expression levels directly correlate with viral replication and the respiratory and systemic symptoms of influenza disease [48]. The expression levels of pro-inflammatory cytokines have been found to be higher in chickens infected with HPAI compared to LPAI [49]. In the present study, the intense expression of these pro-inflammatory cytokine genes by MDCK-clone in the trachea and spleen suggests that its tropism is associated with the upper respiratory tract and lymphoid organs. In contrast, the locally deficient inflammatory response by the CEF-clone may be due to its replicative inability in these tissues (trachea, duodenum, and spleen).
Another dominant difference in response to CEF-clone and MDCK-clone is the expression of IFN-γ. In response to MDCK-clone infection, IFN-γ was significantly elevated in trachea and spleen. Remarkably, the immunosuppression observed on the corresponding splenocytes positively correlated with induction of IFN-γ in the spleen in vivo, suggesting the pivotal role of IFN-γ on the higher and longer immunosuppressive effect induced by MDCK-clone. This view is supported by the previous insights regarding infectious bursal disease (IBDV) infection [50]. The immunosuppressive effect of H9N2 could elucidate its exacerbating role in numerous co-infection cases noticed in the field [51,52,53] and under experimental conditions [27]. However, the duration and severity of immunosuppression depend on infection dose and age and breed of infected chickens [54, 55].
These data also emphasize that host cells from which the purified clones were obtained can impact their viral pathogenesis. Both CEF and MDCK are widely used cell-cultures as a substrate for the propagation of influenza A viruses [56]. As the AIVs belong to a RNA virus family, they have been shown to have a stable complex distribution of mutant genomes in a population due to error-prone replication, described as quasispecies [57]. The heterogeneity in an influenza virus would persist in one host where variants have equal growth potential but would also be destroyed when introduced into a new host. As a result, when the stock virus is seeded in different cell cultures, different subpopulations may be selected depending on the best host-adaptation condition. The generation of distinct H9N2 clones after a single round of purification on different cell cultures suggests the existence and selection of virus quasispecies with variant virulence in the original H9N2 stock. However, as no next generation sequencing was performed, the proportion of the two cloned variants as quasispecies populations in the original virus stock could not be determined. It should also be pointed out that the presence as quasispecies in the original isolate is not the only explanation for the outcome of two distinct clones from a single field isolate. Also, post-translational modifications, depending on the cell-culture systems used for viral purification, may affect the virulence of the cloned viruses. It was reported that the cells in which the virus was grown, can affect the binding properties of the influenza A hemagglutinin by adding differently structured oligosaccharides [58,59,60]. Receptor distribution analysis demonstrated a high prevalence of SA 2,3-Gal linkages in CEF cells that validates the high replication of avian influenza viruses. Whereas, MDCK cells express a high amount of both SA α2,6-Gal and α 2,3-Gal linkages on their surface supporting the growth of a broader range of influenza A viruses [61]. In this context, the unique ability of some H9N2 viruses to bind both SA α2,6-Gal and α 2,3-Gal linkages [62, 63] is of importance to explain the multi-tropism of the MDCK-clone G1-H9N2. These hypotheses would merit more in-depth investigations, as the propagation in kidney cell cultures resulted in increased virulence of the H9N2 corresponding with the observation that the infections with some LPAI were nephrotropic during the simulated systemic infection of chickens [64,65,66]. The biological properties of two H9N2 clones should be explored through the analysis of the plaque morphology, growth kinetics, and receptor binding profiles. Finally, molecular changes such as amino acid substitutions are responsible for phenotypical changes such as virulence-shifts. Strikingly, comparison of the genomic information of the two selected clones revealed only four substitutions, including the residues 198, 234 (equivalent to residue 226 in H3 numbering [8]) and 502 of the HA protein, and residue 33 of the NA protein. All, a combination of some or just one of these substitutions might be responsible for the detected different phenotype in chickens. Residues 198 and 234 are located in the receptor-binding pocket of HA and directly involved in the binding of SA receptors. Any changes here would be predicted to affect linkage specificity and strength. However, no virulence markers have previously been associated with these positions in H9N2. The two other residues in HA and NA are not associated with any functional regions of these glycoproteins and the detected mutations HA-D502E and NA-V33 M have so far not been described as virulence marker.
To our knowledge, this study is the first description of how the purification of an H9N2 isolate on different cell cultures results in the generation of two clones with distinct pathogenic characteristics, induction of different immune profiles and distinct immunosuppression profiles in SPF chickens. Overall, these results suggest that deficient TLR-7 and type I IFNs expression mediates the pathogenicity profile of H9N2 in chickens. In addition, only four mutations that differentiate the two clones. Further research is required to investigate the function of genetic mutations responsible for the major phenotypical differences observed between these two clones by using reverse genetics approach. A full-length cDNA copy of genome from avirulent H9N2 (CEF-clone) could be used as a backbone in which each mutation or a combination of mutations would be introduced and the phenotype of recombinant viruses could be investigated. Furthermore, the quasispecies of the H9N2 original isolate and clones will need to be determined by NGS approaches. Finally, any post-translational modifications that might have occurred will be investigated.
Abbreviations
- AIV:
-
avian influenza virus
- BHI:
-
brain–heart infusion
- BSL-3:
-
biosecurity level 3
- CEF:
-
chicken embryo fibroblast
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- (c)DNA:
-
(complementary)deoxyribonucleic acid
- EID50 :
-
50% egg infectious dose
- dpi:
-
days post-infection
- ELISA:
-
enzyme-linked immunosorbent assay
- FBS:
-
fetal bovine serum
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- HA:
-
hemagglutinin
- HI:
-
hemagglutinin inhibition
- HPAI:
-
highly pathogenic avian influenza
- IBDV:
-
infectious bursal disease virus
- (ch)IFN:
-
(chicken)interferon
- IL:
-
interleukin
- LPAI:
-
low pathogenic avian influenza
- MDCK:
-
Madin–Darby canine kidney
- NA:
-
neuraminidase
- (v)NDV:
-
(virulent)Newcastle disease virus
- NGS:
-
new generation sequencing
- OD:
-
optical density
- PBS:
-
phosphate-buffered saline
- PWM:
-
pokeweed mitogen
- (m)RNA:
-
(messenger)ribonucleic acid
- RRT-PCR:
-
real-time reverse transcription polymerase chain reaction
- SA:
-
sialic acid
- SPF:
-
specific-pathogen-free
- TPCK:
-
tolylsulfonyl phenylalanyl chloromethyl ketone
- TLR:
-
toll-like receptor
References
Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO (2013) New world bats harbor diverse influenza A viruses. PLoS Pathog 9:e1003657
Stallknecht DE, Shane SM (1988) Host range of avian influenza virus in free-living birds. Vet Res Commun 12:125–141
Slemons RD, Easterday BC (1976) The natural history of type-A influenza viruses and wild waterfowl. Wildlife diseases. Springer, Boston, pp 215–224
Vahlenkamp TW, Harder TC (2006) Influenza virus infections in mammals. Berl Munch Tierarztl Wochenschr 119:123–131
Senne DA, Panigrahy B, Kawaoka Y, Pearson JE, Suss J, Lipkind M, Kida H, Webster RG (1996) Survey of the hemagglutinin (HA) cleavage site sequence of H5 and H7 avian influenza viruses: amino acid sequence at the HA cleavage site as a marker of pathogenicity potential. Avian Dis 40:425–437
Homme PJ, Easterday BC (1970) Avian influenza virus infections. I. Characteristics of influenza A-Turkey-Wisconsin-1966 virus. Avian Dis 14:66–74
Jiang W, Liu S, Hou G, Li J, Zhuang Q, Wang S, Zhang P, Chen J (2012) Chinese and global distribution of H9 subtype avian influenza viruses. PLoS One 7:e52671
Butt AM, Siddique S, Idrees M, Tong Y (2010) Avian influenza A (H9N2): computational molecular analysis and phylogenetic characterization of viral surface proteins isolated between 1997 and 2009 from human population. Virol J 7:319
Nagy A, Mettenleiter TC, Abdelwhab EM (2017) A brief summary of the epidemiology and genetic relatedness of avian influenza H9N2 virus in birds and mammals in the Middle East and North Africa. Epidemiol Infect 145:3320–3333
Chen BL, Zhang Z, Chen WB (1994) The study of avian influenza: I. The isolation and preliminary serological identification of avian influenza virus in chicken. Chin J Vet Med 20:3–5
Nili H, Asasi K (2002) Natural cases and an experimental study of H9N2 avian influenza in commercial broiler chickens of Iran. Avian Pathol 31:247–252
Lee YJ, Shin JY, Song MS, Lee YM, Choi JG, Lee EK, Jeong OM, Sung HW, Kim JH, Kwon YK, Kwon JH, Kim CJ, Webby RJ, Webster RG, Choi YK (2007) Continuing evolution of H9 influenza viruses in Korean poultry. Virology 359:313–323
Gharaibeh S (2008) Pathogenicity of an avian influenza virus serotype H9N2 in chickens. Avian Dis 52:106–110
Bijanzad P, Momayez R, Bozorgmehri Fard MH, Hablolvarid MH, Mahmoodzadeh M, Jeyrani Moghaddam AR, Kaboli K, Azizpour A, Eshratabadi F (2013) Study on clinical aspects of SPF chickens infected with H9N2 subtype of avian influenza virus. Ann Biol Res 4:81–85
Kwon HJ, Cho SH, Kim MC, Ahn YJ, Kim SJ (2006) Molecular epizootiology of recurrent low pathogenic avian influenza by H9N2 subtype virus in Korea. Avian Pathol 35:309–315
Mosleh N, Dadras H, Mohammadi A (2009) Molecular quantitation of H9N2 avian influenza virus in various organs of broiler chickens using TaqMan real time PCR. J Mol Genet Med 3:152–157
Vasfi MM, Pazani J, Helan A, Houssein S, Ghods F (2008) Evaluation of the pathogenicity of A/Chicken/Iran/ZMT-173/99(H9N2) strain of avian influenza virus in serologically Mycoplasma galisepticum positive and negative broiler chickens. Ir J Virol 2:6–13
Perdue ML (2009) Molecular determinants of pathogenicity for avian influenza viruses. In: Swayne DE (ed) avian influenza. Blackwell Publishing, Ames, pp 23–41
Samy A, Naguib MM (2018) Avian respiratory coinfection and impact on avian influenza pathogenicity in domestic poultry: field and experimental findings. Vet Sci 5:E23
Davidson I, Fusaro A, Heidari A, Monne I, Cattoli G (2014) Molecular evolution of H9N2 avian influenza viruses in Israel. Virus Genes 48:457–463
Pantin-Jackwood MJ, Costa-Hurtado M, Miller PJ, Afonso CL, Spackman E, Kapczynski DR, Shepherd E, Smith D, Swayne DE (2015) Experimental co-infections of domestic ducks with a virulent Newcastle disease virus and low or highly pathogenic avian influenza viruses. Vet Microbiol 177:7–17
Banet-Noach C, Perk S, Simanov L, Grebenyuk N, Rozenblut E, Pokamunski S, Pirak M, Tendler Y, Panshin A (2007) H9N2 influenza viruses from Israeli poultry: a five-year outbreak. Avian Dis 51(1 Suppl):290–296
Ellakany HF, Gado AR, Elbestawy AR, Abd El-Hamid HS, Hafez HM, Abd El-Hack ME, Swelum AA, Al-Owaimer A, Saadeldin IM (2018) Interaction between avian influenza subtype H9N2 and Newcastle disease virus vaccine strain (LaSota) in chickens. BMC Vet Res 14:358
Fazel P, Mehrabanpour M (2018) Evaluation of the viral interference between lentogenic Newcastle disease virus (Lasota) and avian influenza virus (H9N2) using real-time reverse transcription polymerase chain reaction in SPF chicken. Braz J Poult Sci 20:537–546
Qiang F, Youxiang D (2011) The effects of H9N2 influenza A on the immune system of broiler chickens in the Shandong Province. Transbound Emerg Dis 58:145–151
Allawe A (2008) Study of effect of immunosuppression caused by H9N2 in broiler. Iraqi J Vet Sci 32:80–89
Bonfante F, Cattoli G, Leardini S, Salomoni A, Mazzetto E, Davidson I, Haddas R, Terregino C (2017) Synergy or interference of a H9N2 avian influenza virus with a velogenic Newcastle disease virus in chickens is dose dependent. Avian Pathol 46:488–496
Xing Z, Cardona CJ, Li J, Dao N, Tran T, Andrada J (2008) Modulation of the immune responses in chickens by low-pathogenicity avian influenza virus H9N2. J Gen Virol 89:1288–1299
Brauer R, Chen P (2015) Influenza virus propagation in embryonated chicken eggs. J Vis Exp 97:e52421
Reed LJ, Muench H (1938) A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497
Hernandez R, Brown DT (2010) Growth and maintenance of chick embryo fibroblasts (CEF). Curr Protoc Microbiol Appendix 4:4I
Xue J, Chambers BS, Hensley SE, Lopez CB (2016) Propagation and characterization of influenza virus stocks that lack high levels of defective viral genomes and hemagglutinin mutations. Front Microbiol 7:326
Rauw F, Gardin Y, Palya V, van Borm S, Gonze M, Lemaire S, van den Berg T, Lambrecht B (2009) Humoral, cell-mediated and mucosal immunity induced by oculo-nasal vaccination of one-day-old SPF and conventional layer chicks with two different live Newcastle disease vaccines. Vaccine 27:3631–3642
Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL (2002) Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol 40:3256–3260
OIE (2008) Manual of diagnostic tests and vaccines for terrestrial animals. Office International Des Epizooties, Paris
Lambrecht B, Gonze M, Meulemans G, van den Berg TP (2004) Assessment of the cell-mediated immune response in chickens by detection of chicken interferon-gamma in response to mitogen and recall Newcastle disease viral antigen stimulation. Avian Pathol 33:343–350
Ingrao F, Rauw F, Steensels M, van den Berg T, Lambrecht B (2018) Early immune responses and profiling of cell-mediated immunity-associated gene expression in response to rHVT-IBD vaccination. Vaccine 36:615–623
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Post J, de Geus ED, Vervelde L, Cornelissen JB, Rebel JM (2013) Systemic distribution of different low pathogenic avian influenza (LPAI) viruses in chicken. Virol J 10:23
Kwon JS, Lee HJ, Lee DH, Lee YJ, Mo IP, Nahm SS, Kim MJ, Lee JB, Park SY, Choi IS, Song CS (2008) Immune responses and pathogenesis in immunocompromised chickens in response to infection with the H9N2 low pathogenic avian influenza virus. Virus Res 133:187–194
Rouse BT, Horohov DW (1986) Immunosuppression in viral infections. Rev Infect Dis 8:850–873
Sareneva T, Matikainen S, Kurimoto M, Julkunen I (1998) Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J Immunol 160:6032–6038
Cornelissen JB, Vervelde L, Post J, Rebel JM (2013) Differences in highly pathogenic avian influenza viral pathogenesis and associated early inflammatory response in chickens and ducks. Avian Pathol 42:347–364
Cauthen AN, Swayne DE, Sekellick MJ, Marcus PI, Suarez DL (2007) Amelioration of influenza virus pathogenesis in chickens attributed to the enhanced interferon-inducing capacity of a virus with a truncated NS1 gene. J Virol 81:1838–1847
Thepmalee C, Sanguansermsri P, Suwanankhon N, Chamnanpood C, Chamnanpood P, Pongcharoen S, Niumsap PR, Surangkul D, Sanguansermsri D (2013) Changes in the NS1 gene of avian influenza viruses isolated in Thailand affect expression of type I interferon in primary chicken embryonic fibroblast cells. Indian J Virol 24:365–372
Dankar SK, Miranda E, Forbes NE, Pelchat M, Tavassoli A, Selman M, Ping J, Jia J, Brown EG (2013) Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol J 10:243
Hale BG, Randall RE, Ortin J, Jackson D (2008) The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89:2359–2376
Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG (2001) Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol 64:262–268
Adams SC, Xing Z, Li J, Cardona CJ (2009) Immune-related gene expression in response to H11N9 low pathogenic avian influenza virus infection in chicken and Pekin duck peripheral blood mononuclear cells. Mol Immunol 46:1744–1749
Rauw F, Lambrecht B, van den Berg T (2007) Pivotal role of ChIFNgamma in the pathogenesis and immunosuppression of infectious bursal disease. Avian Pathol 36:367–374
Pan Q, Liu A, Zhang F, Ling Y, Ou C, Hou N, He C (2012) Co-infection of broilers with Ornithobacterium rhinotracheale and H9N2 avian influenza virus. BMC Vet Res 8:104
Chu J, Zhang Q, Zhang T, Han E, Zhao P, Khan A, He C, Wu Y (2016) Chlamydia psittaci infection increases mortality of avian influenza virus H9N2 by suppressing host immune response. Sci Rep 6:29421
Motamed N, Mayahi M, Seifi MR, Jafari RA (2013) Effect of infectious bursal disease virus on pathogenicity of avian influenza virus subtype H9N2 in broiler chicks. J Vet Med Anim Health 5:276–280
Spencer JL (1984) Progress towards eradication of lymphoid leukosis viruses—a review. Avian Pathol 13:599–619
Wilkins BS, Davis Z, Lucas SB, Delsol G, Jones DB (2003) Splenic marginal zone atrophy and progressive CD8+ T-cell lymphocytosis in HIV infection: a study of adult post-mortem spleens from Côte d’Ivoire. Histopathology 42:173–185
WHO (2007) Recommendations and laboratory procedures for detection of avian influenza A (H5N1) virus in specimens from suspected human cases. https://www.who.int/influenza/resources/documents/RecAIlabtestsAug07.pdf?ua=1
Domingo E, Menendez-Arias L, Quinones-Mateu ME, Holguin A, Gutierrez-Rivas M, Martinez MA, Quer J, Novella IS, Holland JJ (1997) Viral quasispecies and the problem of vaccine-escape and drug-resistant mutants. Prog Drug Res 48:99–128
Crecelius DM, Deom CM, Schulze IT (1984) Biological properties of a hemagglutinin mutant of influenza virus selected by host cells. Virology 139:164–177
Nakamura K, Compans RW (1979) Host cell- and virus strain-dependent differences in oligosaccharides of hemagglutinin glycoproteins of influenza A viruses. Virology 95:8–23
Deom CM, Caton AJ, Schulze IT (1986) Host cell-mediated selection of a mutant influenza A virus that has lost a complex oligosaccharide from the tip of the hemagglutinin. Proc Natl Acad Sci U S A 83:3771–3775
Seo SH, Goloubeva O, Webby R, Webster RG (2001) Characterization of a porcine lung epithelial cell line suitable for influenza virus studies. J Virol 75:9517–9525
Butt KM, Smith GJ, Chen H, Zhang LJ, Leung YH, Xu KM, Lim W, Webster RG, Yuen KY, Peiris JS, Guan Y (2005) Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J Clin Microbiol 43:5760–5767
Matrosovich MN, Krauss S, Webster RG (2001) H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Virology 281:156–162
Slemons RD, Swayne DE (1990) Replication of a waterfowl-origin influenza virus in the kidney and intestine of chickens. Avian Dis 34:277–284
Slemons RD, Swayne DE (1992) Nephrotropic properties demonstrated by A/chicken/Alabama/75 (H4N8) following intravenous challenge of chickens. Avian Dis 36:926–931
Swayne DE, Alexander DJ (1994) Confirmation of nephrotropism and nephropathogenicity of three low-pathogenic chicken-origin influenza viruses for chickens. Avian Pathol 23:345–352
Nang NT, Lee JS, Song BM, Kang YM, Kim HS, Seo SH (2011) Induction of inflammatory cytokines and Toll-like receptors in chickens infected with avian H9N2 influenza virus. Vet Res 42:64
Hangalapura BN, Kaiser MG, Poel JJ, Parmentier HK, Lamont SJ (2006) Cold stress equally enhances in vivo pro-inflammatory cytokine gene expression in chicken lines divergently selected for antibody responses. Dev Comp Immunol 30:503–511
Cornax I, Miller PJ, Afonso CL (2012) Characterization of live LaSota vaccine strain-induced protection in chickens upon early challenge with a virulent Newcastle disease virus of heterologous genotype. Avian Dis 56:464–470
Wang J, Tang C, Wang Q, Li R, Chen Z, Han X, Xu X (2015) Apoptosis induction and release of inflammatory cytokines in the oviduct of egg-laying hens experimentally infected with H9N2 avian influenza virus. Vet Microbiol 177:302–314
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GTN participated in the design of the study, carried out laboratory activities and drafted the manuscript. ID and FB participated in the preparation of the viruses used in this study. FI participated in the cytokine expression data analysis. FR carried out the statistical analysis. FR, MS, BL provided consultation and participated in the revision of the manuscript. BL conceived the study, participated in the design of the study and coordinated all the activities of the project. All authors read and approved the final manuscript.
Acknowledgements
We acknowledge M. Boschmans for technical assistance and C. Delgrange and M. Vandenbroeck for the animal work they performed.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Animal experiments were conducted with the authorization and supervision of the Biosafety and Bioethics Committees at Sciensano-Avian Virology and Immunology unit (Brussels, Belgium) according to National and European regulations.
Funding
This work was supported by ANIHWA-NADIV Project and CODA/CERVA, Belgium.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nguyen, G.T., Rauw, F., Steensels, M. et al. Study of the underlying mechanisms and consequences of pathogenicity differences between two in vitro selected G1-H9N2 clones originating from a single isolate. Vet Res 50, 18 (2019). https://doi.org/10.1186/s13567-019-0635-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-019-0635-1