
Abstract
Viral diseases are among the main challenges in farming of Atlantic salmon (Salmo salar). The most prevalent viral diseases in Norwegian salmon aquaculture are heart and skeletal muscle inflammation (HSMI) caused by Piscine orthoreovirus (PRV), and pancreas disease (PD) caused by Salmonid alphavirus (SAV). Both PRV and SAV target heart and skeletal muscles, but SAV additionally targets exocrine pancreas. PRV and SAV are often present in the same locations and co-infections occur, but the effect of this crosstalk on disease development has not been investigated. In the present experiment, the effect of a primary PRV infection on subsequent SAV infection was studied. Atlantic salmon were infected with PRV by cohabitation, followed by addition of SAV shedder fish 4 or 10 weeks after the initial PRV infection. Histopathological evaluation, monitoring of viral RNA levels and host gene expression analysis were used to assess disease development. Significant reduction of SAV RNA levels and of PD specific histopathological changes were observed in the co-infected groups compared to fish infected by SAV only. A strong correlation was found between histopathological development and expression of disease related genes in heart. In conclusion, experimentally PRV infected salmon are less susceptible to secondary SAV infection and development of PD.
Similar content being viewed by others


Introduction
Virus infections are a continuous challenge in large-scale aquaculture of Atlantic salmon (Salmo salar). Environmental factors, high intensity production and infectious agents affect both welfare and production [1–3]. The two most prevalent viral diseases in Norwegian Atlantic salmon aquaculture are heart and skeletal muscle inflammation (HSMI) and pancreas disease (PD) [4]. Piscine orthoreovirus (PRV) is associated with HSMI, is ubiquitous in sea reared Atlantic salmon in Norway and often detected without any signs of disease [5, 6]. Pancreas disease is caused by Salmon pancreas disease virus, more commonly known as Salmonid alphavirus (SAV). The two viral diseases have overlapping geographic distributions [4, 7], both target heart and skeletal muscle and may co-infect Atlantic salmon [8–10].
PRV is a non-enveloped virus with a segmented, double stranded RNA genome, belonging to the genus Orthoreovirus in the family Reoviridae [5, 11]. Salmonid erythrocytes are major target cells for PRV and more than 50% of these cells may be infected in the peak phase of the infection [12]. In later stages of the infection, PRV infects myocytes of the heart and skeletal muscles [13]. The histopathological changes in heart and skeletal muscle gave the condition its name in the late 1990s, and later the association with PRV was established [5, 14].
SAV is an enveloped virus with a single-stranded positive sense RNA genome of the family Togaviridae [15]. Pancreas, heart and skeletal muscle are the main target tissues. The disease is recognized by growth retardation, reduced slaughter quality and increased mortality [9, 16, 17]. Histopathological changes are characterized by acute necrosis of exocrine pancreas, myocardial and skeletal muscle necrosis with subsequent inflammation [9]. The pancreatic lesions are a hallmark of PD and hence used for diagnostic differentiation from HSMI and cardiomyopathy syndrome (CMS) [9]. However, PRV and SAV have common target tissues in heart and skeletal muscles, making interactions between the two viral infections possible.
SAV is divided into six different phylogenetic subtypes [18] and subtypes 2 and 3 are present in Norwegian aquaculture [19, 20]. The two subtypes show approximately 7% differences in nucleotide sequence [19], are endemically present in separate geographic areas and differ in virulence [20–22]. The mechanisms behind the difference in virulence are unknown. No stereotypical difference is described between subtype 2 and 3 [23]. PD outbreaks vary in duration, severity and accumulated mortality [24], indicating that factors other than SAV influences disease development. Interaction with other infectious agents may be such a factor.
Protection to a secondary virus infection induced by an unrelated primary virus infection has been recognized since the 1950s [25], and has also been described for several viruses infecting salmonid fish [26–30]. However, the duration of the protection of rainbow trout to infectious hematopoietic necrosis after primary infection with the non-virulent cutthroat trout virus was found to be no more than 4 weeks [26]. In addition, some viral infections in terrestrial animals are shown to aggravate disease development of a secondary viral infection [31, 32].
The purpose of this study was to determine if a primary PRV infection alters the outcome of a subsequent SAV infection. Experimental infection trials were performed to compare disease development, viral kinetics and expression of disease associated genes between PRV-SAV co-infected and SAV infected fish.
Materials and methods
Fish
Sea water adapted Atlantic salmon (N = 987) of a SalmoBreed strain (Bergen, Norway) were used in the study (VESO Vikan, Namsos, Norway). The post smolts were transferred to sea water two weeks before PRV challenge. Prior to challenge, the fish were screened and found to be negative for PRV, infectious pancreatic necrosis virus (IPNV) and SAV by reverse transcriptase (RT) qPCR. PRV shedders (N = 5) sampled four weeks after PRV challenge were confirmed negative for Atlantic salmon calicivirus [33]. During the challenge trial the fish were kept in filtered and UV-radiated seawater (34 ‰ salinity), 12 °C (±1 °C) and on 24 h light. The fish were fed 1% of total biomass per day and starved for 24 h prior to handling and sampling. Before sampling, the fish were euthanized by bath immersion containing benzocaine chloride (1 g/5L water) (Apotekproduksjon AS, Oslo, Norway) for 5 min. The challenge trial was approved by the Norwegian Animal Research Authority and performed in accordance with the recommendations of the current animal welfare regulations: FOR-1996-01-15-23 (Norway).
Experimental challenge
PRV challenge
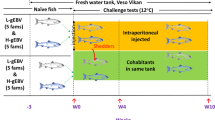
The inoculum (consisting of pelleted blood cells) was collected in a previous cohabitation trial (VESO Vikan), i.e. the second passage in experimental fish, and originated from a Norwegian field outbreak of HSMI in 2012. RTqPCR was performed as earlier described [34] and a high level of PRV RNA was indicated (Ct 17.3, using a total RNA input of 100 ng in the RTqPCR). The blood pellet was dissolved 1:1 in PBS and stored at −80 °C. On Day 0 of the trial, the blood pellet and PBS solution was thawed on ice, diluted 1:2 in PBS and 0.1 mL of the inoculum was i.p. injected into the anesthetized shedders. The inoculum was confirmed negative for IPNV, infectious salmon anemia virus (ISAV), SAV, piscine myocarditis virus (PMCV) by RTqPCR. After i.p. inoculation, the shedders (N = 363) were marked by adipose fin removal and placed in a tank containing naïve fish (N = 396). Four weeks post PRV shedder introduction (WPC-PRV), the PRV shedders were removed and the cohabitants were distributed into four tanks. As displayed in Figure 1, at 4 WPC-PRV, two tanks containing PRV cohabitants (N = 80) were supplied with either SAV2 (N = 20) or SAV3 (N = 20) shedders (1:4 ratio, shedder:cohabitant) starting the early co-infection. While the two other tanks contained PRV cohabitants until 10 WPC-PRV (indicated by “PRV” in Figure 1) and were sampled as PRV controls at 7 and 10 WPC-PRV. At 10 WPC-PRV, the PRV cohabitants (N = 80) in the tanks, were supplied with SAV2 (N = 20) or SAV3 (N = 20) shedders, initiating the late co-infection. Hence, cohabitants in the early and late co-infection were challenged with SAV shedders 6 and 12 weeks post sea water transfer, respectively.

The co-infection challenge trial. The timeline above indicates weeks post PRV shedder introduction (WPC-PRV). Blue down arrow indicates transfer to sea water (SW). Yellow down arrow and yellow up arrow indicates introduction and removal, respectively, of PRV shedder fish. Red down arrow indicates introduction of SAV shedders. “Naïve fish 1” indicate the experimental fish before virus challenge, “Naïve fish 2” indicate fish dedicated to be SAV shedders and SAV controls at 4 WPC-PRV and “Naïve fish 3” indicate fish dedicated to be SAV shedders at 10 WPC-PRV. Blue box named “PRV cohabitation” indicates cohabitant fish exposed to 50% PRV shedders from 0 to 4 WPC-PRV. Blue box named “PRV” indicates PRV cohabitants from 4 to 10 WPC-PRV without PRV shedders. Boxes indicating a 6 week period of exposure to 20% SAV shedders are colored red (SAV only) and green (PRV-SAV). Co-infection induced at 4 WPC-PRV and 10 WPC-PRV is denoted PRV-SAV-early and PRV-SAV-late, respectively.
SAV challenge
Naïve fish (N = 218) were kept in a separate tank (“Naïve fish 2”, Figure 1). SAV2 or SAV3 shedders were i.p. injected with 0.1 mL of cell culture medium containing SAV2 or SAV3 at a concentration of 104 TCID50/mL. The SAV inocula were prepared as described earlier [21]. SAV shedders were marked by maxilla cutting. After injection, the SAV shedders were kept in separate tanks for four days before being introduced to the cohabitants. SAV2 or SAV3 shedders (N = 13) were placed in tanks with respective SAV subtype control tanks (N = 52 in each tank) (Figure 1). Naïve fish to be i.p. injected with SAV at 10 WPC-PRV were kept in a separate tank from 4 to 10 WPC-PRV (“Naïve fish 3” in Figure 1). Time after introduction of SAV shedders will be referred to as weeks post SAV shedder introduction (WPC-SAV). Organ samples from heart on RNAlater™ (Ambion Inc., USA) and heparinized blood were collected from naïve fish (N = 4) before SAV challenge. These were confirmed negative for SAV and PRV by RTqPCR. Due to differences in virulence and geographic distribution [21, 22], both SAV2 and SAV3 were included in the study.
Sampling
Eight cohabitants were sampled from the PRV only and PRV-SAV co-infected groups, whereas six cohabitants were sampled from the SAV control groups at each sampling. Eight fish sampled prior to PRV challenge served as negative controls. Weight and fork length was registered for all sampled cohabitants and Fulton’s condition factor (k-factor) was calculated (k-factor = weight in grams/length in cm3 × 100).
Tissue samples for histopathological evaluation (heart, pyloric caeca including exocrine pancreas and red and white skeletal muscle including the lateral line) were collected and fixed in 10% phosphate buffered formalin. After 24 h, the formalin was replaced with 70% ethanol and stored at 4 °C until further handling.
Two pieces of 2 mm3 from heart were collected on prefilled 1.0 mL tubes (FluidX® Ltd, UK) with 0.5 mL RNAlater™ for RTqPCR analysis. Heparinized blood was collected from the caudal vein.
Histopathology and immunohistochemistry
Samples for histopathology were processed and stained with hematoxylin and eosin following standard procedures. The sections from heart, pyloric caeca and skeletal muscle were examined blind and scored for pathological changes in an ordinal system (0, 1, 2, and 3), based on Taksdal et al. and McLoughlin et al. [21, 35]. The scoring performed in this study was modified and extended to include a separate score for acute myocardial necrosis and epicarditis as the former is a hallmark in PD development and the latter is observed in both PD and HSMI [9]. The scoring criteria for exocrine pancreas, myocardial degeneration and inflammation, acute myocardial necrosis, epicarditis and inflammation in skeletal muscle are displayed in Additional file 1.
Immunohistochemistry for detection of PRV and SAV in heart tissue were performed as described earlier for detection of PRV [13]; polyclonal rabbit anti-σ1 serum (1:2500) [13] for PRV and monoclonal murine anti-E2 (17H23) (1:2000) [36] for SAV were used as primary antibodies. Both were incubated overnight in a humidity chamber, SAV at room temperature and PRV at 4 °C. Biotinylated goat anti-rabbit (1:200) and biotinylated rabbit anti-mouse (1:300) were used as secondary antibodies (Dako, Agilent Technologies, Glostrop, Denmark). Vectastain ABC-AP kit (Vector, Laboratories, Burlingame, CA, USA) was used for visualization. Heart tissues from double infected fish tissues were investigated. As negative and positive controls, slides with SAV or PRV single infected tissues were included.
RNA isolation and RTqPCR
All samples, including heparinized blood, were shipped cool (5–10 °C) and arrived within 24 h to the Norwegian Veterinary Institute laboratory after sampling. Tissue samples on RNAlater™ were placed at −20 °C until further analysis. A sub-sample of 200 µL from each heparinized blood sample was subsequently shipped cold, together with heart samples on RNAlater™, to PatoGen AS for virus analysis.
PatoGen AS performed RNA extraction and RTqPCR analysis for PRV and SAV transcripts in heart and heparinized blood. The RTqPCR assay targeting PRV is validated to ISO17025 standards and was described by Glover et al. [37]. The SAV assay is validated and accredited to ISO17025 standards and was performed as described earlier [38]. Samples were defined as positive when having a PRV or SAV Ct lower than 37.0. Elongation factor 1α (EF1α) served as an internal reference gene [39] for all RTqPCR assays performed. PRV and SAV Ct values were normalized to EF1α Ct values (ΔCt = Cttarget − CtEF1α). After finalizing the virus analyses, RNA (in RNase free H2O) extracted from heart and blood by PatoGen AS was shipped frozen on dry-ice, overnight to the NVI. RNA quantification and purity was determined using NanoDrop 2000 UV–Vis Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Finally, 1 µL RNase Out (0.5 U/µL, Life technologies) was added and the RNA was stored at −80 °C until gene expression analysis.
For gene expression analysis, cDNA was produced from 600 ng total RNA using QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. Quantitative PCR was performed using 15 ng (5 µL of 3 ng/µL) cDNA input per reaction using Maxima SYBR Green (Thermo Scientific) with 10 µM of both primers. Primers are listed in Table 1. The cycling conditions used were 95 °C for 10 min, then 40 cycles of 95 °C/15 s, 60 °C/30 s and 72 °C/30 s in a Mx3005P (Stratagene, La Jolla, CA, USA). A seven-point concentration grade standard curve (40–0.675 ng) was run during testing of the primer pairs.
Microarray analyses
The analyses were carried out using NOFIMA’s Atlantic salmon oligonucleotide microarray SIQ-6 and bioinformatic package STARS [40]. The platform includes 15 k unique probes to protein encoding transcripts; the genes were annotated by functions (GO), pathways (KEGG) and custom vocabulary. Microarrays were manufactured by Agilent Technologies (Santa Clara, CA, USA) and unless indicated otherwise, the reagents and equipment were purchased from the same source. The microarray analyses were performed on RNA from heart tissue that was shipped overnight from NVI to NOFIMA on dry ice. RNA from uninfected hearts, sampled at Day 0, was used as a common reference in all hybridizations. RNA amplification and labelling were performed with a Two-Color Quick Amp Labelling Kit and a Gene Expression Hybridization kit was used for fragmentation of labelled RNA. Total RNA input for each reaction was 500 ng. After overnight hybridization in an oven (17 h, 65 °C, rotation speed 0.01 g), arrays were washed with Gene Expression Wash Buffers 1 and 2 and scanned with a GenePix 4100A (Molecular Devices, Sunnyvale, CA, USA). GenePix Pro 6.0 was used for spot to grid alignment, assessment of spot quality, feature extraction and quantification. Subsequent data analyses were performed with STARS. After filtration of low quality spots flagged by GenePix, Lowess normalization of log2-expression ratios (ER) was performed. Genes that passed the quality control in more than half of the samples were included in the subsequent analyses. Differential expression was assessed by criteria: ER > 1.75-fold and p < 0.05.
Data analysis and statistics
The statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software inc., USA). Differences in viral RNA levels were calculated based on ΔCt values using the non-parametric Mann–Whitney unpaired rank test. Differences in gene transcript levels in heart tissue and histopathological scores between the groups were examined using the non-parametric Mann–Whitney unpaired rank test. An unpaired student t test was used to examine differences in k-factor and weight. Spearman’s rank correlation was calculated using STATA 13.1 (StataCorp, USA), for associations between viral RNA levels in SAV cohabitants and histopathology score of acute myocardial necrosis. In addition, association between gene expression (ΔCt) and histopathology score of both myocardial degeneration and inflammation and acute myocardial necrosis was calculated. In all calculations of differences, a p < 0.05 was considered statistically significant.
Results
The cohabitant challenge experiment is displayed in Figure 1. Weeks post PRV shedder introduction are abbreviated as WPC-PRV and weeks post SAV shedder introduction as WPC-SAV. The challenge trial consisted of 4 or 10 weeks of PRV infection alone (WPC-PRV) and a subsequent 6-week PRV-SAV co-infection period. The different time-points for SAV shedder introduction, i.e. 4 and 10 WPC-PRV, are termed early and late co-infection, respectively. Hence, the terms used are PRV-SAV-early or PRV-SAV-late. The SAV control fish were challenged simultaneously with the early co-infection. The results originate from cohabitant fish unless specified otherwise.
Mortality and growth
Mortality was low during the experiment. Three (0.76%) PRV infected fish died during the first 4 weeks, while one fish died in the PRV-SAV2-early group (1.25%) between 4 and 10 WPC-PRV. The accumulated mortality at 6 WPC-SAV was 2.0 and 5.7% in the SAV2 and SAV3 controls, respectively. In the late co-infection there were no mortalities.
On Day 0, the mean weight, length and condition (k)-factor was 105.6 g (range 70.6–160.4 g), 21.2 cm (range 18.5–24.5 cm) and 1.09 (range 1.02–1.17), respectively. At the time of SAV shedder introduction, the mean weight at 4 WPC-PRV was 114.7 g (N = 16, PRV early group) and 109.3 g (N = 4, naïve fish) and at 10 WPC-PRV, the mean weight was 157.4 g (N = 16, PRV late group). At 10 WPC-PRV, all groups had increased average weight and length. However, the mean k-factor was reduced in the PRV-SAV2-early (1.06), SAV2 (1.02) and SAV3 (1.02) groups and increased in PRV controls (1.10) and the PRV-SAV3-early group (1.11). The difference between the SAV control groups compared to the PRV controls and the PRV-SAV3-early group were significant (p < 0.05) at 10 WPC-PRV. At the end of the trial, i.e. 16 WPC-PRV, the PRV-SAV2-late and PRV-SAV3-late groups had a mean k-factor of 1.17 and 1.11, respectively. Additional file 2 shows detailed range of weight, length and k-factor.
PRV infection kinetics
At day 0, the fish were confirmed negative for both PRV and SAV by RTqPCR. Successful transmission and infection of the cohabitants with PRV were confirmed by detection of viral RNA in blood and heart (Figures 2A–D). PRV was first detected in cohabitant fish at 3 WPC-PRV and the level of PRV RNA peaked at 5 WPC-PRV in blood (mean Ct 14.2) and 6 WPC-PRV in heart (mean Ct 18.3). High PRV RNA levels were present in the fish until the end of the experiment at 16 WPC-PRV (Figures 2C and D).

PRV RNA kinetics. PRV ΔCt values (PRV Ct–EF1α Ct) in blood and heart from individual co-challenged fish (black and grey dots) and PRV control fish (red x) assayed by RTqPCR targeting PRV RNA segments. Red arrow indicates introduction of SAV shedders early (A, B) or late (C, D). Weeks post PRV shedder introduction (WPC-PRV) are indicated at the x-axis. N positive individuals (raw Ct <37.0) of total sampled fish is tabulated under each sub-figure. Asterisk indicates significant (p < 0.05) differences in PRV RNA levels.
Histopathological changes including epicarditis and myocardial degeneration and inflammation in red skeletal muscle were in accordance to experimentally induced HSMI (Additional file 3), as previously described [13]. There were no differences in ΔCt values of PRV RNA or histopathological changes between the PRV controls and PRV-SAV groups. Likewise, there were no differences between the co-infected groups when comparing ΔCt values of PRV transcripts except a significantly lower ΔCt value in PRV-SAV3-late group compared to PRV-SAV2-late group at 12 WPC-PRV, p < 0.05 (Figures 2C and D).
SAV infection kinetics
SAV2 and SAV3 shedders were confirmed SAV positive by RTqPCR in heart and blood. The histopathological changes were characteristic for PD, i.e. loss of exocrine pancreatic tissue, acute myocardial necrosis and myocarditis.
SAV challenge at 4 WPC-PRV (early co-infection)
In the PRV-SAV2-early group, the levels of SAV RNA were significantly lower in blood at 3, 4 and 6 WPC-SAV compared to the SAV2 controls (p < 0.05) and in heart at 4 and 6 WPC-SAV (p < 0.05). At both time points, 3/8 fish were SAV negative in heart in the co-infected group.
The SAV RNA levels in the SAV3 control group peaked 3 WPC-SAV, however the PRV-SAV3-early group did not reach the same level (Figures 3C and D). At 3 and 4 WPC-SAV, the SAV RNA levels in blood was undetectable in more than 50% of the fish in the co-infected group and significantly lower than in the SAV3 control group (p < 0.05). The SAV RNA level in heart was significantly lower in the co-infected fish at 4 and 6 WPC-SAV compared to the SAV3 controls (p < 0.05).

SAV RNA kinetics, early co-infection. SAV ΔCt values (SAV Ct–EF1α Ct) in blood and heart from individual co-challenged fish (black dots) and SAV control fish (red x) assayed by RTqPCR targeting SAV RNA segments. Red arrow indicates introduction of SAV2 shedders (A, B) or SAV3 shedders (C, D). Weeks post SAV shedder introduction (WPC-SAV) are indicated at the x-axis. N positive individuals (raw Ct < 37.0) of total sampled fish is tabulated under each sub-figure. Asterisk indicates significant (p < 0.05) differences between co-challenged groups and SAV control group.
SAV challenge at 10 WPC-PRV (late co-infection)
In the late co-infection, i.e. addition of SAV shedders at 10 WPC-PRV, SAV RNA was first detected in the PRV-SAV2-late group in blood and heart at 3 WPC-SAV and peaked at week 4 post SAV introduction, which was one week later than the control group (Figures 4A and B). The SAV RNA level in blood was significantly lower in the co-infected group at 3 WPC-SAV compared to the SAV control groups (p < 0.05). Furthermore, the co-infected group had a significantly lower SAV RNA level in heart compared to the controls at 3 WPC-SAV (p < 0.05).

SAV RNA kinetics, late co-infection. SAV ΔCt values (SAV Ct–EF1α Ct) in blood and heart from individual co-challenged fish (black dots) assayed by RTqPCR targeting SAV RNA segments. For illustration, SAV ΔCt values from SAV control fish (red x) obtained during the early co-infection, was included in the figure. Red arrow indicate introduction of SAV2 shedders (A, B) or SAV3 shedders (C, D). Weeks post SAV shedder introduction (WPC-SAV) are indicated at the x-axis. N positive individuals (raw Ct < 37.0) of total sampled fish is tabulated under each sub-figure. Asterisk indicates significant (p < 0.05) differences between co-challenged groups and SAV control fish.
In the PRV-SAV3-late group the SAV RNA levels did not show the same delay as observed for the PRV-SAV2-late group (Figures 4C and D). SAV RNA was first detected 2 WPC-SAV and reached peak levels 4 WPC-SAV in both heart and blood. However, at 3 and 4 WPC-SAV the SAV RNA level was significantly lower in heart in the co-infected group (p < 0.05). In blood, the SAV RNA level was significantly lower at 3 WPC-SAV in the PRV-SAV3-late group compared to the SAV3 controls (p < 0.05).
During the co-infection, the SAV shedders also got infected with PRV (Figure 5). Although not significant, the PRV RNA levels in the SAV2 shedders after 6 WPC-SAV, were higher in heart when compared to SAV3 shedders.

PRV RNA levels in SAV2 and SAV3 shedder fish after 6 weeks in the tank with PRV infected fish. PRV ΔCt values (PRV Ct–EF1α Ct) assessed by RTqPCR targeting PRV RNA in blood (A) and heart (B). WPC-SAV indicates weeks post SAV2 or SAV3 shedder introduction.
Histopathology in co-infected groups
Histopathological scoring of changes in pancreas and acute myocardial necrosis showed a reduction in the co-infected groups compared with SAV control groups (Figures 6 and 7). Using a non-parametric rank test of the ordinal histopathological score, changes in pancreas were found to be significantly lower at 4 and 6 WPC-SAV in both early and late co-infection compared to the SAV control groups (p < 0.05), except at 4 WPC-SAV in the PRV-SAV3-late group (Figures 6 and 7). The PRV-SAV2-late group had also a significantly lower score in pancreas compared to the SAV2 control group at 3 WPC-SAV (p < 0.05). The prevalence of acute myocardial necrosis was significantly reduced at 4 WPC-SAV in the co-infected groups compared to the SAV controls (p < 0.05). At 6 WPC-SAV, the prevalence of acute myocardial necrosis was significantly lower in the PRV-SAV3-late and PRV-SAV2-early groups compared to the SAV controls (p < 0.05).

Histopathological evaluation of PRV-SAV2 co-infected fish. Histopathological scoring of PD associated changes in pancreas and acute necrosis in myocardium detected during the 6 week co-infection. Asterisk indicates significant (p < 0.05) differences between co-infected groups and SAV2 control fish. Week 0 represent uninfected control fish.

Histopathological evaluation of PRV-SAV3 coinfected fish. Histopathological scoring of PD associated changes in pancreas and acute necrosis in myocardium during the 6 week co-infection. Asterisk indicates significant (p < 0.05) differences between co-infected groups and SAV3 control fish. Week 0 represent uninfected control fish.
The SAV RNA levels (ΔCt) and histopathology score of acute myocardial necrosis showed a strong positive Spearman’s rank correlation (rs = 0.81) in the SAV3 control group, p < 0.05 (N = 28), whereas a weaker correlation (rs = 0.59) was seen in the SAV2 controls, p < 0.05 (N = 28).
Immunohistochemistry (IHC) was performed on sections of heart tissue from single infected and co-infected fish. The fish were selected based on high viral levels (indicated by low Ct levels) of both viruses. The fish presented in Figure 8 were sampled 7 WPC-PRV, which for the SAV controls and co-infected groups correspond to three weeks post SAV introduction (3 WPC-SAV) in the early co-infection. Virus Ct values in the respective heart tissues are noted in Figure 8. In the SAV3 infected fish, sparse but distinct staining restricted to single cells was observed using SAV antibodies. The PRV antibodies yield a weak pink background color in both PRV infected and SAV infected heart tissues. However, distinct staining was only observed in PRV infected heart. Staining of both SAV and PRV antibodies were achieved in two separate sections of a co-infected heart. In the co-infected heart tissue more diffuse staining was observed in epicard, interpreted as unspecific binding for both SAV and PRV antibodies. When staining the control tissue with PRV antibodies, a weak pink background color was observed. Additional files 4, 5 and 6 include more detailed pictures. The IHC demonstrates the presence of PRV and SAV in the same areas of a co-infected heart section. However, no individual cells could be defined as co-infected.

Immunohistochemical detection of PRV and SAV in a co-infected heart. Sections of heart tissue from SAV infected, PRV infected and PRV-SAV co-infected fish were stained using polyclonal rabbit antiserum targeting PRV σ1 (left panel) and monoclonal murine anti-SAV E2 (right panel). The fish were sampled at the same time point, 7 WPC-PRV which corresponds to 3WPC-SAV in the early co-infection. The small internal panels are enlargements to show specificity of the staining.
PRV-SAV co-infection and gene expression linked to heart pathology
Microarray analysis was performed on hearts sampled at 4 and 6 WPC-SAV from the late co-infection and differences between the SAV3 control and PRV-SAV3-late group were analyzed (Table 2). Genes were selected based on their correlation with severity of pathological changes in heart induced by SAV infection, as previously reported [41]. To confirm the array results, RT-qPCR assays were run for seven selected genes, of which six showed significant differences (Figure 9). The gene regulation relative to 4 WPC-PRV (set to zero) is shown in Figure 9. At this time point the heart appeared healthy by histopathological evaluation. Gene expression at 4 and 6 WPC-SAV in both the early and the late co-infected group were compared to 4 and 6 WPC-SAV in the SAV3 control group. Significant differences were found between PRV-SAV3-late and SAV3 control for calsequestrin, neuropeptide Y-1 and interleukin 1-receptor accessory protein-like 2 (IL1R-2) at both 4 and 6 WPC-SAV, at 4 WPC-SAV for mitochondrial arginase-2 and at 6 WPC-SAV for arginase-1 and serum amyloid A5-protein (SAA5) (p < 0.05). The early co-infected groups differed significantly from the SAV controls for IL1R-2, mitochondrial arginase-2 and SAA5 at 4 WPC-SAV, and for calsequestrin, IL1R-2, arginase-1 and SAA5 at 6 WPC-SAV (p < 0.05). No significant differences between the groups were found for matrix metalloproteinase 13 (MMP13) expression.

Expression of potential pancreas disease marker genes in heart. Fold induction or repression of genes identified as potential PD associated genes assessed in SAV3 infected (blue bars) and co-infected (red bars: PRV-SAV3-early, orange bars: PRV-SAV3-late) 4 or 6 weeks after SAV3 shedder introduction. Significant differences between SAV3 controls and co-infected groups are indicated with *p < 0.05. Boxes indicate mean fold change relative to mean levels at 4 WPC-PRV.
Histopathology scores (ordinal variable 0, 1, 2 and 3) of myocardial degeneration and inflammation and acute myocardial necrosis were correlated with gene expression levels from the qPCR, using Spearman’s rank correlation (Table 3). The groups investigated (N = 6/group) were PRV controls (4 and 10 WPC-PRV), PRV-SAV3-early and -late (4 and 6 WPC-SAV) and SAV3 controls (4 and 6 WPC-SAV), making a total of N = 46 fish evaluated. Significant correlation was found for all gene expression levels toward the score of acute necrosis and inflammation in myocardial tissue, except for MMP13 against inflammation (Table 3). When corrected for multiple comparisons by Bonferroni-adjusted significant level, IL1R2, neuropeptide Y-1, SAA5, mitochondrial arginase-2 against necrosis and calsequestrin, IL1R-2, neuropeptide Y-1, arginase-1 against inflammation remained significant.
Discussion
This study demonstrates that a primary PRV infection reduces disease development of a subsequent SAV infection with either SAV2 or SAV3, as evidenced by lower levels of SAV RNA, less severe PD pathological lesions and higher condition factors in the co-infected groups. The lack of parallel groups must be accounted for when evaluating the presented results. Nevertheless, the observation of a similar reduction in disease development for both SAV subtypes in co-infected fish strengthens the validity of the results.
The most pronounced evidence of protection was a reduction in PD specific pathological lesions in exocrine pancreas. Lesions in the pancreas are a hallmark of SAV infection and used diagnostically for separation of PD and HSMI [9]. The heart is a target organ for both PRV and SAV, with myocarditis and epicarditis observed in both diseases [8, 9, 13]. This may possibly mask the protective effect on SAV induced myocarditis and epicarditis in this study. Cardiomyocytic necrosis is a typical pathological finding of early stages of SAV infection [9], and is not considered a specific feature of HSMI. In our study we found that the early co-infected groups had a significantly lower degree of acute myocardial necrosis compared to SAV controls. A recent study indicate a possible difference in susceptibility to SAV infection depending on time following sea water transfer [42]. Therefore, we cannot exclude that the size of the fish and time after sea transfer may have an impact on the difference in protection observed between the SAV only control groups and the late co-infected groups in our study.
Co-detection of PRV and SAV in hearts have been shown by RTqPCR in farmed Atlantic salmon escapees in Norway [10]. Here, we demonstrate the presence of both viruses in neighboring cells in heart tissue by immunohistochemistry along with co-detection by RTqPCR. However, since the protective effect was observed in pancreas which is not a target organ for PRV, this indicates that the PRV mediated protection is due to systemic responses and not to interaction through co-infection in the same tissue. PRV utilizes erythrocytes for replication and dissemination in the fish [34], and the salmon erythrocytes can mount innate antiviral responses after PRV infection [12, 43].
The PRV control group had a significantly higher k-factor 10 WPC-PRV compared to the SAV control groups at the same time point. The k-factors in the SAV2 and SAV3 controls were lower compared to the corresponding double infected groups (PRV-SAV2-early and PRV-SAV3-early), although only significant for the PRV-SAV3-early group. The higher k-factor supports the observed less severe histopathological changes in the co-infected groups, confirming the reduced impact of SAV after a preceding PRV infection.
Our results are in accordance with previous co-infections in fish where reduced mortality was observed. Pre-exposure of rainbow trout with either the non-virulent cutthroat trout virus (CTV) (Hepeviridae), chum salmon reovirus (CSV), or IPNV, gave a four week protection to a subsequent IHNV infection [26–30, 44, 45]. A similar effect against ISAV, lasting eight weeks, was observed for IPNV infected fish. However in that study the ISAV challenge was given intraperitoneally [46]. This indicates that long-lasting cross-protection between non-related viruses in fish is a general phenomenon, although the duration of protection may vary. Our study suggest an inhibitory effect of PRV on a secondary SAV infection which may last for at least 10 weeks post PRV challenge, and thus a longer duration of protection compared to other interfering virus infections reported in salmonids [26, 46].
Activation of the antiviral innate immune response, where the type 1 interferon (IFN) system is central [47, 48], is a possible explanation for protection against unrelated viruses after a primary virus infection. Studies on SAV infection in cell cultures and in vivo have shown upregulation of IFNα and a number of interferon induced genes [49, 50]. However, these studies revealed a complex antiviral innate response after SAV infection leading to reduced SAV propagation. PRV infection of Atlantic salmon RBC also cause strong up-regulation of antiviral genes of the innate immune response [43]. Two recent studies show that Mx expression is upregulated in heart tissue for 11 weeks [41], and in erythrocytes for at least eight weeks after PRV infection [43]. Interferon type I production was induced at the transcriptional level in erythrocytes for up to 7 weeks [43]. This suggests that circulating PRV infected erythrocytes could play an important role in the observed suppressive effect on SAV propagation and PD development by inducing interferon-regulated antiviral responses in most organs prior to SAV infection [51].
Therefore, at the time of early SAV shedder introduction in our trial, i.e. at 4 WPC-PRV, an upregulation of innate antiviral genes is expected. A possible long lasting innate immune response may contribute to the protection seen during the late co-infection. SAV RNA levels in heart were significantly lower 6 WPC-SAV in the early co-infected groups compared to the SAV controls. This difference was not present at the same time point in the late co-infected hearts, which could indicate a decreased protection caused by a reduction of innate immunity. However, since fish IgM have lower specificity and antigen affinity compared to mammalian serum antibodies, at least up to 15 weeks post infection, a mechanism of low affinity polyreactive natural antibodies cannot be ruled out [52, 53].
A possible difference was observed between the two SAV subtypes in the ability to handle the consequences of the preceding PRV infection. The SAV2 and SAV3 RNA levels differed in the co-infected groups. SAV2 replicated more efficiently than SAV3 during the early co-infection, whereas the peak phase of both viruses was lost in the late co-infection with PRV, either by a delay (SAV2) or by a reduction (SAV3). There were large individual variations in levels of SAV RNA and in prevalence of SAV positive fish in the co-infected groups, which makes it difficult to conclude if the apparent differences in SAV2 and SAV3 kinetics show true different properties of the virus subtypes.
SAV RNA kinetics in the heart tissue of both SAV2 and SAV3 controls were similar when assessed by RTqPCR, which is in accordance with a previous SAV challenge trial where several isolates were tested [21]. An interesting finding, that should be addressed in a suitable study, was the strong correlation between acute myocardial necrosis and SAV3 RNA level (rs = 0.81) compared to SAV2 (rs = 0.59) in the individual infected fish. If SAV3 is a stronger inducer of acute myocardial necrosis, this may partly explain the observed higher mortality associated with SAV3 compared to SAV2 [21, 22, 54].
A previous study using salmon microarray and RTqPCR analysis have reported that changes in the expression levels of certain genes are specifically associated with SAV mediated pathological changes in heart [41]. We found a similar regulation of these genes in the SAV controls in our study and that the expression levels of these genes were less affected in the co-challenge groups. This is in tune with the protective effects of a primary PRV infection, supporting histopathological observations and virus kinetics. RTqPCR run on a selected number of genes confirmed the microarray results. In general, the gene expression pattern was more affected in the SAV controls compared to the co-infected group. A previous study indicated a difference in gene expression patterns between the two diseases [41]. Our study found that the expression differences between the co-infected and SAV3 control groups changed in line with score of histopathological lesions, with a strong correlation of neuropeptide Y-1 and arginase-1 expression to the score of myocardial degeneration and inflammation. In mammals, neuropeptide Y has been shown to have several effects on inflammatory responses and cardiomyopathy [55, 56]. Furthermore, expression of IL1R-2, SAA5 and mitochondrial arginase-2 show a strong and significant correlation towards acute myocardial necrosis. Thus, genetic analysis may prove to be an additional tool for evaluation of the severity of salmon heart disease and tissue damage.
A peculiar finding was the secondary PRV infection detected in the SAV shedders who resided with the PRV cohabitants. This was observed in both the early and late co-infection. During the early co-infection, SAV2 shedders had a higher PRV RNA level, although not significant, in heart when compared to SAV3 shedders six weeks after introduction to PRV cohabitants. This may suggest that SAV3 yields a stronger protection against PRV than SAV2. A possible explanation is that SAV3, reported by others to be more virulent [21, 22, 54], yield a stronger immune response than SAV2. However, the lack of parallel tanks and number of fish per group (N = 6) must be accounted for when interpreting these results. The strong correlation between SAV RNA levels and myocardial necrosis may be a novel step towards understanding the observed virulence differences between the subtypes. An interesting observation in this perspective is the higher prevalence of HSMI in the geographically separated endemic areas of SAV2 in mid-Norway compared to those of SAV3 further south [4, 7]. These field observations could potentially be linked to a higher possibility of cross-infection between SAV2 and PRV and more prevalent development of both diseases in PRV-SAV2 dual infected fish.
Interactions between viral diseases may be part of the explanation for the large variation in severity described for SAV infections in the field [24]. PRV is found to infect fish in fresh water facilities and is ubiquitous in sea farms [6]. The protective effect in this study could affect the outcome of a PRV-SAV co-infection after sea transfer. In this study, the experimental fish had high levels of PRV RNA and developed HSMI. A more subtle PRV infection, where there is no HSMI development may cause a difference in strength and duration of the protection. Further research should address various field conditions when assessing the implications of PRV-SAV co-infection.
In conclusion, we found that a primary PRV infection partially protects against the outcomes of SAV infection and PD pathological lesions.
References
Krkošek M (2010) Host density thresholds and disease control for fisheries and aquaculture. Aquac Environ Interact 1:21–32
Wheatley SB, McLoughlin MF, Menzies FD, Goodall EA (1995) Site management factors influencing mortality rates in Atlantic salmon (Salmo salar L.) during marine production. Aquaculture 136:195–207
Aunsmo A, Bruheim T, Sandberg M, Skjerve E, Romstad S, Larssen RB (2008) Methods for investigating patterns of mortality and quantifying cause-specific mortality in sea-farmed Atlantic salmon Salmo salar. Dis Aquat Organ 81:99–107
Bornø G, Lie Linaker M (2015) The health situation in Norwegian aquaculture 2014. Norwegian Veterinary Institute. http://www.eng.vetinst.no/eng/Publications/Fish-Health-Report/Fish-Health-Report-2014.html
Palacios G, Lovoll M, Tengs T, Hornig M, Hutchison S, Hui J, Kongtorp RT, Savji N, Bussetti AV, Solovyov A, Kristoffersen AB, Celone C, Street C, Trifonov V, Hirschberg DL, Rabadan R, Egholm M, Rimstad E, Lipkin WI (2010) Heart and skeletal muscle inflammation of farmed salmon is associated with infection with a novel reovirus. PLoS One 5:e11487
Løvoll M, Alarcón M, Bang Jensen B, Taksdal T, Kristoffersen AB, Tengs T (2012) Quantification of piscine reovirus (PRV) at different stages of Atlantic salmon Salmo salar production. Dis Aquat Organ 99:7–12
Hjortaas MJ, Bang Jensen B, Taksdal T, Olsen AB, Lillehaug A, Trettenes E, Sindre H (2016) Genetic characterization of salmonid alphavirus in Norway. J Fish Dis 39:249–257
Kongtorp RT, Taksdal T, Lyngøy A (2004) Pathology of heart and skeletal muscle inflammation (HSMI) in farmed Atlantic salmon Salmo salar. Dis Aquat Organ 59:217–224
McLoughlin MF, Graham DA (2007) Alphavirus infections in salmonids—a review. J Fish Dis 30:511–531
Madhun AS, Karlsbakk E, Isachsen CH, Omdal LM, Eide Sørvik AG, Skaala O, Barlaup BT, Glover KA (2015) Potential disease interaction reinforced: double-virus-infected escaped farmed Atlantic salmon, Salmo salar L., recaptured in a nearby river. J Fish Dis 38:209–219
Markussen T, Dahle MK, Tengs T, Løvoll M, Finstad ØW, Wiik-Nielsen CR, Grove S, Lauksund S, Robertsen B, Rimstad E (2013) Sequence analysis of the genome of piscine orthoreovirus (PRV) associated with heart and skeletal muscle inflammation (HSMI) in Atlantic Salmon (Salmo salar). PLoS One 8:e70075
Wessel Ø, Olsen CM, Rimstad E, Dahle MK (2015) Piscine orthoreovirus (PRV) replicates in Atlantic salmon (Salmo salar L.) erythrocytes ex vivo. Vet Res 46:26
Finstad ØW, Falk K, Løvoll M, Evensen Ø, Rimstad E (2012) Immunohistochemical detection of piscine reovirus (PRV) in hearts of Atlantic salmon coincide with the course of heart and skeletal muscle inflammation (HSMI). Vet Res 43:27
Kongtorp RT, Kjerstad A, Taksdal T, Guttvik A, Falk K (2004) Heart and skeletal muscle inflammation in Atlantic salmon, Salmo salar L.: a new infectious disease. J Fish Dis 27:351–358
Weston JH, Welsh MD, McLoughlin MF, Todd D (1999) Salmon pancreas disease virus, an alphavirus infecting farmed Atlantic salmon, Salmo salar L. Virology 256:188–195
Aunsmo A, Valle PS, Sandberg M, Midtlyng PJ, Bruheim T (2010) Stochastic modelling of direct costs of pancreas disease (PD) in Norwegian farmed Atlantic salmon (Salmo salar L.). Prev Vet Med 93:233–241
Pettersen JM, Rich KM, Bang-Jensen B, Aunsmo A (2015) The economic benefits of disease triggered early harvest: a case study of pancreas disease in farmed Atlantic salmon from Norway. Prev Vet Med 121:314–324
Fringuelli E, Rowley HM, Wilson JC, Hunter R, Rodger H, Graham DA (2008) Phylogenetic analyses and molecular epidemiology of European salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J Fish Dis 31:811–823
Hodneland K, Bratland A, Christie KE, Endresen C, Nylund A (2005) New subtype of salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis Aquat Organ 66:113–120
Hjortaas MJ, Skjelstad HR, Taksdal T, Olsen AB, Johansen R, Bang-Jensen B, Ørpetveit I, Sindre H (2013) The first detections of subtype 2-related salmonid alphavirus (SAV2) in Atlantic salmon, Salmo salar L., in Norway. J Fish Dis 36:71–74
Taksdal T, Jensen BB, Böckerman I, McLoughlin MF, Hjortaas MJ, Ramstad A, Sindre H (2015) Mortality and weight loss of Atlantic salmon, Salmo salar L., experimentally infected with salmonid alphavirus subtype 2 and subtype 3 isolates from Norway. J Fish Dis 38:1047–1061
Jansen MD, Bang-Jensen B, Brun E (2014) Clinical manifestations of pancreas disease outbreaks in Norwegian marine salmon farming—variations due to salmonid alphavirus subtype. J Fish Dis 38:343–353
Graham DA, Rowley HR, Frost P (2014) Cross-neutralization studies with salmonid alphavirus subtype 1-6 strains: results with sera from experimental studies and natural infections. J Fish Dis 37:683–691
Stormoen M, Kristoffersen AB, Jansen PA (2013) Mortality related to pancreas disease in Norwegian farmed salmonid fish, Salmo salar L. and Oncorhynchus mykiss (Walbaum). J Fish Dis 36:639–645
Wagner R (1960) Viral interference. Some considerations of basic mechanisms and their potential relationship to host resistance. Bacteriol Rev 24:151–166
Hedrick RP, LaPatra SE, Yun S, Lauda KA, Jones GR, Congleton JL, de Kinkelin P (1994) Induction of protection from IHNV in rainbow trout Oncorhynchus mykiss by pre-exposure to the avirulent cutthroat trout virus (CTV). Dis Aquat Organ 20:111–118
LaPatra SE, Lauda KA, Jones GR (1995) Aquareovirus interference mediated resistance to infectious hematopoietic necrosis virus. Vet Res 26:455–459
Tafalla C, Rodriguez Saint-Jean S, Pérez-Prieto S (2006) Immunological consequences of the coinfection of brown trout (Salmo trutta) with infectious hematopoietic necrosis virus (IHNV) and infectious pancreatic necrosis virus (IPNV). Aquaculture 256:15–22
Byrne N, Castric J, Lamour F, Cabon J, Quentel C (2008) Study of the viral interference between infectious pancreatic necrosis virus (IPNV) and infectious haematopoietic necrosis virus (IHNV) in rainbow trout (Oncorhynchus mykiss). Fish Shellfish Immunol 24:489–497
Brudeseth B, Castric J, Evensen Ø (2002) Studies on pathogenesis following single and double infection with viral hemorrhagic septicemia virus and infectious hematopoietic necrosis virus in rainbow trout (Oncorhynchus mykiss). Vet Pathol 189:180–189
Jung K, Renukaradhya GJ, Alekseev KP, Fang Y, Tang Y, Saif LJ (2009) Porcine reproductive and respiratory syndrome virus modifies innate immunity and alters disease outcome in pigs subsequently infected with porcine respiratory coronavirus: implications for respiratory viral co-infections. J Gen Virol 90:2713–2723
Renukaradhya GJ, Alekseev K, Jung K, Fang Y, Saif LJ (2010) Porcine reproductive and respiratory syndrome virus-induced immunosuppression exacerbates the inflammatory response to porcine respiratory coronavirus in pigs. Viral Immunol 23:457–466
Mikalsen AB, Nilsen P, Frøystad-Saugen M, Lindmo K, Eliassen TM, Rode M, Evensen Ø (2014) Characterization of a novel calicivirus causing systemic infection in Atlantic salmon (Salmo salar L.): proposal for a new genus of Caliciviridae. PLoS One 9:e107132
Finstad ØW, Dahle MK, Lindholm TH, Nyman IB, Løvoll M, Wallace C, Olsen CM, Storset AK, Rimstad E (2014) Piscine orthoreovirus (PRV) infects Atlantic salmon erythrocytes. Vet Res 45:35
McLoughlin MF, Graham DA, Norris A, Matthews D, Foyle L, Rowley HM, Jewhurst H, MacPhee J, Todd D (2006) Virological, serological and histopathological evaluation of fish strain susceptibility to experimental infection with salmonid alphavirus. Dis Aquat Organ 72:125–133
Moriette C, LeBerre M, Boscher SK, Castric J, Brémont M (2005) Characterization and mapping of monoclonal antibodies against the Sleeping disease virus, an aquatic alphavirus. J Gen Virol 86:3119–3127
Glover KA, Sørvik AGE, Karlsbakk E, Zhang Z, Skaala Ø (2013) Molecular genetic analysis of stomach contents reveals wild Atlantic cod feeding on piscine reovirus (PRV) infected Atlantic salmon originating from a commercial fish farm. PLoS One 8:e60924
Hodneland K, Endresen C (2006) Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). J Virol Methods 131:184–192
Løvoll M, Austbø L, Jørgensen JB, Rimstad E, Frost P (2011) Transcription of reference genes used for quantitative RT-PCR in Atlantic salmon is affected by viral infection. Vet Res 42:8
Krasnov A, Timmerhaus G, Afanasyev S, Jørgensen SM (2011) Development and assessment of oligonucleotide microarrays for Atlantic salmon (Salmo salar L.). Comp Biochem Physiol Part D Genomics Proteomics 6:31–38
Johansen L-H, Thim HL, Jørgensen SM, Afanasyev S, Strandskog G, Taksdal T, Fremmerlid K, McLoughlin MF, Jørgensen JB, Krasnov A (2015) Comparison of transcriptomic responses to pancreas disease (PD) and heart and skeletal muscle inflammation (HSMI) in heart of Atlantic salmon (Salmo salar L). Fish Shellfish Immunol 46:612–623
Jarungsriapisit J, Moore LJ, Taranger GL, Nilsen TO, Morton HC, Fiksdal IU, Stefansson S, Fjelldal PG, Evensen Ø, Patel S (2016) Atlantic salmon (Salmo salar L.) post-smolts challenged two or nine weeks after seawater-transfer show differences in their susceptibility to salmonid alphavirus. Virol J 13:66
Dahle MK, Wessel Ø, Timmerhaus G, Nyman IB, Jørgensen SM, Rimstad E, Krasnov A (2015) Transcriptome analyses of Atlantic salmon (Salmo salar L.) erythrocytes infected with piscine orthoreovirus (PRV). Fish Shellfish Immunol 45:780–790
Alonso M, Rodríguez S, Pérez-Prieto SI (1999) Viral coinfection in salmonids: infectious pancreatic necrosis virus interferes with infectious hematopoietic necrosis virus. Arch Virol 144:657–673
Saint-Jean SR, Pérez-Prieto SI (2007) Effects of salmonid fish viruses on Mx gene expression and resistance to single or dual viral infections. Fish Shellfish Immunol 23:390–400
Johansen LH, Sommer AI (2001) Infectious pancreatic necrosis virus infection in Atlantic salmon Salmo salar post-smolts affects the outcome of secondary infections with infectious salmon anaemia virus or Vibrio salmonicida. Dis Aquat Organ 47:109–117
Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147:258–267
Workenhe ST, Rise ML, Kibenge MJT, Kibenge FSB (2010) The fight between the teleost fish immune response and aquatic viruses. Mol Immunol 47:2525–2536
Grove S, Austbø L, Hodneland K, Frost P, Løvoll M, McLoughlin MF, Thim HL, Braaen S, König M, Syed M, Jørgensen JB, Rimstad E (2013) Immune parameters correlating with reduced susceptibility to pancreas disease in experimentally challenged Atlantic salmon (Salmo salar). Fish Shellfish Immunol 34:789–798
Xu C, Guo TC, Mutoloki S, Haugland Ø, Marjara IS, Evensen Ø (2010) Alpha interferon and not gamma interferon inhibits salmonid alphavirus subtype 3 replication in vitro. J Virol 84:8903–8912
Langevin C, Aleksejeva E, Passoni G, Palha N, Levraud JP, Boudinot P (2013) The antiviral innate immune response in fish: evolution and conservation of the IFN system. J Mol Biol 425:4904–4920
Ye J, Kaattari IM, Kaattari SL (2011) The differential dynamics of antibody subpopulation expression during affinity maturation in a teleost. Fish Shellfish Immunol 30:372–377
Baumgarth N (2014) How specific is too specific? B-cell responses to viral infections reveal the importance of breadth over depth. Immunol Rev 255:82–94
Graham DA, Frost P, McLaughlin K, Rowley HM, Gabestad I, Gordon A, McLoughlin MF (2011) A comparative study of marine salmonid alphavirus subtypes 1-6 using an experimental cohabitation challenge model. J Fish Dis 34:273–286
Dimitrijević M, Stanojević S (2011) The intriguing mission of neuropeptide Y in the immune system. Amino Acids 45:41–53
Dvorakova MC, Kruzliak P, Rabkin SW (2014) Role of neuropeptides in cardiomyopathies. Peptides 61:1–6
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MKD, VA and ER launched the project idea. MKD and ER participated in the overall design and coordination of the study, interpretation of data and drafting the manuscript. AK and GT performed and interpreted the data from the micro array and revised the manuscript. IBN performed gene expression analysis and revised the manuscript. VA participated in the coordination of the study and revised the manuscript. ML coordinated the challenge design and study, all practicalities regarding sample logistics, gathering and interpretation of data and drafted the manuscript. MVR coordinated the challenge design and study, carried out the histopathological scoring, performed immunohistochemical staining, interpretation of data and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The research was supported by Grants #235788/E40 and #241107/O30 of the Research Council of Norway and #901001 of the Norwegian Seafood Research Fund. The SAV inoculum was kindly provided by Hilde Sindre. Thanks to Sven Martin Jørgensen (NOFIMA), Øystein Wessel (NMBU) and Torunn Taksdal (Norwegian Veterinary Institute) for expert advises on the challenge design and Turhan Markussen (NMBU) for valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Morten Lund and Magnus Vikan Røsæg shared first authorship
Additional files
13567_2016_389_MOESM1_ESM.pdf
Additional file 1. Scoring criteria for histopathological changes. The table explains the categories of inflammatory changes that was used in the histopathological scoring.
13567_2016_389_MOESM2_ESM.pdf
Additional file 2. Mean weight and k-factor is presented with range in brackets. Accumulated mortality is presented in N with% in brackets. Number indicates weeks post PRV challenge. PRV-SAV2/3e and PRV-SAV2/3l indicates early and late co-infection, respectively.
13567_2016_389_MOESM3_ESM.pdf
Additional file 3. Histopathology evaluation of epicarditis, pancreas pathology, acute necrosis in myocardium, inflammation in myocardium, inflammation in white and red skeletal muscle in PRV cohabitants. Weeks after PRV challenge is shown. Week 0 represents uninfected control fish.
13567_2016_389_MOESM4_ESM.jpg
Additional file 4. Immunohistochemistry of co-infected heart tissue stained using polyclonal rabbit antiserum targeting PRV σ1 (right panel) and monoclonal murine anti-SAV E2 (left panel). The heart tissue had Ct values of 14.2 and 15.9 for SAV and PRV, respectively.
13567_2016_389_MOESM5_ESM.jpg
Additional file 5. Immunohistochemistry of SAV-infected heart tissue stained using polyclonal rabbit antiserum targeting PRV σ1 (right panel) and monoclonal murine anti-SAV E2 (left panel). The heart tissue had a SAV Ct-value of 17.1.
13567_2016_389_MOESM6_ESM.jpg
Additional file 6. Immunohistochemistry of PRV infected heart tissue stained using polyclonal rabbit antiserum targeting PRV σ1 (right panel) and monoclonal murine anti-SAV E2 (left panel). The heart tissue had a PRV Ct- value of 17.7.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Lund, M., Røsæg, M.V., Krasnov, A. et al. Experimental Piscine orthoreovirus infection mediates protection against pancreas disease in Atlantic salmon (Salmo salar). Vet Res 47, 107 (2016). https://doi.org/10.1186/s13567-016-0389-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-016-0389-y