
Abstract
The poultry-associated bacterium Mycoplasma iowae colonizes multiple sites in embryos, with disease or death resulting. Although M. iowae accumulates in the intestinal tract, it does not cause disease at that site, but rather only in tissues that are exposed to atmospheric O2. The activity of M. iowae catalase, encoded by katE, is capable of rapid removal of damaging H2O2 from solution, and katE confers a substantial reduction in the amount of H2O2 produced by Mycoplasma gallisepticum katE transformants in the presence of glycerol. As catalase-producing bacteria are often beneficial to hosts with inflammatory bowel disease, we explored whether M. iowae was exclusively protective against H2O2-producing bacteria in a Caenorhabditis elegans model, whether its protectiveness changed in response to O2 levels, and whether expression of genes involved in H2O2 metabolism and virulence changed in response to O2 levels. We observed that M. iowae was in fact protective against H2O2-producing Streptococcus pneumoniae, but not HCN-producing Pseudomonas aeruginosa, and that M. iowae cells grown in 1% O2 promoted survival of C. elegans to a greater extent than M. iowae cells grown in atmospheric O2. Transcript levels of an M. iowae gene encoding a homolog of Mycoplasma pneumoniae CARDS toxin were 5-fold lower in cells grown in low O2. These data suggest that reduced O2, representing the intestinal environment, triggers M. iowae to reduce its virulence capabilities, effecting a change from a pathogenic mode to a potentially beneficial one.
Similar content being viewed by others

Introduction
H2O2 is a dangerous reactive oxygen species (ROS) involved in both pathogenesis and defense against infectious agents. Bacteria may be exposed to multiple sources of H2O2 during infection, including macrophages, which produce a variety of ROS that damage and degrade bacteria. A variety of bacterial pathogens can also produce H2O2 as a means of causing damage to host tissues [1,2], and some disease states stem from exposure of tissues to ROS including H2O2. In particular, in inflammatory bowel disease (IBD), a growing problem in humans in developed countries with a prevalence of 10-20% [3], a direct correlation between increased ROS production and damage to gut epithelial cells has been reported [4-7].
The enzyme catalase catalyzes degradation of H2O2 [8]. The production of catalase by microbes can benefit both the microorganisms themselves and a host organism with which they associate. Catalase can protect catalase-producing bacteria from environmental H2O2 and prolong infection. As for the host, several reports have examined the ability of catalase-expressing probiotic bacteria to decrease IBD symptoms [9]. In a murine trinitrobenzenesulfonic acid-induced Crohn’s disease model, administration of Lactobacillus casei engineered to express catalase results in faster recovery from initial weight loss, increased gut enzyme activity, and decreased intestinal inflammation as compared with mice infected with wild-type or no bacteria [10]. Similar bacteria decrease cecal and colonic inflammation in mice treated with dextran sodium sulfate to induce moderate colitis [11].
Mycoplasma iowae is a catalase-positive bacterium that infects poultry animals, primarily turkeys but also occasionally chickens [12,13]. The most common outcome of naturally-occurring M. iowae infection in turkeys is late embryo mortality, with a 2-5% reduction in hatchability, and leg abnormalities in offspring [14-16]. Symptoms commonly associated with experimental infection include airsacculitis, stunting, poor feathering, and leg and joint problems [14,16]. The M. iowae catalase gene, katE, confers both catalase activity and a significant reduction in H2O2 production upon Mycoplasma gallisepticum, a robust H2O2 producer that elaborates no other known toxins [13,17,18]. It is unclear how M. iowae causes disease, but in addition to its attachment organelle-mediated adherence and motility functions [19], its genome encodes potential virulence factors, including two closely linked genes encoding proteins similar to Mycoplasma pneumoniae CARDS toxin [13], an ADP-ribosylating toxin that is associated with many of the symptoms of M. pneumoniae infection [20-22].
Interestingly, M. iowae colonizes a variety of body sites with a range of O2 concentrations. It has been isolated from the cloacae of healthy poults and mature turkeys [23,24], but also infects the chorioallantoic membrane and a variety of organs in embryos [25], which accounts for the morbidity and mortality associated with M. iowae. Significantly, M. iowae also has a pronounced tendency to colonize the gut with no ill effects. Following yolk sac inoculation of eight-day-old turkey embryos, M. iowae can be detected in the small intestines, with bacteria most often attaching to microvilli [25]. Oral inoculation of day-old poults with M. iowae results in bacteria present in both the feces and intestinal wall for at least 21 days post-inoculation, and no differences in fecal appearance or cloaca temperature as compared to control birds [26]. The gut is a very low-O2 environment [27], whereas in ovo embryos, which are commonly damaged by M. iowae, are exposed to atmospheric O2 due to eggshell permeability [28]. It is intriguing that M. iowae infection at aerobic sites, but apparently not the gut, causes disease, leading us to suspect that O2 might play a role in regulating expression of M. iowae virulence factors, resulting in different outcomes at different body sites.
Differential gene expression regulation exists in a variety of mycoplasma species despite their reduced genomes. The M. pneumoniae promoters for acetate kinase and lactate dehydrogenase are strongly induced in the presence of glucose and glycerol, respectively [29]. M. pneumoniae lipoprotein genes are also differentially expressed in response to exposure to host cells, H2O2, and low pH [30]. Multiple forms of regulation occur in Mycoplasma hyopneumoniae in response to heat shock [31], iron deprivation [32], H2O2 treatment [33], and infection of pigs [34]. The means by which most of these regulatory events occur is unknown.
In this study we examined the ability of M. iowae, by virtue of its catalase activity, to offer protection to a model host from H2O2 stress, and to test whether M. iowae experiences differential regulation of catalase as well as possible virulence-associated genes in response to changes in O2 level. We used Caenorhabditis elegans bioassays, developed previously as a model for use with M. iowae [13], and reverse transcription-quantitative polymerase chain reaction (RT-qPCR) of selected candidate virulence-associated genes. Our results indicate that catalase contributes to M. iowae being protective toward the host against H2O2-producing organisms, even though catalase activity is reduced under conditions of low O2. They also indicate that M. iowae grown in low O2 results in increased survival of C. elegans upon co-incubation as compared with M. iowae grown in atmospheric conditions, accompanied by a 5-fold decrease in expression of at least one of the CARDS toxin-like protein genes.
Materials and methods
Bacterial strains and growth conditions
Mycoplasma strains used include M. iowae serovar K strain DK-CPA, M. gallisepticum Rlow, and M. gallisepticum Rlow transformant 56A. For M. gallisepticum and for aerobic growth of M. iowae, all strains were grown at 37 °C in 175-cm2 tissue culture flasks containing 50 mL of SP-4 broth [35] to mid-log phase. Transformant 56A was grown in the presence of 4 μg mL−1 tetracycline. SP-4 broth for samples grown in low O2 was allowed to equilibrate with a 1% O2/99% N2 gas mix overnight. Following equilibration, 50 mL of broth was aliquotted into glass bottles that were capped with rubber stoppers in which cultures were grown to mid-log phase at 37 °C.
Escherichia coli DH5α was grown in Luria broth (LB) with 100 μg mL−1 ampicillin at 37 °C in a shaking incubator. A Streptococcus pneumoniae obtained from the Miami University Department of Microbiology stock collection was grown at 37 °C without shaking in brain heart infusion (BHI) broth. Pseudomonas aeruginosa strain PAO1, also from the Miami University Department of Microbiology stock collection, was grown at 37 °C with shaking in LB broth.
Preparation of cell lysates and protein analysis
Fifty- mL cultures of mycoplasma cells were collected by centrifugation at 20 000 × g for 20 min and washed three times with cold phosphate-buffered saline (PBS). Cells were resuspended in 1 mL cold PBS containing 1% sodium dodecyl sulfate using a 25-gauge syringe and incubated at 37 °C for 30 min. Cell lysates were stored at −80 °C. Protein concentration in cell lysates was determined using bicinchoninic acid assays (Pierce Biotechnology Inc.).
Catalase enzyme activity
Catalase activity was measured in whole cell lysates of M. iowae using the Amplex Red Catalase Assay kit (Invitrogen). Catalase activity was normalized to total protein concentration in cell lysate samples. Statistical significance of results was calculated using unpaired Student’s T-test. Results represent two biological replicates from each condition with four technical replicates each.
H2O2 assays
Methods were adapted from Hames et al. [36]. For S. pneumoniae assays, colonies grown on BHI agar plates were picked and grown overnight in 5 mL of BHI broth at 37 °C without shaking. Cultures were diluted 1:100 in pre-warmed BHI broth and grown to mid-log phase at 37 °C without shaking (OD620 = 0.2-0.3). Cells were collected by centrifugation at 10 000 × g for 6 min and washed three times in cold HNM buffer (67.6 mM HEPES, pH 7.3, 140 mM NaCl, and 7 mM MgCl2. H2O2 levels were measured using colorimetric test strips (EM Quant, range 0.5-25 mg L−1). Four biological replicates were examined. Statistical significance was calculated using unpaired Student’s T-test.
To determine H2O2 production by mycoplasmas alone, 50-mL cultures of mycoplasma cells were grown to mid-log phase. Cells were collected by centrifugation at 20 000 × g for 20 min and washed three times in cold HNM buffer. Following resuspension in the same buffer to an OD550 = 1.0, aliquots of 1 mL were added to 24-well plates with 500 μM or 1 mM sucrose and incubated at room temperature for 24 h. H2O2 levels were measured and statistical analysis was performed as described above. Two biological replicates, each with 2 technical replicates, were examined.
To determine H2O2 production by S. pneumoniae when using mycoplasma cells as a source of carbohydrates, both bacteria were grown independently and collected as described above. One-mL aliquots were placed in 24-well plates that contained S. pneumoniae at an OD620 = 0.05 and mycoplasmas at an OD550 = 1.0 with no sucrose. Samples were incubated at room temperature for 24 h. H2O2 levels were measured and statistical analysis was performed as described above. Three biological replicates were examined.
C. elegans growth conditions
All assays were performed with C. elegans strain N2 (Bristol). Nematodes were cultured using standard practices [37]. Briefly, worms were cultured on nematode growth media plates seeded with E. coli OP50 as a food source at room temperature on the benchtop.
C. elegans survival assays
Plates containing large, gravid nematodes were treated with hypochlorite solution to obtain sterile eggs using standard procedures [37]. Eggs were hatched overnight in 10 mL of M9 buffer with gentle shaking to obtain L1 larvae. L1 larvae were washed with M9 buffer and aliquotted into 24-well plate wells. The number of live larvae per well (indicated by movement) was counted prior to the addition of samples to a final volume of 1 mL. Plates were incubated at room temperature for the designated time, at which time live nematodes were counted again to measure survival. They were considered dead if no movement was observed in response to shaking or tapping the plate. Following the 24-h incubation period, H2O2 levels were also recorded with the use of colorimetric test strips as described above. For long-term assays, plates were wrapped in parafilm to prevent evaporation of liquid during the extended incubation time required for the experiment. Long-term assay results represent two biological replicates from each condition with six technical replicates each. Statistical significance of results was calculated using unpaired Student’s T-test, with p < 0.05 being regarded as significant.
For mycoplasma samples, cells were collected and washed as described for H2O2 assays. M. iowae cells tested alone were resuspended to various OD550 values and incubated with C. elegans larvae in the presence of the indicated concentrations of H2O2. Assays examining protection from abiotic H2O2 were performed with mycoplasma cells resuspended to an OD550 = 1.0 in the presence of 8 mg L−1 H2O2. Three biological replicates, each with 3–4 technical replicates, were examined.
For assays with S. pneumoniae, bacteria were grown and harvested as described for H2O2 assays. Samples were added to C. elegans larvae with S. pneumoniae resuspended to OD620 = 0.05 (approximately 1.1 × 107 CFUs), mycoplasmas resuspended to OD550 = 1.0 (approximately 1.2 × 109 CFUs for M. iowae), and 500 μM sucrose. Three biological replicates, each with 2 technical replicates, were examined.
For assays with P. aeruginosa, colonies grown on LB agar plates were picked and grown overnight in 5 mL of LB broth at 37 °C in a shaking incubator. Cultures were diluted 1:100 in pre-warmed LB broth and grown to mid-log phase at 37 °C and 200 rpm (OD600 = 0.4). Cells were pelleted by centrifugation at 10 000 × g for 10 min and washed three time with cold buffer containing 20 mM L-glutamate, 5 mM K2HPO4, 5 mM NaH2PO4, 2 mM MgSO4 · 7H2O, 0.02 mM FeCl3, 12.5 mM glycine, and 50 mM Tris, pH 7.5 [38]. P. aeruginosa samples tested alone with worms were resuspended to OD650 = 0.1, 0.05, and 0.01 in the same buffer. For assays performed in combination with mycoplasmas, P. aeruginosa was used at OD650 = 0.1 (approximately 1.1 × 108 CFUs). Mycoplasmas were washed and resuspended in the same buffer as P. aeruginosa and used at an OD550 = 1.0. Three biological replicates, each with 2–4 technical replicates, were examined.
Sequence analysis
Predicted amino acid sequences for M. iowae CARDS1 and CARDS2 [13] and M. pneumoniae CARDS toxin [39] were aligned to one another with BLAST.
RNA isolation and quantification
RNA was isolated from M. iowae cells using TRI reagent (Sigma). Briefly, cells were collected by centrifugation at 20 000 × g for 20 min. Following resuspension of cell pellets in TRI reagent, RNA was extracted with chloroform, pelleted with isopropanol, and washed with 75% ethanol. RNA was resuspended in 100–300 μL DEPC-treated water and stored at −20 °C. To eliminate DNA contamination, samples were treated with DNase I (Invitrogen or QIAGEN) according to the manufacturer’s instructions. RNA was then cleaned up using the RNeasy Mini Kit (QIAGEN). Elimination of DNA contamination was confirmed by PCR with glpF primers (Table 1). RNA quality was determined by analysis on an RNA Pico Chip (Agilent Technologies) using a Bioanalyzer 2100 (Agilent Technologies).
Reverse transcription (RT)
RT was carried out with the Verso cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions. One hundred ng of RNA was used as starting material and random hexamers were used as primers in reactions with a final volume of 20 μL. Reactions were incubated at 70 °C for 5 min, 42 °C for 30 min, 95 °C for 2 min, and then chilled on ice. cDNA synthesis was confirmed by PCR with glpF primers (Table 1). Controls were performed to exclude the possibility of DNA contamination in which the same reactions were performed with water substituted for Verso Enzyme Mix and RT Enhancer. Control reactions were treated and stored identically to other samples.
Genomic DNA isolation and standard generation
Genomic DNA was isolated from a 50-mL culture using the QIAamp® DNA Mini Kit (QIAGEN). To improve quantification, plasmids were constructed that contained a copy of each gene target. Plasmids were constructed from PCR products amplified from M. iowae genomic DNA using gene-specific primers (Table 2) that were ligated into pCR®2.1 (Invitrogen) and transformed into competent E. coli DH5α. Plasmids were isolated from clones with the Zyppy™ Plasmid Miniprep Kit (Zymo Research). Insertion was confirmed with digestion by EcoRI and sequence was confirmed using vector-specific primers M13F and M13R (Table 2). Gene copy numbers were calculated using the concentration of each plasmid assuming 1.096 × 10−12 g bp−1 [40]. Standard curves were generated from ten-fold dilutions of DNA with known copy numbers and were analyzed by qPCR in triplicate.
RT-quantitative PCR (qPCR)
Each RT-qPCR reaction was done in at least triplicate using two biological replicates. Reactions were performed with PerfeCTa® SYBR® Green Supermix (Quanta Biosciences), 1 μL of cDNA or DNA template (5 ng), and 300 nM gene-specific primers (Table 1) in a final volume of 25 μL. Amplification conditions were 5 min at 95 °C for 3 min followed by 40 cycles of 10 s at 95 °C and 45 s at 52 °C. To determine the melting temperature and PCR product specificity, a melting curve was obtained after every run by heating from 50 °C (2 °C below Ta) to 95 °C. Primer specificity was determined by melting curve analysis and agarose gel electrophoresis of PCR products. Controls with no template or reverse transcriptase were included for each sample during each run. Runs were performed using the CFX Connect (Bio-Rad) and analysis was performed using Bio-Rad CFX Manager 3.0 software (Bio-Rad). The 16S rRNA gene was used for normalization. Statistical analysis of results was calculated using unpaired Student’s T-test. MIQE guidelines were followed for performing and reporting experiments [41].
Results
Catalase confers host protection from peroxigenic bacteria
M. iowae has an active catalase protein, enabling reduction of H2O2 by this organism [13]. C. elegans is a well-established model for the study of H2O2-mediated bacterial pathogenicity [1,2,42], and has recently been adapted for use with mycoplasma cells [13]. M. gallisepticum naturally lacks catalase activity, but M. gallisepticum katE transformant 56A, which produces M. iowae catalase, produces less H2O2 and kills fewer C. elegans larvae in the presence of the peroxigenic molecule glycerol than wild-type M. gallisepticum Rlow, suggesting a role for catalase in protection from killing [13]. Incubation with M. iowae at OD550 = 0.01 increased survival of C. elegans from 30% in the absence of bacteria to 50% (not shown), a reasonable set of conditions for subsequent assays.
In the vertebrate host, bacteria can come into contact with multiple sources of H2O2, including host immune defense mechanisms and other microbial pathogens [1,2,43]. To mimic more closely conditions of H2O2 exposure in the host and test whether M. iowae was protective under these conditions, C. elegans survival assays were performed with the use of H2O2 produced continuously by a biotic source. S. pneumoniae is a bacterial pathogen that produces H2O2 from a variety of substrates including sucrose [44-46], a carbohydrate that M. iowae and M. gallisepticum are unable to metabolize [17]. When S. pneumoniae resuspended to OD620 = 0.05 was incubated with 500 μM sucrose at room temperature for 24 h, H2O2 accumulated rapidly, reaching maximum levels after 8 h of incubation, at which point H2O2 levels remained constant for the duration of the 24 h period (Figure 1). M. iowae or wild-type M. gallisepticum resuspended to OD550 = 1.0 and incubated under the same conditions produced less than 1 mg L−1 H2O2, respectively, and the same outcome occurred when mycoplasma cells were co-incubated with S. pneumoniae in the absence of sucrose (data not shown; see Figure 2B). Based upon these results, C. elegans survival assays were performed with S. pneumoniae at OD620 = 0.05, mycoplasmas at OD550 = 1.0, and 500 μM sucrose.

H 2 O 2 accumulation by S. pneumoniae . Accumulation of H2O2 over time by S. pneumoniae at an OD620 of 0.05 in the presence of 500 μM sucrose. Error bars, SD.

Host protection from S. pneumoniae -produced H 2 O 2 by catalase-positive mycoplasmas. Survival of C. elegans (A) and amount of H2O2 remaining (B) at 24 h upon incubation with S. pneumoniae at OD620 = 0.05 in the presence of 500 μM sucrose. Experiments were performed in triplicate. Results shown are from one representative experiment (B). Error bars, SD. *, statistically significantly different from S. pneumoniae alone with 500 μM sucrose (p < 0.05).
When incubated with S. pneumoniae alone, approximately 6% of C. elegans larvae survived for 24 h in the presence of sucrose (Figure 2A), correlating with an accumulation of 8 mg L−1 H2O2 (Figure 2B). In the absence of sucrose, very little H2O2 accumulated (Figure 2B) and almost all C. elegans larvae survived upon incubation with S. pneumoniae (Figure 2A), demonstrating that under these conditions the toxicity was associated primarily with H2O2. Inclusion of wild-type M. gallisepticum with S. pneumoniae and sucrose did not significantly alter the amount of C. elegans survival as compared to S. pneumoniae alone with sucrose (Figure 2A). However, H2O2 accumulation decreased to 4.5 mg L−1 (Figure 2B), possibly because of loss of H2O2 upon reaction with the high number of M. gallisepticum cells in suspension. On the other hand, inclusion of catalase-producing M. iowae or M. gallisepticum transformant 56A resulted in significantly increased amounts of C. elegans survival (Figure 2A) and no detectable H2O2 (Figure 2B). Taken together, these results suggest that catalase enables M. iowae to offer protection from H2O2 being continually produced by other organisms.
Reasoning that protection by M. iowae is due to catalase and therefore might be specific to H2O2-mediated stress, C. elegans survival assays were repeated with a non-H2O2 producing pathogen. P. aeruginosa PAO1 produces the toxic molecule HCN [47], to which C. elegans larvae are highly susceptible; incubation with this bacterial strain typically results in complete killing of larvae [48,49]. At OD650 = 0.1, P. aeruginosa caused almost complete killing of larvae at 24 h (data not shown). Under these conditions, co-incubation of C. elegans with P. aeruginosa and any of the mycoplasma strains resulted in no difference in the survival of larvae at 24 h (Figure 3).

Absence of host protection from P. aeruginosa by catalase-positive mycoplasmas. Survival of C. elegans at 24 h upon co-incubation with P. aeruginosa at OD650 = 0.1. Experiments were performed in triplicate. Error bars, SD. *, no statistically significant difference from P. aeruginosa alone (p > 0.05).
O2-dependent changes in catalase activity and long-term survival of C. elegans
M. iowae lives in both aerobic and reduced-oxygen environments in its natural host, but its pathogenicity appears to be limited or non-existent in the gut, where it accumulates but where O2 is low. Therefore, we explored whether M. iowae grown under low O2 conditions might be less toxic to host cells. Interestingly, catalase activity of M. iowae significantly decreased by 24% (p < 0.05) in low O2 (Figure 4), suggesting that O2 exposure has an impact on catalase activity in M. iowae. M. gallisepticum 56A, which offered protection comparable to that of M. iowae [13] had 63% less catalase activity (p < 0.05) than M. iowae under atmospheric conditions (Figure 4). Therefore the decreased catalase activity in M. iowae cells grown in 1% O2 is unlikely to result in a significant decrease in protection.

Catalase activity in M. iowae and M. gallisepticum transformed with katE . Extracts from cells grown under atmospheric conditions or in 1% O2 were assayed for catalase activity as described in Material and methods. Error bars, SD.
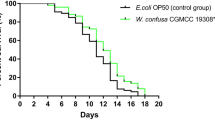
Whereas diffusible molecules such as H2O2 are responsible for damage to C. elegans within the first 48 h of incubation, the impact of toxins becomes apparent after 48 h as bacteria are able to establish an infection within the nematode gut [2]. To examine differences in the ability of M. iowae to cause damage to its host, long-term C. elegans assays were performed with bacterial cells grown under different O2 concentrations. It is important to consider that the C. elegans assay conditions are carried out in a buffer, preventing mycoplasma growth, and at room temperature, which is well below the temperature range at which M. iowae can grow. It is therefore very likely that by the end of the incubation with C. elegans, M. iowae cells have no more catalase or putative toxins than they had at the beginning. A repeated measures ANOVA revealed significant differences between the two groups [F(1,17) = 27.39, p < 0.05]. Cells grown aerobically caused nematodes to die much more quickly, with 50% survival achieved after approximately 5 days as compared to approximately 9 days with cells grown in the presence of 1% O2, and death of all larvae by 15 days with aerobically-grown M. iowae cells as opposed to 20 days with low O2-grown mycoplasmas (Figure 5). These data suggest that M. iowae cells are less harmful in low-O2 environments.

Long-term survival of C. elegans when fed M. iowae grown aerobically or in the presence of 1% O 2 . Error bars, SD. *, statistically significantly different from aerobically-grown M. iowae (p < 0.05).
O2 regulation of potential M. iowae virulence genes
Because it is unlikely that a relatively small O2-dependent decrease in M. iowae catalase activity was related to increased survival of C. elegans, we hypothesized that M. iowae genes more directly linked to virulence experienced O2-dependent regulation. Candidate virulence-associated genes were selected for analysis of regulation of expression by O2. Transcription of the potential virulence factors glpF and cards1 was examined as well as katE and another antioxidant enzyme, sodA. glpF, which allows uptake of glycerol for production of H2O2, was chosen as a representative of the presumptive glycerol catabolism operon that includes glpK and glpO. Additionally, M. iowae serovar K has two copies of genes homologous to M. pneumoniae CARDS toxin [13]. We examined the predicted amino acid sequences for the proteins encoded by these genes. There are three common motifs shared by many ADP-ribosylating toxins: a conserved arginine for NAD+ binding, a serine-threonine-serine motif to maintain structural integrity of the NAD+ binding site, and a catalytic glutamate [20]. These amino acids are all present in M. pneumoniae CARDS toxin, which has ADP-ribosyltransferase activity [20]. When both M. iowae CARDS toxin homologs were examined (named CARDS1 and CARDS2), some deviations were observed (Figure 6). CARDS2 has a conservative change of the second serine residue in the STS motif to a threonine. CARDS1 contains multiple changes, with substitutions of like charge at the conserved arginine and glutamate residues and less conservative substitutions for two of the three amino acids in the serine-threonine-serine motif. These M. iowae homologues have 28% and 25% identity, respectively, to the M. pneumoniae CARDS toxin sequence. Each M. iowae gene also has 99% identity to its respective homolog in M. iowae serovar I strain 695 [50]. cards1 was chosen despite the greater degree of disparity from M. pneumoniae CARDS toxin since only 20 bases separate cards1 and cards2, likely making both genes transcriptionally linked.

Alignment of CARDS toxin-like sequences from M. pneumoniae and M. iowae . Residues generally conserved in other ADP-ribosylating toxins are bolded and enlarged.
To examine transcript levels in M. iowae cells grown under different O2 concentrations, qPCR was performed (Table 3). Expression of 16S rRNA was used as a control. The putative toxin gene cards1 and the catalase gene katE underwent statistically significant down-regulation in M. iowae cells grown in the presence of 1% O2, with decreases of 4.9- and 5.4-fold, respectively (Table 3). In contrast, glpF and sodA did not exhibit significant changes in expression in response to O2 availability. Taken together, these data suggest that M. iowae undergoes differential regulation of select genes in response to low O2 conditions. The reduction in cards1 expression is consistent with reduced pathogenicity in the gut.
Discussion
Both genome sequences currently available for M. iowae [13,50] reveal the presence of a gene for catalase, an H2O2-degrading enzyme absent from all other published mycoplasma genomes, which produces an active protein [13]. Bacteria can encounter exposure to H2O2 from a variety of different sources, including other bacterial pathogens as well as the host immune response [1,2,43]. Bacteria bearing enzymes that detoxify ROS, including catalase, have been demonstrated to provide benefit to animal hosts in the context of probiotics [9]. Our results support a model wherein protection by M. iowae is specific to catalase-mediated reduction of H2O2 stress, but other mechanisms are also possible. Catalase is specifically implicated in this protection because M. gallisepticum, which does not normally have catalase activity, becomes protective upon transformation with M. iowae katE [13], despite 2.7-fold less catalase activity in the M. gallisepticum transformant 56A as compared with M. iowae.
Host-associated bacteria live in the presence of a consortium of other microorganisms, some of which may be H2O2 producers. We used S. pneumoniae as a model pathogen that produces H2O2 as an important virulence factor that not only can cause damage to host cells but has also been studied in the context of C. elegans [51-53]. Furthermore, S. pneumoniae can inhibit the growth of other bacterial competitors by virtue of its H2O2 production [54]. The ability of M. iowae to protect against H2O2-mediated damage caused by S. pneumoniae, albeit not against other kinds of damage such as that caused by P. aeruginosa, suggests that M. iowae could be beneficial to its host provided it is not also producing molecules harmful to its host.
M. iowae causes damage at multiple sites throughout the body of its natural poultry hosts, including legs, joints, air sac, and feathers [14-16]. However, despite many accounts of detection in and isolation from the gut, no clear-cut reports of disease at this site due to M. iowae have been documented [23-26]. Because low O2 concentration is a hallmark of the gut environment that distinguishes it from other sites at which M. iowae causes damage and disease [27], we examined the impact of growth in 1% O2 on M. iowae with regard to activities that might be associated with both disease and protection. Our finding that M. iowae has an ability to prolong survival when fed to C. elegans larvae following growth in 1% O2 supports the notion that M. iowae is not harmful in the gut and might in fact be a beneficial component of the gut microflora. Significantly, at the transcriptional level, this environment causes a significant down-regulation in not only the gene encoding catalase, but also a gene encoding a homolog of CARDS toxin, a causative agent of host damage by M. pneumoniae [20,55] and M. penetrans [56]. These data suggest that M. iowae undergoes differential regulation of select genes in response to growth in low O2 environments.
The down-regulation of katE in 1% O2 could be a response to a decreased threat of damage from ROS. Despite this reduction, it is important to consider that the catalase activity in low O2-grown M. iowae cells is still 2.1-fold greater than the amount present in M. gallisepticum transformant 56A, which itself still confers significant protection of C. elegans exposed to H2O2 [13]. Therefore, the reduction in M. iowae catalase activity in a gut-like environment is not necessarily associated with decreased protection. It is possible that production of catalase facilitates colonization of the gut by M. iowae, in parallel with Campylobacter jejuni, which expresses an active catalase that is important for colonization of the poultry intestinal tract [57]. The degree of down-regulation of katE at the transcription level does not closely match the change in catalase activity in whole cell lysates, with a smaller decrease observed in the latter. The high level of catalase activity might not decrease linearly as the concentration of catalase decreases.
The down-regulation of cards1, a homolog of CARDS toxin, which is likely accompanied by similar down-regulation of the very closely linked cards2, may be associated with decreased pathogenicity of M. iowae in the gastrointestinal tract. The amount of CARDS toxin produced correlates positively with the amount of host damage caused by M. pneumoniae [55]. Differential expression of toxins in different environments is well-established in diverse bacteria, including clostridia [58], Vibrio cholerae [59], and cyanobacteria [60]. It is conceivable that the environment of the intestine is sufficiently nutritious for M. iowae that severe damage to host cells is unwarranted, favoring a strategy in which virulence factors may be used principally in environments in which nutrients are less readily available. In this model, low O2 provides a cue to M. iowae that it is in the gut, where it can conserve energy by down-regulating virulence genes, including those encoding the CARDS toxin-like proteins. The similar down-regulation of both cards1 and katE transcript levels when grown in the presence of 1% O2, on the order of 5-fold, raises the possibility that a common regulatory mechanism may be acting on both genes, but further work is necessary to elucidate such a mechanism.
Abbreviations
- ROS:
-
Reactive oxygen species
- IBD:
-
Inflammatory bowel disease
- RT-qPCR:
-
Reverse transcription-quantitative polymerase chain reaction
- BHI:
-
Brain heart infusion
- PBS:
-
Phosphate-buffered saline.
References
Bolm M, Jansen WTM, Schnabel R, Chhatwal G (2004) Hydrogen peroxide-mediated killing of Caenorhabditis elegans: a common feature of different streptococcal species. Infect Immun 72:1192–1194
Moy TI, Mylonakis E, Calderwood SB, Ausubel FM (2004) Cytotoxicity of hydrogen peroxide produced by Enterococcus faecium. Infect Immun 72:4512–4520
Whelan K, Quigley EMM (2013) Probiotics in the management of irritable bowel syndrome and inflammatory bowel disease. Curr Opin Gastroenterol 29:184–189
Simmonds NJ, Allen RE, Stevens TR, Van Someren RN, Blake DR, Rampton DS (1992) Chemiluminescence assay of musical reactive oxygen metabolites in inflammatory bowel disease. Gastroenterology 103:186–196
Sedghi S, Fields JZ, Klamut M, Urban G, Durkin M, Winship D, Fretland D, Olyaee M, Keshavarzian A (1993) Increased production of luminol and chemiluminescence by the inflamed colonic mucosa in patients with ulcerative colitis. Gut 34:1191–1197
Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Weissman GS, Katz S, Floyd RA, McKinley MJ, Fisher SE, Mullin GE (1996) Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci 41:2078–2086
Keshavarzian A, Banan A, Farhadi A, Komanduri S, Mutlu E, Zhang Y, Fields JZ (2003) Increases in free radicals and cytoskeletal protein oxidation and nitration in the colon of patients with inflammatory bowel disease. Gut 52:720–728
Switala J, Loewen PC (2002) Diversity of properties among catalases. Arch Biochem Biophys 401:145–154
Zhu H, Li YR (2012) Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med 237:474–480
LeBlanc JG, del Carmen S, Miyoshi A, Azevedo V, Sesma F, Langella P, Bermúdez-Humarán LG, Watterlot L, Perdigon G, de Moreno de LeBlanc A (2011) Use of superoxide dismutase and catalase producing lactic acid bacteria in TNBS induced Crohn’s disease in mice. J Biotechnol 151:287–293
Rochat T, Bermúdez-Humarán L, Gratadoux J-J, Fourage C, Hoebler C, Corthier G, Langella P (2007) Anti-inflammatory effects of Lactobacillus casei BL23 producing or not a manganese-dependent catalase on DSS-induced colitis in mice. Microb Cell Fact 6:22
Al-Ankari AR, Bradbury JM (1996) Mycoplasma iowae: a review. Avian Pathol 25:205–229
Pritchard RE, Prassinos AJ, Osborne JD, Raviv Z, Balish MF (2014) Reduction of hydrogen peroxide accumulation and toxicity by a catalase from Mycoplasma iowae. PLoS One 9:e105188
Bradbury JM, Ideris A, Oo TT (1988) Mycoplasma iowae infection in young turkeys. Avian Pathol 17:149–171
Trampel DW, Goll F, Jr (1994) Outbreak of Mycoplasma iowae infection in commercial turkey poults. Avian Dis 38:905–909
Ley DH, Marusak RA, Vivas EJ, Barnes HJ, Fletcher OJ (2010) Mycoplasma iowae associated with chondrodystrophy in commercial turkeys. Avian Pathol 39:87–93
Taylor RR, Mohan K, Miles RJ (1996) Diversity of energy-yielding substrates and metabolism in avian mycoplasmas. Vet Microbiol 51:291–304
Papazisi L, Gorton TS, Kutish G, Markham PF, Browning GF, Nguyen DK, Swartzell S, Madan A, Mahairas G, Geary SJ (2003) The complete genome sequence of the avian pathogen Mycoplasma gallisepticum strain Rlow. Microbiology 149:2307–2316
Jurkovic DA, Newman JT, Balish MF (2012) Conserved terminal organelle morphology and function in Mycoplasma penetrans and Mycoplasma iowae. J Bacteriol 194:2877–2883
Kannan TR, Baseman JB (2006) ADP-ribosylating and vacuolating cytotoxin of Mycoplasma pneumoniae represents unique virulence determinant among bacterial pathogens. Proc Natl Acad Sci U S A 103:6724–6729
Hardy RD, Coalson JJ, Peters J, Chaparro A, Techasaensiri C, Cantwell AM, Kannan TR, Baseman JB, Dube PH (2009) Analysis of pulmonary inflammation and function in the mouse and baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS One 4:e7562
Medina JL, Coalson JJ, Brooks EG, Winter VT, Chaparro A, Principe MF, Kannan TR, Baseman JB, Dube PH (2012) Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am J Respir Cell Mol Biol 46:815–822
Jordan FTW, Amin MM (1980) A survey of mycoplasma infections in domestic poultry. Res Vet Sci 28:96–100
Jordan FTW (1983) Recovery and identification of avian mycoplasmas. In: Tully JG, Razin S (ed) Methods in Mycoplasmology. Diagnostic Mycoplasmology, vol II. Academic, New York, pp 69–79
Mirsalimi SM, Rosendal S, Julian RJ (1989) Colonization of the intestine of turkey embryos exposed to Mycoplasma iowae. Avian Dis 33:310–315
Shah-Majid M, Rosendal S (1986) Oral challenge of turkey poults with Mycoplasma iowae. Avian Dis 31:365–369
He G, Shankar RA, Chzhan M, Samouilov A, Kuppusamy P, Zweier JL (1999) Noninvasive measurement of anatomic structure and intraluminal oxygenation in the gastrointestinal tract of living mice with spatial and spectral ER imaging. Proc Natl Acad Sci U S A 96:4586–4591
Wangensteen OD, Rahn H (1970–1971) Respiratory gas exchange by the avian embryo. Respir Physiol 11:31–45
Halbedel S, Eiler H, Jonas B, Busse J, Hecker M, Engelmann S, Stülke J (2007) Transcription in Mycoplasma pneumoniae: analysis of the promoters of the ackA and ldh genes. J Mol Biol 371:596–607
Hallamaa KM, Tang S-L, Ficorilli N, Browning GF (2008) Differential expression of lipoprotein genes in Mycoplasma pneumoniae after contact with human lung epithelial cells, and under oxidative and acidic stress. BMC Microbiol 8:124
Madsen ML, Nettleton D, Thacker EL, Edwards R, Minion FC (2006) Transcriptional profiling of Mycoplasma hyopneumoniae during heat shock using microarrays. Infect Immun 74:160–166
Madsen ML, Nettleton D, Thacker EL, Minion FC (2006) Transcriptional profiling of Mycoplasma hyopneumoniae during iron depletion using microarrays. Microbiology 152:937–944
Schafer ER, Oneal MJ, Madsen ML, Minion FC (2007) Global transcriptional analysis of Mycoplasma hyopneumoniae following exposure to hydrogen peroxide. Microbiology 153:3785–3790
Madsen ML, Puttamreddy S, Thacker EL, Carruthers MD, Minion FC (2008) Transcriptome changes in Mycoplasma hyopneumoniae during infection. Infect Immun 76:658–663
Tully JG, Rose DL, Whitcomb RF, Wenzel RP (1979) Enhanced isolation of Mycoplasma pneumoniae from throat washings with a newly-modified culture medium. J Infect Dis 139:478–482
Hames C, Halbedel S, Hoppert M, Frey J, Stülke J (2009) Glycerol metabolism is important for cytotoxicity of Mycoplasma pneumoniae. J Bacteriol 191:747–753
Lewis JA, Fleming JT (1995) Basic culture methods. In: Epstein HF, Shakes DC (ed) Methods in Cell Biology. Caenorhabditis elegans: Modern Biological Analysis of an Organism, vol 48. Academic, Boston, pp 3–29
Castric PA (1983) Hydrogen cyanide production by Pseudomonas aeruginosa at reduced oxygen levels. Can J Microbiol 29:1344–1349
Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li B-C, Herrmann R (1996) Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res 24:4420–4449
Kong W, Nakatsu CH (2010) Optimization of RNA extraction for PCR quantification of aromatic compound degradation genes. Appl Environ Microbiol 76:1282–1284
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubist M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Jansen WTM, Bolm M, Balling R, Chhatwal GS, Schnabel R (2002) Hydrogen peroxide-mediated killing of Caenorhabditis elegans by Streptococcus pyogenes. Infect Immun 70:5202–5207
Slauch JM (2011) How does the oxidative burst of macrophages kill bacteria? Still an open ended question. Mol Microbiol 80:580–583
Avery OT, Neill JM (1924) Studies on oxidation and reduction by pneumococcus. II. The production of peroxide by sterile extracts of pneumococcus. J Exp Med 39:357–366
Spellerberg B, Cundell DR, Sandros J, Pearce BJ, Idänpään-Heikkilä I, Rosenow C, Masure HR (1996) Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol Microbiol 19:803–813
Iyer R, Camilli A (2007) Sucrose metabolism contributes to in vivo fitness of Streptococcus pneumoniae. Mol Microbiol 66:1–13
Castric PA (1974) Hydrogen cyanide, a secondary metabolite of Pseudomonas aeruginosa. Can J Microbiol 21:613–618
Darby C, Cosma CL, Thomas JH, Manoil C (1999) Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96:15202–15207
Gallagher LA, Manoil C (2001) Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183:6207–6214
Wei S, Guo Z, Li T, Zhang T, Li X, Zhou Z, Li Z, Lui M, Luo R, Bi D, Chen H, Zhou R, Jin H (2012) Genome sequence of Mycoplasma iowae strain 695, an unusual pathogen causing deaths in turkeys. J Bacteriol 194:547–548
Duane PG, Rubins JB, Weisel HR, Janoff EN (1993) Identification of hydrogen peroxide as a Streptococcus pneumoniae toxin for rat alveolar epithelial cells. Infect Immun 61:4392–4397
Hirst RA, Sikand KS, Rutman A, Mitchell TJ, Andrew PW, O’Callaghan C (2000) Relative roles of pneumolysin and hydrogen peroxide from Streptococcus pneumoniae in inhibition of ependymal ciliary beat frequency. Infect Immun 68:1557–1562
Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE, Calderwood SB, Ausubel FM (2001) A simple model host for identifying Gram-positive virulence factors. Proc Natl Acad Sci U S A 98:10892–10897
Pericone CD, Overweg K, Hermans PW, Weiser JN (2000) Inhibitory and bactericidal effects of hydrogen peroxide production by Streptococcus pneumoniae on other inhabitants of the upper respiratory tract. Infect Immun 68:3990–3997
Techasaensiri C, Tagliabue C, Cagle M, Iranpour P, Katz K, Kannan TR, Coalson JJ, Baseman JB, Hardy RD (2010) Variation in colonization, ADP-ribosylating and vacuolating cytotoxin, and pulmonary disease severity among Mycoplasma pneumoniae strains. Am J Respir Crit Care Med 182:797–804
Johnson C, Kannan TR, Baseman JB (2009) Characterization of a unique ADP-ribosyltransferase of Mycoplasma penetrans. Infect Immun 77:4362–4370
Flint A, Sun Y-Q, Stintzi A (2012) Cj1386 is an ankyrin-containing protein involved in heme trafficking to catalase in Campylobacter jejuni. J Bacteriol 194:334–345
Carter GP, Cheung JK, Larcombe S, Lyras D (2014) Regulation of toxin production in the pathogenic clostridia. Mol Microbiol 91:221–231
Matson JS, Withey JH, DiRita VJ (2007) Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun 75:5542–5549
Neilan BA, Pearson LA, Muenchhoff J, Moffitt MC, Dittmann E (2013) Environmental conditions that influence toxin biosynthesis in cyanobacteria. Environ Microbiol 15:1239–1253
Acknowledgments
This work was supported by the National Institutes of Health (Public Health Service grant R15 AI073994) to MFB and by an award from the Sigma Xi foundation to REP. We thank A. Bollmann and members of her research group for assistance with low-O2 growth of M. iowae. We thank A. Kiss of the Miami University Center for Bioinformatics and Functional Genomics for training in RNA quantification. We thank C.N. Bialorucki for assistance with quantification of the relationship among OD550 values, colony-forming units, and protein concentrations for mycoplasma species. This work was done in partial fulfillment of REP’s doctoral dissertation requirements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
REP carried out all the experimentation, participated in the design of the study, participated in the analysis of the results, and drafted the manuscript. MFB conceived of the study, participated in the design of the study, the analysis of the results, and the writing of the manuscript. Both authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pritchard, R.E., Balish, M.F. Mycoplasma iowae: relationships among oxygen, virulence, and protection from oxidative stress. Vet Res 46, 36 (2015). https://doi.org/10.1186/s13567-015-0170-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-015-0170-7