
Abstract
Background
We have recently shown that treatment of mice with the neutral endopeptidase (NEP) inhibitor phosphoramidon (PA) improves the bioavailability and tumor uptake of biodegradable radiopeptides. For the truncated gastrin radiotracer [111In-DOTA]MG11 ([(DOTA)DGlu10]gastrin(10–17)), this method led to impressively high tumor-to-kidney ratios. Translation of this concept in the clinic requires the use of certified NEP inhibitors, such as thiorphan (TO) and its orally administered prodrug racecadotril (Race). Besides NEP, angiotensin-converting enzyme (ACE) has also been implicated in the catabolism of gastrin analogs. In the present study, we first compared the effects induced by NEP inhibition (using PA, TO, or Race) and/or by ACE inhibition (using lisinopril, Lis) on the biodistribution profile of [111In-DOTA]MG11 in mice. In addition, we compared the efficacy of PA and TO at different administered doses to enhance tumor uptake.
Methods
[111In-DOTA]MG11 was coinjected with (a) vehicle, (b) PA (300 μg), (c) TO (150 μg), (d) Lis (100 μg), (e) PA (300 μg) plus Lis (100 μg), or (f) 30–40 min after intraperitoneal (ip) injection of Race (3 mg) in SCID mice bearing AR42J xenografts. In addition, [111In-DOTA]MG11 was coinjected with vehicle, or with progressively increasing amounts of PA (3, 30, or 300 μg) or TO (1.5, 15, and 150 μg) in SCID mice bearing twin A431-CCK2R(+/−) tumors. In all above cases, biodistribution was conducted at 4 h postinjection (pi).
Results
During NEP inhibition, the uptake of [111In-DOTA]MG11 in the AR42J tumors impressively increased from 1.8 ± 1.0 % ID/g (controls) to 15.3 ± 4.7 % ID/g (PA) and 12.3 ± 3.6 % ID/g (TO), while with Race tumor values reached 6.8 ± 2.8 % ID/g. Conversely, Lis had no effect on tumor uptake and no additive effect when coinjected with PA. During the dose dependence study in mice, PA turned out to be more efficacious in enhancing tumor uptake of [111In-DOTA]MG11 in the CCK2R-positive tumors compared to equimolar amounts of TO. In all cases, renal accumulation remained low, resulting in notable increases of tumor-to-kidney ratios.
Conclusions
This study has confirmed NEP as the predominant degrading enzyme of [111In-DOTA]MG11 and ruled out the involvement of ACE in the in vivo catabolism of the radiotracer. NEP inhibition with the clinically tested NEP inhibitors TO and Race resulted in significant enhancement of tumor-to-kidney ratios vs. controls. However, compared with PA, TO and its prodrug Race induced less potent increases of tumor uptake, highlighting the significance of inhibitor type, administration route, and dose for implementing a first proof-of-principle study in human.
Similar content being viewed by others

Background
Cholecystokinin subtype 2 receptors (CCK2Rs) have attracted much attention in nuclear oncology due to their overexpression in various human tumors, including medullary thyroid carcinoma (MTC), small cell lung cancer, astrocytomas, and stromal ovarian cancers [1–4]. Accordingly, peptide radioligands based on gastrin or on cholecystokinin (CCK) have been proposed for diagnostic imaging and radionuclide therapy of CCK2R-positive tumors [5, 6].
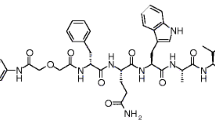
Human gastrin I is a C-terminally amidated heptadecapeptide characterized by a penta-Glu repeat in positions 6 through 10 (pGlu-Gly-Pro-Trp-Leu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2; Fig. 1) [7]. This highly polar and negatively charged amino acid sequence determines important biological properties of gastrin and its radiolabeled analogs. Thus, Glu6–10-containing radioligands show high receptor affinity and are metabolically robust. As a result, they efficiently localize in CCK2R-rich lesions in rodents and in patients, but their application in the clinic has been hitherto restricted by their unfavorably high renal retention. Conversely, truncated desGlu6–10 gastrin analogs, as for example [111In-DOTA]MG11 ([(DOTA)DGlu10]gastrin(10–17); Fig. 1), display favorably low renal uptake but also severely impaired ability to target CCK2R-positive tumors in vivo, owing to their poor metabolic stability [6, 8].

Human gastrin I; [111In-DOTA]MG11; the NEP inhibitors PA, TO, and its prodrug Race; and the ACE inhibitor Lis
According to previous reports, proteases that might be implicated in the degradation of CCK and gastrin analogs include (a) aminopeptidases [9], (b) angiotensin-converting enzyme (ACE, EC 3.4.15.1) [10], and (c) neutral endopeptidase (NEP, neprilysin, EC 3.4.24.11) [11, 12]. Given that N-terminal capping is an effective means to prevent peptide degradation by aminopeptidases [9, 13], it is reasonable to assume that [111In-DOTA]MG11 with a bulky radiometal chelate attached at the N-terminus becomes aminopeptidase-resistant. The role of ACE in the degradation of CCK and different-length gastrin analogs has been previously investigated in vitro [10]. Interestingly, minigastrin (MG, gastrin(5–17)) was shown to be resistant to ACE, whereas successive shortening of the penta-Glu chain to truncated gastrin analogs with less than two Glu residues led to in vitro degradation by ACE. Accordingly, [111In-DOTA]MG11 is expected to be resistant to ACE by virtue of the polar 111In-DOTA9-DGlu10 sequence, mimicking the Glu9–10 construct of ACE-resistant gastrin(9–17). However, the role of ACE in the in vivo fate of gastrin analogs, including [111In-DOTA]MG11, has not been elucidated yet. The third protease implicated in the metabolism of gastrin is NEP, an ectoenzyme with broad substrate specificity and a wide distribution in the body. While scarcely found in plasma, NEP is abundantly expressed in most tissues, anchored on the endothelial cell surface of the vasculature compartment, and in major organs, such as kidneys, liver, lungs, and the gastrointestinal tract [14]. The involvement of NEP in the in vitro and in vivo degradation of gastrin has been well described in previous reports [11, 12].
We have recently proposed an effective strategy to improve the bioavailability and tumor uptake of biodegradable radiopeptides, involving in situ inhibition of the degrading protease(s) (e.g., NEP) by coinjection of a suitable inhibitor (e.g., the NEP inhibitor phosphoramidon, PA) [15–17]. For the truncated gastrin radiotracer [111In-DOTA]MG11, this method led to impressive in vivo stabilization, which translated into remarkably increased uptake in CCK2R-positive tumors in mice and into high tumor-to-kidney ratios [15, 18].
The above promising results have prompted further research toward clinical translation of the proposed concept in a first “proof-of-principle” study in man, using [111In-DOTA]MG11 as a paradigm. The success of this step largely relies on the identification of the peptidase(s) actually responsible for the proteolytic degradation of [111In-DOTA]MG11 after its entry in the circulation by intravenous injection. Rapid in vivo degradation deteriorates the radiotracer supply to target sites and eventually compromises its localization on tumor CCK2R-positive lesions. Therefore, we were first interested to study the potential involvement of ACE on the tumor uptake of [111In-DOTA]MG11. For this purpose, the effect of in situ ACE inhibition by lisinopril (Lis) [19] alone vs. controls was compared in mice bearing AR42J tumors. Furthermore, the combination of dual ACE/NEP inhibition after injection of a Lis and PA mixture vs. single NEP inhibition by PA was compared in the same mouse model.
Another important step toward clinical translation is the assessment of biosafety and efficacy of the NEP inhibitor intended for human application. It should be noted that so far, no extensive toxicity studies have been reported for PA. This bacterial origin and potent NEP inhibitor has been administered in human only in homeopathic amounts [16, 20]. Consequently, we decided to compare the in vivo efficacy of the clinically certified NEP inhibitor thiorphan (TO) [21, 22] vs. PA to enhance the tumor uptake of [111In-DOTA]MG11 in the same mouse model. Racecadotril (Race) is a prodrug of TO administered orally and a registered antidiarrheal drug (Fig. 1) [23, 24], which has also been included in our study. Race was administered intraperitoneally (ip) due to its poor water solubility prior to [111In-DOTA]MG11 iv injection.
In the last part of the present study, we have directly compared the efficacy of TO and PA iv-injected in equimolar and progressively increasing doses to enhance the uptake of [111In-DOTA]MG11 in a double A431-CCK2R(+/−) tumor mouse model.
Methods
Chemistry and radiochemistry
Chemicals and radionuclides
All chemicals were reagent grade and used without further purification. DOTA-MG11 (DOTA-minigastrin 11, DOTA-DGlu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) and DG2 (Demogastrin 2, N4-Gly-DGlu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) [25] were purchased from PiChem (Graz, Austria). PA (phosphoramidon disodium dehydrate, N-(α-rhamnopyranosyloxyhydroxyphosphinyl)-L-leucyl-L-tryptophan × 2Na × 2H2O) was provided by PeptaNova GmbH (Sandhausen, Germany). TO (DL-thiorphan, DL-3-mercapto-2-benzylpropanoylglycine) was a kind gift of Prof. B. Roques (Université René Descartes, Paris, France). Lis (lisinopril dehydrate, ((S)1-1-[N2-(1-carboxy-3-phenylpropyl)-lysyl-proline dehydrate, MK 521) and Race (racecadotril, (RS)-benzyl N-[3-(acetylthio)-2-benzylpropanoyl]glycinate) were purchased from Sigma-Aldrich (Fig. 1).
Indium-111 used for labeling was purchased in the form of 111InCl3 in a solution of 0.05 M HCl (0.5 mL) from Mallinckrodt Medical B.V. (Petten, the Netherlands).
Preparation of [111In-DOTA]MG11
Lyophilized DOTA-MG11 was dissolved in water to a final concentration of 1 mM, and 50 μL aliquots were stored at −20 °C. Labeling with 111In was conducted in an Eppendorf vial containing 0.1 M sodium acetate buffer pH 4.6 in the presence of excess methionine (Met) to prevent oxidation of Met15 in DOTA-MG11 [26]. Freshly prepared sodium ascorbate buffer (10 mM) was added in the vial, followed by 111InCl3 solution (37–74 MBq), Met (1000 nmol), and DOTA-MG11 (10 nmol). The mixture was left to react at 90 °C for 20 min. Prior to performing quality control by HPLC, EDTA in 0.1 M acetate buffer (pH 4.6) was added to a final concentration of 1 mM to the labeling reaction mixture as a “free” 111In3+ scavenger.
Quality control of [111In-DOTA]MG11
For quality control of the labeled reaction mixture, RP-HPLC was performed using system 1: A Waters Chromatograph (Waters, Vienna, Austria) based on a 600 solvent delivery system coupled to a Waters 996 photodiode array UV detector and a Gabi gamma detector (Raytest, RSM Analytische Instrumente GmbH, Germany) employing a 20 μL injection loop was applied. The Millennium Software (Waters, USA) was used for data processing and chromatographic control, and an XTerra RP-18 (5 μm, 4.6 mm × 150 mm) cartridge column (Waters, Germany) was eluted at 1 mL/min flow rate with a linear gradient starting from 0 % B and advancing to 40 % B within 40 min (solvent A = 0.1 % aqueous trifluoroacetic acid (TFA) and B = MeCN). For metabolism studies, HPLC analysis was performed using system 2: A Waters Chromatograph (Waters, Vienna, Austria) with a 600E multisolvent delivery system coupled to a Gabi gamma detector (Raytest, Germany) employing a 0.5-mL injection loop was applied. Data processing and chromatography run were controlled with Empower Software, and a Symmetry Shield RP-18 (5 μm, 3.9 mm × 20 mm) column (Waters, Germany) was eluted adopting linear gradient starting from 0 % B and advancing to 40 % B within 40 min (solvent A = 0.1 % aqueous TFA and B = MeCN) with a flow rate of 1 mL/min. Radioactivity measurements were conducted in an automated well-type gamma counter (NaI(Tl) crystal, Canberra Packard Auto-Gamma 5000 series instrument) calibrated for 111In.
Biology
Cell lines and cell culture
The rat pancreatic tumor cell line AR42J, endogenously expressing the CCK2R [27], was kindly provided by Prof. C. Decristoforo (University Clinic Innsbruck, Austria). The human epidermoid carcinoma A431 cell line transfected to stably express the CCK2R (A431-CCK2R(+)) or devoid of CCK2R expression (A431-CCK2R(−)) was a gift from Prof. O. Boerman (Department of Nuclear Medicine, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands) and Prof. L. Aloj (Istituto di Biostrutture e Bioimmagini, Consiglio Nazionale delle Ricerche, Naples, Italy) [28].
All culture media were purchased from Gibco BRL, Life Technologies (Grand Island, NY, USA), and supplements were supplied by Biochrom KG Seromed (Berlin, Germany). AR42J cells were cultured in F-12K Nutrient Mixture (Kaighn’s Modification), supplemented with 10 % (v/v) fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1 mM L glutamine. A431-CCK2R(+/−) cells were grown in Dulbecco’s Modified Eagle medium with GlutaMAX-I supplemented with 10 % (v/v) fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 4500 mg/L D-glucose, and 250 μg/mL G418. Cells were kept in a controlled humidified air containing 5 % CO2 at 37 °C. Splitting of cells with a ratio of 1:2 to 1:5 was performed when approaching confluency using a trypsin/EDTA solution (0.05/0.02 % w/v) [29].
Metabolism in blood
Animal experiments were carried out in compliance with European and national regulations and were approved by national authorities. For metabolic studies, in-house male Swiss albino mice (30 ± 5 g) were used. A bolus containing [111In-DOTA]MG11 (100 μL, 11–22 MBq, 3 nmol of total peptide, normal saline/EtOH 9/1 v/v) was injected in the tail vein of mice, together with (a) vehicle (100 μL; control), (b) PA (100 μL of vehicle containing 300 μg PA; PA), or (c) (100 μL of vehicle containing 150 μg TO; TO). Additional animals intraperitoneally (ip) received a fine dispersed suspension of Race (3 mg Race dissolved in 0.025 mL DMSO and freshly mixed with 0.375 mL saline) 30–40 min prior to radioligand injection. The animals were kept for 5 min in cages with free access to water. They were sacrificed by cardiac puncture under ether anesthesia, and blood was withdrawn with a syringe and immediately placed in a pre-chilled polypropylene vial on ice containing EDTA and Met. Blood samples were centrifuged at 2000g at 4 °C for 10 min. The plasma was collected, an equal volume of MeCN was added, and the mixture was centrifuged for 10 min at 15,000g at 4 °C. The supernatant was collected and concentrated under a gentle N2-flux at 40 °C to a volume of ≈0.1 mL; the concentrate was diluted with physiological saline (0.4 mL) and passed through a Millex-GV syringe-driven filter unit (0.22 μm/13 mm; Millipore, Milford, USA). Suitable aliquots of the filtrate were analyzed by RP-HPLC [15, 29]. The t R of the parent radiopeptide in the applied chromatographic conditions (system 2) was established by coinjection of samples with [111In-DOTA]MG11.
Biodistribution in AR42J tumor-bearing SCID mice
In-house male SCID mice (NCSR “Demokritos” Animal House) of 6 weeks of age at the time of arrival (18 ± 2 g body weight) were inoculated subcutaneously (sc) in their flanks with a suspension of freshly harvested AR42J cells (1 × 107 cells in ~150 μL saline). Animals were kept in aseptic conditions for 14 days when well-palpable tumors developed at the inoculation sites (0.31 ± 0.17 g) [29]. At the day of biodistribution, animals received a bolus of [111In-DOTA]MG11 (100 μL, 37–74 kBq, 10 pmol total peptide, in saline/EtOH 9/1 v/v) through the tail vein, coinjected with (a) vehicle (100 μL; control group, n = 10), (b) PA (100 μL of vehicle containing 300 μg PA; PA group, n = 10), (c) TO (100 μL of vehicle containing 150 μg TO; TO group, n = 4), (d) Lis (100 μL of vehicle containing 100 μg Lis; Lis group, n = 4), (e) PA plus Lis (100 μL vehicle containing 300 μg PA and 100 μg Lis; PA+Lis group, n = 5), or (f) 30–40 min after ip injection of Race (3 mg Race dissolved in 0.025 mL DMSO and freshly mixed with 0.375 mL saline; Race group, n = 4). In a separate animal group, the mice were coinjected with 100 μL vehicle containing both 300 μg PA and 100 μg DG2 [25] to assess non-specific tumor uptake during in situ NEP inhibition (in vivo CCK2R blockade; block group, n = 4).
Mice had access to drinking water ad libitum until they were euthanized at 4 h pi. Blood samples, organs of interest, and tumors were collected immediately after dissection, weighted, and measured for radioactivity in the gamma counter; only stomachs were emptied of their contents prior to measurements. Biodistribution data were calculated as percent of injected dose per gram tissue (% ID/g) with the aid of suitable standards of the injected dose, using the GraphPad Prism Software (San Diego, CA).
PA and TO dose dependence study in SCID mice bearing twin A431-CCK2R(+/−) tumors
Inocula of freshly harvested A431-CCK2R(+/−) cells (1.6 × 107/1.4 × 107 cell suspensions in 150 μL saline) were sc-injected in the right and left flanks of the SCID mice (male SCID mice, 6 weeks of age and of 18 ± 2 g body weight on arrival day; NCSR “Demokritos” Animal House). The animals were kept in aseptic conditions for 8 days till well-palpable tumors (0.26 ± 0.08 g) developed at the inoculation sites [29]. At the day of the experiment, the mice received a bolus of [111In-DOTA]MG11 (100 μL, 37–74 kBq, 10 pmol total peptide, in saline/EtOH 9/1 v/v) through the tail vein, coinjected with (a) vehicle (100 μL; control group, n = 5), (b) three different doses of PA (3 μg, n = 5; 30 μg, n = 4; or 300 μg PA, n = 10, dissolved in 100 μL vehicle), and (c) three different doses of TO (1.5, 15, or 150 μg TO dissolved in 100 μL vehicle, all groups of n = 4). Mice had free access to drinking water until they were sacrificed at 4 h pi, and biodistribution was conducted as described above.
Statistical analysis
The in vivo data were statistically analyzed with the Student’s t test (Prism™ 2.01, GraphPad Software, San Diego, CA). Analyses were two-tailed and a P value <0.05 was considered statistically significant.
Results
Radiolabeling
The radiolabeling of DOTA-MG11 with 111In was straightforward following published methods and involved 20-min incubation of the peptide conjugate in acidic medium at 90 °C in the presence of 111InCl3 [26]. A >96 % radiometal incorporation was typically demonstrated by RP-HPLC analysis (system 1). The radiochemical purity was >97 %, verifying that the addition of Met in the labeling reaction mixture prevented the oxidation of Met15 in the peptide chain (Additional file 1: Figure S1).
Metabolism in blood
As shown by HPLC analysis of mouse blood collected 5 min pi, [111In-DOTA]MG11 was very rapidly degraded in vivo in agreement with previous findings (Fig. 2a) [8, 15, 26]. Treatment of mice with PA, TO, and its ip pre-administered prodrug Race notably increased the amount of intact radiotracer in peripheral blood from <5 to >70 % (Fig. 2b, c, d, respectively). Conversely, coinjection of [111In-DOTA] MG11 with the ACE inhibitor Lis produced no change on radiotracer stability in mouse circulation up to 5 min pi (results not shown), in support of the assumption that [111In-DOTA]MG11 is resistant to ACE [18].

Stability of [111In-DOTA]MG11 in peripheral mouse blood at 5 min pi. Analysis by HPLC (system 2) of murine blood collected 5 min after injection of [111In-DOTA] MG11 with a vehicle (<4 % intact radiotracer), b PA (300 μg; >70 % intact radiopeptide), c TO (150 μg; >70 % intact radiopeptide), or d 30–40 min after ip injection of Race (2 mg; >70 % intact radiopeptide). The t R of [111In-DOTA]MG11 is indicated by the arrow
Biodistribution in AR42J tumor-bearing mice—impact of NEP/ACE inhibitors
Results of [111In-DOTA]MG11 biodistribution in SCID mice bearing AR42J tumors at 4 h pi are summarized in Table 1 and in Fig. 3, as % ID/g (n ≥ 4). In addition to the values obtained by coinjection of the radiotracer with vehicle (controls), Table 1 includes values obtained during treatment of mice with one of the NEP inhibitors (PA, TO, and Race) and/or the ACE inhibitor Lis. In all cases, the radiotracer rapidly cleared from background tissues via the kidneys and the urinary tract with consistently low renal values. Uptake in the AR42J tumors remarkably increased from 1.82 ± 1.25 %ID/g in controls to 15.32 ± 4.71 % ID/g in the PA group (P < 0.001). Most importantly, this increase was shown to be receptor specific, as suggested by the significantly reduced tumor values determined during coinjection of excess DG2 [25] and PA (0.34 ± 0.04 % ID/g, P < 0.001). In contrast, coinjection of the ACE inhibitor Lis did not provoke any raise in tumor values (1.80 ± 0.74 % ID/g) vs. controls (P > 0.05), whereas coinjection of both peptidase inhibitors PA and Lis did not provoke any additional increase in the PA group values (14.51 ± 4.73 % ID/g, P > 0.05). This finding is a strong indication that ACE is not involved in the in vivo catabolism of [111In-DOTA]MG11.

a Uptake of [111In-DOTA]MG11 in kidneys and AR42J tumors at 4 h pi in SCID mice. Data are given for kidneys and tumors as mean of %ID/g ± SD, n ≥ 4; [111In-DOTA]MG11 iv-coinjection with vehicle ( ), with 100 μg Lis (
), with 100 μg Lis ( ), with 300 μg PA (
), with 300 μg PA ( ), with a mixture of 100 μg Lis and 300 μg PA (
), with a mixture of 100 μg Lis and 300 μg PA ( ), with 150 μg TO (
), with 150 μg TO ( ), or 30–40 min after ip injection of 3 mg Race (
), or 30–40 min after ip injection of 3 mg Race ( ); for CCK2R blockade, 40 nmol DG2 [25] was iv-coinjected along with 300 μg PA (
); for CCK2R blockade, 40 nmol DG2 [25] was iv-coinjected along with 300 μg PA ( ). b Corresponding tumor-to-kidney ratios (Tu/Ki)
). b Corresponding tumor-to-kidney ratios (Tu/Ki)
On the other hand, coinjection of TO, at a dose equimolar to PA, produced similar albeit slightly less potent increase of AR42J tumor uptake (12.32 ± 3.66 % ID/g, P > 0.05). Interestingly, the TO prodrug Race although administered at a ten times higher dose than TO induced half as high an increase in the tumor uptake of [111In-DOTA]MG11 (6.81 ± 2.79 %ID/g, P < 0.001).
Biodistribution in A431-CCK2R(+/−) tumor-bearing mice—impact of PA and TO dose
The efficacy of the two NEP inhibitors PA and TO in improving the in vivo targeting of [111In-DOTA]MG11 was compared at 4 h pi in SCID mice bearing double A431-CCK2R(+/−) tumors. Three gradually increasing inhibitor doses and equimolar for PA (3, 30, and 300 μg) and TO (1.5, 15, and 150 μg) were tested, and the respective results are summarized in Table 2 and Fig. 4. It is interesting to note that in all cases, a significant and receptor-specific increase was recorded in the uptake of the radiotracer in the CCK2R-expressing tumors (P < 0.001), whereas no change was observed in the tumors devoid of CCK2R expression. Stepwise increase of TO dose resulted in significant and gradual increase of tumor uptake vs. controls. In the case of PA, however, increasing the dose from 30 to 300 μg did not further improve tumor uptake. Overall, PA clearly exerted a more potent tumor enhancement effect compared to the respective equimolar doses of TO (P < 0.001).

a Uptake of [111In-DOTA]MG11 in kidneys and A431-CCK2R(+) and A431-CCK2R(−) tumors at 4 h pi in SCID mice for controls and increasing amounts of the NEP inhibitors TO and PA. Data are given as mean of %ID/g ± SD, n ≥ 4. [111In-DOTA]MG11 iv-coinjection with vehicle ( ), with 1.5 μg TO (
), with 1.5 μg TO ( ), with 15 μg TO (
), with 15 μg TO ( ), with 150 μg TO (
), with 150 μg TO ( ), with 3 μg PA (
), with 3 μg PA ( ), with 30 μg PA (
), with 30 μg PA ( ), or 300 μg PA (
), or 300 μg PA ( ) in SCID mice bearing twin A431-CCK2R(+) (Tu+) and naïve A431-CCK2R(−) (Tu−) human xenografts. b Corresponding tumor-to-kidney ratios (Tu/Ki)
) in SCID mice bearing twin A431-CCK2R(+) (Tu+) and naïve A431-CCK2R(−) (Tu−) human xenografts. b Corresponding tumor-to-kidney ratios (Tu/Ki)
Discussion
The advent of radiolabeled somatostatin analogs, like OctreoScan® ([111In-DTPA]octreotide) and Lutathera® ([177Lu-DOTA]Tate), in the management of sst2-expressing neuroendocrine tumors [30], has established the application of peptide radioligands for molecular imaging and radionuclide therapy of tumors. This success has largely depended on the metabolic robustness of the cyclic-octapeptide analogs applied. The evolution of peptide receptor-targeted diagnosis and therapy beyond the boundaries of sst2-positive neuroendocrine tumors is valid based on the overexpression of other peptide receptors in a variety of human tumors [31]. However, progress in this direction has been greatly restricted by the sub-optimal metabolic stability of peptide radioligands. Structural interventions to improve metabolic stability often deteriorate other important biological features of radiopeptides, as for example receptor affinity or pharmacokinetics [9].
We have recently proposed the in situ stabilization of biodegradable radiopeptides by coinjection of a suitable protease inhibitor, leading to significant improvement of tumor uptake [15]. This concept has indeed led to impressive diagnostic signal amplification on tumor lesions in preclinical models and was recently shown to improve the outcome of radionuclide therapy as well [32]. Especially in the case of [111In-DOTA]MG11 and other radiolabeled gastrins, high enhancement of tumor uptake combined with a preserved low kidney retention were observed, resulting in particularly appealing tumor-to-kidney ratios [18, 29]. These preclinical results, obtained after coinjection of [111In-DOTA]MG11 with the NEP inhibitor PA, warrant further validation in a “proof-of-principle” study in patients.
It should be noted, that our studies on in situ radiopeptide stabilization and enhancement of tumor uptake have primarily been focused on NEP inhibition by PA. Remarkably, a great variety of radioligands originating from different peptide families, including somatostatin, bombesin, and gastrin, have profited by application of this simple methodology, revealing a central role of NEP in initiating the in vivo degradation of these analogs [15]. The impact of in situ NEP inhibition relies on the abundant and ubiquitous presence of this ectoenzyme in the body combined with its broad substrate specificity [14]. However, NEP may not always exclusively determine the in vivo fate of radiopeptides. For example, ACE has often been implicated in the degradation of peptide ligands, and its role in the in vitro degradation of different-length gastrin analogs has been previously reported [10]. It should be stressed that the impact of in situ ACE inhibition on the bioavailability and tumor localization of [111In-DOTA]MG11 in vivo has not been thus far investigated.
For this purpose, in our study we have coinjected [111In-DOTA]MG11 together with the potent ACE inhibitor Lis, a registered antihypertensive drug [19]. As a result, we were not able to observe any change in the radioligand uptake in AR42J tumors in mice treated with Lis vs. controls at 4 h pi. In addition, dual NEP/ACE inhibition by combined coinjection of both Lis and PA inhibitors with [111In-DOTA]MG11 induced no additive effect vs. single NEP inhibition by PA (Table 1, Fig. 3; P > 0.05). These findings strongly suggest that ACE does not contribute to the in vivo processing of [111In-DOTA]MG11 and verify NEP as the major degrading protease.
Owing to the fast kinetics of radiopeptide localization to target sites upon entry in the bloodstream, only rapidly occurring degradation events—as those related to NEP—are relevant for tumor-targeting efficacy. Hence, the onset of NEP inhibition should occur and reach its maximum quite rapidly but not last longer than the time needed for the radioactivity to clear from blood and access the target. In this respect, the inhibitor should preferably induce rapid, complete, and transient NEP inhibition. In addition, it should be potent, water-soluble, chemically stable, and available at a reasonable cost. PA combines most of the above desirable properties; however, it has not been thoroughly investigated in terms of biosafety [17]. Only very low amounts of PA have been administered in human, not sufficient for in vivo effective inhibition of NEP [20].
Aiming at clinical translation, our interest has been attracted by two clinically certified NEP inhibitors, TO and its prodrug Race. TO is a potent, reversible NEP inhibitor with a relative rapid onset of action [21], but its water solubility is not ideal for iv injection as a bolus together with the radiopeptide. It has been previously tested in high doses in human, as for example by iv infusion of 150 mg in 250 mL of isotonic glucose in 42 patients showing excellent hemodynamic tolerance [22]. The impact of TO on the bioavailability and tumor uptake of our paradigm compound [111In-DOTA]MG11 was compared vs. PA 4 h after coinjection of the radiotracer with equimolar amounts of either inhibitor in AR42J tumor-bearing mice. As a result, we observed comparable and impressive enhancement of tumor values (Table 1, Fig. 3). On the other hand, Race is a registered antidiarrheal drug for oral use and a prodrug for TO [23, 24]. Due to its poor water solubility, it cannot be iv-administered in mice and was instead ip-injected in high molar excess (10-fold than the TO dose) 30–40 min prior to the iv injection of [111In-DOTA]MG11. As a result, tumor values significantly increased compared to controls but half as efficiently compared to the iv-injected hydrophilic inhibitors PA and TO. This finding highlights the significance of administration route and actual concentration of the inhibitor in the blood at the time of the radiotracer injection to achieve maximum enhancement of uptake on tumor lesions.
In the last part of this study, the efficacy of TO to enhance the uptake of [111In-DOTA]MG11 specifically in human CCK2R(+)-expressing tumors was compared vs. PA at progressively increasing administered doses in mice bearing double A431-CCK2R(+/−) tumors (Table 2, Fig. 4). While both NEP inhibitors induced significant and CCK2R-specific tumor uptake enhancement even at the lowest administered doses (1.5 μg TO and 3 μg PA), PA showed consistently superior efficacy at all tested dose levels compared to TO (P < 0.001). Remarkably, PA reached maximum tumor uptake enhancement at the 30 μg dose already, whereas TO reached half as high tumor values at the corresponding 10-fold molar dose of 150 μg. These intriguing findings may be tentatively assigned to the free thiol functionality of TO (Fig. 1). This strong nucleophile is prone to interact in vivo with electrophiles, and hence, the effective dose of TO might be much lower than originally anticipated. In fact, previous reports have demonstrated the fast in vivo inactivation of thiol-based NEP inhibitors, including TO [33, 34]. Another practical disadvantage of thiols is their propensity to oxidize in aqueous solutions to the respective disulfides, and hence, their storage for longer time periods in ready to use formulations for clinical application becomes challenging.
Conclusions
In summary, translation of the concept of in situ stabilization of biodegradable peptide radioligands to improve tumor targeting and hence diagnostic sensitivity and therapeutic efficacy, from the preclinical setting into patients, poses certain challenges that need to be competently addressed. Promising data thus far obtained from mouse models for our paradigm radiotracer [111In-DOTA]MG11 and the NEP inhibitor PA have been directly compared herein with results retrieved after treatment of mice with the ACE inhibitor Lis and the NEP inhibitors TO and Race, all of which have been clinically tested previously. This study has shown that only NEP inhibition is relevant for clinical translation. It has also shown that significant enhancement of the radiotracer tumor uptake is indeed feasible by both TO and Race. However, for maximum efficacy, matching that of PA, further studies need to be performed to optimize dose, administration route, and time of inhibitor injection in respect to radiotracer injection and are currently actively pursued.
Abbreviations
- DOTA:
-
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- DTPA:
-
diethylenetriamine-pentaacetic acid
- N4 :
-
6-(carboxyl)-1,4,8,11-tetraazaundecane
- Octreotide:
-
H2N-DPhe-c[Cys-Phe-DTrp-Lys-Thr-Cys]-Thr(ol)
- PET:
-
positron emission tomography
- SPECT:
-
single photon emission computed tomography
- sst2 :
-
somatostatin subtype 2 receptor
- Tate:
-
H2N-DPhe-c[Cys-Tyr-DTrp-Lys-Thr-Cys]-Thr-OH
References
Reubi JC, Schaer JC, Waser B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57(7):1377–86.
Reubi JC, Waser B. Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int J Cancer. 1996;67(5):644–7.
Reubi JC. CCK receptors in human neuroendocrine tumors: clinical implications. Scand J Clin Lab Invest Suppl. 2001;234:101–4.
Ferrand A, Wang TC. Gastrin and cancer: a review. Cancer Lett. 2006;238(1):15–29.
Béhé M, Behr TM. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers. 2002;66(6):399–418.
Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T, et al. Comparative biodistribution of 12 111In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging. 2011;38(8):1410–6.
Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86(3):805–47.
Ocak M, Helbok A, Rangger C, Peitl PK, Nock BA, Morelli G, et al. Comparison of biological stability and metabolism of CCK2 receptor targeting peptides, a collaborative project under COST BM0607. Eur J Nucl Med Mol Imaging. 2011;38(8):1426–35.
Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15(1-2):40–56.
Dubreuil P, Fulcrand P, Rodriguez M, Fulcrand H, Laur J, Martinez J. Novel activity of angiotensin-converting enzyme. Hydrolysis of cholecystokinin and gastrin analogues with release of the amidated C-terminal dipeptide. Biochem J. 1989;262(1):125–30.
Deschodt-Lanckman M, Pauwels S, Najdovski T, Dimaline R, Dockray GJ. In vitro and in vivo degradation of human gastrin by endopeptidase 24.11. Gastroenterology. 1988;94(3):712–21.
Power DM, Bunnett N, Turner AJ, Dimaline R. Degradation of endogenous heptadecapeptide gastrin by endopeptidase 24.11 in the pig. Am J Physiol. 1987;253(1 Pt 1):G33–9.
Migaud M, Durieux C, Viereck J, Soroca-Lucas E, Fournie-Zaluski MC, Roques BP. The in vivo metabolism of cholecystokinin (CCK-8) is essentially ensured by aminopeptidase A. Peptides. 1996;17(4):601–7.
Roques BP, Noble F, Dauge V, Fournie-Zaluski MC, Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45(1):87–146.
Nock BA, Maina T, Krenning EP, de Jong M. “To serve and protect”: enzyme inhibitors as radiopeptide escorts promote tumor targeting. J Nucl Med. 2014;55(1):121–7.
Suda H, Aoyagi T, Takeuchi T, Umezawa H. Letter: a thermolysin inhibitor produced by actinomycetes: phosphoramidon. J Antibiot (Tokyo). 1973;26(10):621–3.
Oefner C, D'Arcy A, Hennig M, Winkler FK, Dale GE. Structure of human neutral endopeptidase (neprilysin) complexed with phosphoramidon. J Mol Biol. 2000;296(2):341–9.
Kaloudi A, Nock BA, Krenning EP, Maina T, De Jong M. Radiolabeled gastrin/CCK analogs in tumor diagnosis: towards higher stability and improved tumor targeting. Q J Nucl Med Mol Imaging. 2015;59(3):287–302.
Brunner DB, Desponds G, Biollaz J, Keller I, Ferber F, Gavras H, et al. Effect of a new angiotensin converting enzyme inhibitor MK 421 and its lysine analogue on the components of the renin system in healthy subjects. Br J Clin Pharmacol. 1981;11(5):461–7.
Polosa R, Santonocito G, Magri S, Paolino G, Armato F, Pagano C, et al. Neutral endopeptidase inhibition with inhaled phosphoramidon: no effect on bronchial responsiveness to adenosine 5'-monophosphate (AMP) in asthma. Eur Respir J. 1997;10(11):2460–4.
Roques BP, Fournie-Zaluski MC, Soroca E, Lecomte JM, Malfroy B, Llorens C, et al. The enkephalinase inhibitor thiorphan shows antinociceptive activity in mice. Nature. 1980;288(5788):286–8.
Floras P, Bidabe AM, Caille JM, Simonnet G, Lecomte JM, Sabathie M. Double-blind study of effects of enkephalinase inhibitor on adverse reactions to myelography. AJNR Am J Neuroradiol. 1983;4(3):653–5.
Lecomte JM, Costentin J, Vlaiculescu A, Chaillet P, Marcais-Collado H, Llorens-Cortes C, et al. Pharmacological properties of acetorphan, a parenterally active “enkephalinase” inhibitor. J Pharmacol Exp Ther. 1986;237(3):937–44.
Salazar-Lindo E, Santisteban-Ponce J, Chea-Woo E, Gutierrez M. Racecadotril in the treatment of acute watery diarrhea in children. N Engl J Med. 2000;343(7):463–7.
Nock BA, Maina T, Béhé M, Nikolopoulou A, Gotthardt M, Schmitt JS, et al. CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J Nucl Med. 2005;46(10):1727–36.
Breeman WA, Fröberg AC, de Blois E, van Gameren A, Melis M, de Jong M, et al. Optimised labeling, preclinical and initial clinical aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl Med Biol. 2008;35(8):839–49.
Scemama JL, De Vries L, Pradayrol L, Seva C, Tronchere H, Vaysse N. Cholecystokinin and gastrin peptides stimulate ODC activity in a rat pancreatic cell line. Am J Physiol. 1989;256(5 Pt 1):G846–50.
Aloj L, Caracó C, Panico M, Zannetti A, Del Vecchio S, Tesauro D, et al. In vitro and in vivo evaluation of 111In-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med. 2004;45(3):485–94.
Kaloudi A, Nock BA, Lymperis E, Sallegger W, Krenning EP, de Jong M, et al. In vivo inhibition of neutral endopeptidase enhances the diagnostic potential of truncated gastrin 111In-radioligands. Nucl Med Biol. 2015;42(11):824–32.
de Jong M, Breeman WA, Kwekkeboom DJ, Valkema R, Krenning EP. Tumor imaging and therapy using radiolabeled somatostatin analogues. Acc Chem Res. 2009;42(7):873–80.
Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24(4):389–427.
Chatalic KL, Konijnenberg M, Nonnekens J, de Blois E, Hoeben S, de Ridder C, et al. In vivo stabilization of gastrin-releasing peptide receptor antagonist enhances PET imaging and radionuclide therapy of prostate cancer in preclinical studies. Theranostics. 2016;6(1):104–17.
Iyer RA, Mitroka J, Malhotra B, Bonacorsi Jr S, Waller SC, Rinehart JK, et al. Metabolism of [(14)C]omapatrilat, a sulfhydryl-containing vasopeptidase inhibitor in humans. Drug Metab Dispos. 2001;29(1):60–9.
Poras H, Bonnard E, Dange E, Fournie-Zaluski MC, Roques BP. New orally active dual enkephalinase inhibitors (DENKIs) for central and peripheral pain treatment. J Med Chem. 2014;57(13):5748–63.
Acknowledgements
This work has been partially supported by the Greek General Secretariat for Research and Technology and the European Regional Development Fund under the Action “Development Grants for Research Institutions—KRIPIS” of OPCE II.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AK was actively engaged in the biological studies and assisted in the writing of this manuscript (ms). BAN designed the overall study, supervised radiochemical work as well as the generation and final editing of the ms. EL performed radiolabeling and radioanalytical work. RV contributed in the selection of the inhibitors and in editing the ms. MdJ and EPK participated in the design of the study and editing of the ms. TM performed animal studies and drafted most parts of the ms. All authors read and approved the final manuscript.
Additional file
Additional file 1: Figure S1.
Quality control of [111In-DOTA]MG11 labeling reaction product by HPLC. (DOCX 219 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kaloudi, A., Nock, B.A., Lymperis, E. et al. Impact of clinically tested NEP/ACE inhibitors on tumor uptake of [111In-DOTA]MG11—first estimates for clinical translation. EJNMMI Res 6, 15 (2016). https://doi.org/10.1186/s13550-015-0158-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13550-015-0158-3