
Abstract
Background
Limb-girdle muscular dystrophies (LGMDs) are a heterogeneous group of inherited autosomal myopathies that preferentially affect voluntary muscles of the shoulders and hips. LGMD has been clinically described in several breeds of dogs, but the responsible mutations are unknown. The clinical presentation in dogs is characterized by marked muscle weakness and atrophy in the shoulder and hips during puppyhood.
Methods
Following clinical evaluation, the identification of the dystrophic histological phenotype on muscle histology, and demonstration of the absence of sarcoglycan-sarcospan complex by immunostaining, whole exome sequencing was performed on five Boston terriers: one affected dog and its three family members and one unrelated affected dog.
Results
Within sarcoglycan-δ (SGCD), a two base pair deletion segregating with LGMD in the family was discovered, and a deletion encompassing exons 7 and 8 was found in the unrelated dog. Both mutations are predicted to cause an absence of SGCD protein, confirmed by immunohistochemistry. The mutations are private to each family.
Conclusions
Here, we describe the first cases of canine LGMD characterized at the molecular level with the classification of LGMD2F.
Similar content being viewed by others


Background
Limb-girdle muscular dystrophies (LGMDs) are a heterogeneous group of Mendelian disorders affecting voluntary muscles of the shoulders and hips [1]. While proximal limb muscles are primarily affected in LGMD, other muscles may degenerate as well, such as the heart and respiratory muscles [1]. Sarcoglycanopathies are a subset of severe, recessive LGMDs (LGMD2C-F) that present in early childhood [2]. There are six known sarcoglycan genes (SGCA, SGCB, SGCD, SGCG, SGCE, and SGCZ); the first four encode single-pass transmembrane glycoproteins (α-, β-, δ-, γ-sarcoglycans) and, along with sarcospan, make up the tetrameric sarcoglycan-sarcospan complex (SGC). As part of the dystrophin-glycoprotein complex, the SGC is critical for maintaining sarcolemmal stability [3]. Mutations in SGCA, SGCB, SGCD, or SGCG can result in non-assembly of the SGC and, therefore, the absence of all four sarcoglycans from muscle of affected patients [3, 4]. There are only a handful of low-frequency founder alleles in human populations responsible for sarcoglycanopathies [5]; thus, they are most commonly caused by mutations in compound heterozygosity [6].
In the domestic dog (Canis familiaris), selective breeding practices encourage pairing of recessive alleles inherited identical by descent (IBD). Accordingly, dogs have an abundance of recessive disorders [7], including muscular dystrophies [8, 9]. Most canine muscular dystrophies are associated with dystrophin deficiency, and founder alleles have been identified in several breeds [10, 11]. Recently, two independent mutations causing dystrophinopathy were described in Cavalier King Charles spaniels [12, 13].
The first report of LGMD associated with sarcoglycan deficiency in dogs involved three breeds: Chihuahua, Cocker spaniel, and a 7-month-old male Boston terrier from Colorado (case 1), but mutations were not identified [8]. Four years later, sarcoglycanopathy was described again in an unrelated 4-month-old male Boston terrier from Iowa [14] (case 2). All dogs affected with sarcoglycanopathy had a clinical dystrophic phenotype including muscle wasting, gait abnormalities, enlarged tongue, dysphagia, and extremely elevated serum creatine kinase (CK) activities [8, 14]. Pathologic features were consistent with dystrophy, having myofiber degeneration, regeneration, and calcific deposits [8, 14]. Affected dogs lacked muscle α-, β-, and γ-sarcoglycans, confirmed by both western blotting and immunohistochemistry [8, 14]. At the time of evaluation, an antibody reactive with canine δ-sarcoglycan was unavailable.
Here, we describe a sarcoglycanopathy in a third family of Boston terriers from Arkansas in which two puppies (cases 3 and 4) from the same kennel but different litters displayed clinical signs of LGMD, pathological changes consistent with a dystrophic phenotype, and immunohistochemical confirmation of absent or decreased sarcoglycans. To identify the genetic basis for LGMD in the Boston terrier breed, we performed whole exome sequencing (WES) of cases 1 and 3 and related dogs. Evaluation of the sarcoglycan genes revealed, to our surprise, two private deletions in SGCD: a 2-bp deletion in exon 6 and a 19.4-kb deletion encompassing exons 7 and 8. Both cause a lack of SGCD, resulting in LGMD2F.
Methods
Animals
Clinical details of case 1 were previously published [8]. Biological samples from case 2 were not available. Female Boston terriers, ages 12 and 5 months, and from the same breeder in Arkansas (cases 3 and 4), were evaluated for a chronic history of progressive dysphagia, lack of appetite, drooling, muscle wasting, and greatly enlarged tongues. Both dogs were examined by the same veterinarian in a clinical setting.
DNA was extracted from diagnostic muscle biopsies of cases 1 and 3 and whole blood of unaffected relatives of cases 3 and 4 using the DNeasy extraction kit (Qiagen, Hilden, Germany). Muscle for isolation of DNA was unavailable from case 4. Whole-blood samples or buccal swabs from unrelated, healthy Boston terriers were recruited, and DNA was isolated following the Gentra PureGene protocol (Qiagen, Hilden, Germany) or the MagJet Genomic DNA purification kit (ThermoFisher Scientific, Waltham, USA). Genomic DNAs from unaffected dogs from multiple breeds were available from DNA archives at Clemson University and CAG GmbH.
The dogs in this study were examined and tissues collected in a clinical practice setting with the written consent of their owners. Studies on tissue biopsies and blood samples were approved by the Institutional Animal Care and Use Committees (IACUC) of Clemson University, the University of California San Diego, the University of Iowa, and the Animal Experiment Board in Finland (ESAVI/7482/04.10.07/2015), as well as the Baden-Württemberg veterinary office at the Landratsamt Tübingen Abt. 32: Veterinärwesen und Lebensmittelüberwachung, Tübingen, Germany (Registriernummer: DE 08 416 1038 21).
Histology and immunofluorescence
Muscle specimens from case 1 were previously obtained as biopsies and archived at −80 °C at the Comparative Neuromuscular Laboratory, University of California San Diego (CNL). Specimens from limb muscles, heart, and tongue were collected by a veterinarian following humane euthanasia at 1 year of age for case 3 and at 5 months of age for case 4. Muscles were either refrigerated or immersion fixed in buffered formalin and shipped to the CNL. Cryosections from all muscle specimens were processed by a standard panel of histochemical stains and reactions [15].
Antibodies used for immunofluorescence were rabbit antibodies R98 anti-α-sarcoglycan [16], R214 anti-δ-sarcoglycan [17], IIH6 anti-α-dystroglycan [18], R256 anti-sarcospan [19]; mouse antibodies 5B1 anti-β-sarcoglycan [19], 21B5 anti-γ-sarcoglycan [19], AP83 anti-β-dystroglycan [18], anti-dystrophin (AbCam, San Franscisco, CA USA), anti-collagenVI (Fitzgerald Laboratories, Acton, MA USA), and anti-caveolin 3 (BD Transduction Laboratories, San Jose, CA USA); rat anti-perlecan (NeoMarkers, Fremont, CA USA).
For secondary immunofluorescence, tissues were blocked with 10% goat serum in phosphate-buffered saline, incubated in primary antibody overnight, washed, incubated in Alexa Fluor 488-, 594-, or 647-conjugated anti-rat, anti-rabbit, or anti-mouse antibodies (Life Technologies, San Diego, CA USA ), respectively, and mounted using ProLong Gold mounting media (Life Technologies). For α-dystroglycan, sarcospan, δ-sarcoglycan, and γ-sarcoglycan staining, tissues were fixed in 2% paraformaldehyde, followed by incubations in 100 mM glycine and 0.05% SDS prior to processing as described above. Images were acquired using a VS120-S5-FL slide scanner microscope (Olympus) with VS-ASW software.
Parentage testing
The Canine Genotypes Panel 1.1 (ThermoFisher Scientific) was used to verify parentage of the experimental dogs. Samples were amplified according to the manufacturer’s instructions and separated and detected on an ABI 3730XL (Applied Biosystems, ThermoFisher Scientific). GeneMarker (Softgenetics, State College, PA, USA) was used to assign peaks and determine genotypes according to ISAG nomenclature.
Whole exome sequencing
DNA from five Boston terriers (case 1, case 3, and three unaffected relatives of cases 3 and 4) was used for WES performed at CeGaT GmbH (Tübingen, Germany). Genomic DNA (1 μg) from each sample was mechanically sheared to approximately 180–250-bp fragments using a Covaris LE220 Ultrasonicator (Woburn, MA, USA). Fragment sizes were assessed for quality control purposes (Fragment Analyzer, Advanced Analytical Technologics Inc.), and the Agilent SureSelect XT Canine All Exon kit (Santa Clara, CA, USA) supplied the 120-mer biotinylated RNA bases with which the fragment library was hybridized. Magnetic streptavidin beads were used for purification according to the manufacturer’s protocol (Agilent). After amplification of library DNA, adaptors and barcodes for sequencing were added (Illumina), and equimolar amounts of each sample were pooled. Both lanes of a Rapid Flowcell were used to sequence the pool on an Illumina HiSeq2500, generating 2 × 100-bp paired-end sequences, resulting in approximately 6 GB per sample. Illumina bcl2fastq 1.8.2 was used to demultiplex sequencing data, skewer 0.1.116 was used to trim sequencing adapters, and the Burrows-Wheeler Aligner (bwa 0.7.2-r351) was used to map the sequences to the canine genome (CanFam3.1). Samtools 0.1.18 and internal software were used to remove PCR duplicates and low-quality alignments. bcftools (0.1.17) and varscan (2.3.5) and internal software were used to call variants, and a single Variant Call Format (VCF) file was generated for each sample using internal software.
IGV (Integrative Genomics Viewer) [20] and Genome Browse (Golden Helix [21, 22] Inc., USA) were used to visualize data, and the Ensembl dbSNP (Can Fam3.1 version) and whole genome sequences (Clemson) were used to exclude variants.
Variant characterization and genotyping
2-bp deletion
The 2-bp deletion identified in case 3 was verified by Sanger sequencing, using primers designed to amplify SGCD exon 6 (Additional file 1: Table S1). The deletion disrupts a BcoD1 restriction enzyme site, yielding a 406-bp and a 347-bp product, representing mutant and wild-type alleles, respectively. Unrelated Boston terriers and dogs from other breeds were genotyped using either restriction digest or Sanger sequencing.
19.4-kb deletion
To define the break points of the microdeletion encompassing SGCD exons 7 and 8 and the 3′ intergenic sequence, primers were designed in flanking sequences (Additional file 1: Table S1).
For genotyping, primer pairs were designed within the deletion to amplify only wild-type alleles, as well as flanking the deletion for amplification of the mutant allele. Primer pairs were multiplexed for amplification using Phire Hot Start II DNA polymerase (ThermoFisher) and products were resolved by gel electrophoresis. Products were initially verified via Sanger sequencing. The multiplex PCR was used to test unrelated Boston terriers and dogs of other breeds.
Results
Clinical findings
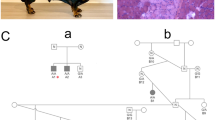
Muscle wasting, dysphagia, exercise intolerance, lethargy, and failure to thrive were accompanied by progressive gait abnormalities including a short, stilted gait in cases 3 (Fig. 1a) and 4. While there was no clinical indication of cardiomyopathy, specific evaluations for heart disease by a veterinary cardiologist were not performed. Clinical chemistry included markedly elevated activities of serum alanine aminotransferase (ALT; 900 IU/L, reference range 10–110 IU/L), aspartate aminotransferase (AST; 920 IU/L, reference range 16–50 IU/L), and creatine kinase (CK; >10,000 IU/L, reference range 50–275 IU/L). Progression of clinical signs necessitated euthanasia at approximately 1 year of age for case 3 and 5 months of age for case 4.

Histopathology of muscle biopsies from a female Boston terrier affected with sarcoglycanopathy (case 3). A hunch back stance was evident in the dog (a). H&E stained cryosections from a representative limb muscle (b) showed degenerative changes and calcific deposits (black arrow). Similar degenerative changes and calcific deposits were observed in the tongue (c). The calcific deposits in the tongue were highlighted bright orange using the alizarin stain for calcium (d)
Histology and immunofluorescence
A dystrophic phenotype including degeneration, regeneration, and calcific deposits was evident in the skeletal muscle (Figs. 1b) and tongue (Fig. 1c, d). Heart muscle was histologically normal (left ventricle, not shown) from cases 3 and 4. Immunofluorescence staining of muscle cryosections showed markedly reduced or absent localization of α-, β-, γ-, and δ-sarcoglycans and sarcospan in cases 3 and 4 (Fig. 2). In contrast, staining for localization of α- and β-dystroglycans, dystrophin, caveolin 3, and perlecan was similar to control muscle (Fig. 3). Staining for collagen VI was increased in the endomysium compared to the control tissue, consistent with endomysial fibrosis. Results of histology, immunofluorescence staining, and western blotting of case 1 were described previously [8]. Staining for localization of δ-sarcoglycan in case 1 was performed on archived muscle cryosections and was similarly absent (not shown).

Loss of SGC staining in cases 3 and 4. Representative H&E and immunofluorescence of cryosections from the muscle of cases 3 and 4, as well as of a control dog muscle. In the control muscle, antibodies to the SGC (α-, β-, δ-, γ-sarcoglycans: αSG, βSG, δSG, γSG), as well as sarcospan (SSPN), localize to the sarcolemma of the muscle fibers. Staining from each of these antibodies is reduced in muscle from cases 3 and 4

Representative immunofluorescence of cryosections from muscle of cases 3 and 4 and control dog muscle. Staining of α-dystroglycan (αDG), β-dystroglycan (βDG), dystrophin (DMD), caveolin 3 (CAV3), collagen VI (COL6), and perlecan (PCAN) in cases 3 and 4. Antibodies to α- and β-dystroglycans, dystrophin, caveolin 3, and perlecan demonstrate sarcolemmal localization and intensity that is comparable to control tissue. An antibody to collagen VI shows increased localization to the endomysium compared to the control tissue, consistent with endomysial fibrosis
Parentage testing
Parentage testing was performed to determine relationships between case 3 and three other dogs obtained from the same breeder. One relative was confirmed to be the dam of case 3 and is referred to hereafter as the obligate carrier. The test excluded the remaining two dogs from being full siblings of case 3 or progeny of the obligate carrier. Their relationship to the other dogs or to one another could not be determined.
Variant identification from WES
Disruption of any one of the sarcoglycans results in reduced immunostaining of the entire SGC, both in LGMD patients and in animal models of sarcoglycanopathy [23,24,25]. Therefore, genetic sequencing was necessary to identify the defective sarcoglycan gene.
2-bp deletion
The candidate genes (SGCA, SGCB, SGCD, SGCG) were sequenced to an approximate depth of 30X. For each gene, we manually screened the VCF file in IGV for variants fitting a pattern of inheritance consistent with a rare recessive allele. We expected both affected dogs to have inherited the causal mutation IBD from a common ancestor; therefore, we searched for variants homozygous in cases 1 and 3, heterozygous in the obligate carrier, and heterozygous or homozygous wild-type in the two relatives. No variants fit these criteria.
Because there was no known relationship between cases 1 and 3, we considered that they may have different genetic causes for LGMD. Thus, we excluded case 1 and searched again for the same pattern. Only one variant fit the pattern: a 2-bp deletion in exon 6 of SGCD (Fig. 4). We validated the deletion through Sanger sequencing and determined that, in addition to the obligate carrier, one relative was heterozygous. The deletion predicts the substitution of an aspartate for a glutamate (E178D) and creates a frameshift, leading to a premature stop codon two amino acids later (P180X) (Fig. 4). We genotyped 199 Boston terriers and 127 dogs from 33 other breeds; none possessed the deletion.

Electropherogram showing the 2-bp SGCD deletion in case 3. The top panel shows the sequence from case 3, while the lower panel shows the sequence from a healthy non-related Boston terrier. The SGCD c.534_535delGA mutation leads to a frameshift and a premature stop codon two amino acids later
19.4-kb deletion
Using BAM files, we reexamined each candidate gene for variants homozygous in case 1 and absent from the other Boston terriers. This approach revealed a complete absence of reads from the final two exons of SGCD (7 and 8) in case 1, which was not apparent from the VCF file. No other variants fit the pattern. We hypothesized that the absence of reads represented a microdeletion and designed three primer pairs flanking exons 6, 7, and 8. PCR amplification yielded a product for exon 6 but not for exon 7 or 8 in case 1, providing further support for the presence of a deletion.
It was not possible to characterize the deletion directly from WES because intergenic and intronic sequences are minimized. Sequence coverage indicated that the deletion was between SGCD exon 6 and TMD4. Furthermore, sporadic intronic and intergenic fragments were present 5′ of exon 7, beginning at chr4:53282570, and in the 3′ UTR, beginning at chr4:53261359, suggesting a maximum deletion size of 21,211 bp. Primer pairs flanking this estimated deletion size yielded large products (~3–5 kb), indicating a deletion approximately 2 kb smaller than suggested by WES. Sanger sequencing of the breakpoint revealed a substitution (chr4:53262018-53262020, ATG > CC), followed by 9 bp that were unchanged before a deletion of 19,403 bp (chr4:53262030-53281432) (Fig. 5). We genotyped 201 Boston terriers and 91 dogs of 19 other breeds and did not find any carriers.

Schematic and sequence showing the breakpoints of the 19,403-bp SGCD deletion in case 1. Note that SGCD is annotated on the minus strand. Whole exome sequence from a healthy dog and case 1 are aligned to the reference genome, visualized in Golden Helix GenomeBrowse ® [21, 22]. Case 1 has no coverage of exons 7 and 8 and flanking regions. Sequence of the wild-type and case 1 alleles show the precise breakpoints. Nucleotides 5′ and 3′ of the breakpoint are in bold blue and orange typeface, respectively. A substitution (chr4:53262020-53262018, CAT > GG) is found 9 bp downstream of the microdeletion and is shown in bold red typeface
Discussion
Sarcoglycanopathies in humans are rare genetic disorders, with an incidence of one in every 178,000 human births [26]. To date, only small animal models are available for study: gene-targeted mouse models for α-, β-, δ-, and γ-sarcoglycanopathy [27] and a spontaneous hamster model for δ-sarcoglycanopathy [28, 29]. Here, we have demonstrated that a naturally occurring muscular dystrophy in a Boston terrier family is a sarcoglycanopathy, consistent with two previously published case reports in the breed. Given that cases have been described in three Boston terrier families, we expected a single recessive allele, present at a very low frequency within the breed, to underlie all cases. Instead, we uncovered independent mutations in the two families studied herein. Unfortunately DNA from case 2 [14] was not available to determine whether this dog shared one of the mutations described herein, a different mutation in SGCD, or a pathogenic variant in another gene.
Both families possessed mutations in SGCD, which encodes δ-sarcoglycan. Canine SGCD is located on CFA 4 and organized into eight exons that form a 1297 bp mRNA transcript [30]. Human (XP_016865213.1) and dog (XP_013968526.1) amino acid sequences share 98% identity. Despite being the largest of the sarcoglycan genes, SGCD least commonly causes sarcoglycanopathy, with the majority of human cases attributed to changes in SGCA [31]. Thus, it is not only surprising that the Boston terriers had independent mutations causing sarcoglycanopathy, but that both had pathogenic alleles of SGCD. Curiously, the only other naturally occurring model of a sarcoglycanopathy, the Syrian hamster, also harbors an SGCD deletion [29].
Mutations of SGCD cause LGMD2F, and although clinical presentation is largely similar among the four sarcoglycanopathies, this is the only subtype not consistently characterized by concomitant cardiomyopathy [1]. The absence of heart involvement in Boston terriers is consistent with this classification; however, because the affected dogs were euthanized at an early age it is unknown if muscle degeneration would have progressed to involve the heart.
Immunohistochemistry illustrated a lack of the SGC in both cases, but provides no indication as to whether SGCD is abnormal or absent altogether. Due to limited sample availability, collected tissues were prioritized for histopathological analysis and genomic DNA sequencing. RNAs were thus unavailable to investigate the consequence of the deletions on SGCD transcripts. The 2-bp pair deletion predicts a premature stop codon in exon 6, possibly causing nonsense-mediated decay. The 19.4-kb microdeletion eliminates the last two exons of SGCD; the complete loss of an SGCD exon is rare [5]. It is hypothesized that exon 6 would splice to one or more cryptic sites, triggering either nonsense-mediated decay and/or the production of mutant protein. It is likely that mutant SGCD would cause assembly of the SGC to fail, resulting in LGMD [3, 32].
WES is a cost-effective method for the sequencing of multiple family members and has been used successfully to identify LGMD mutations in humans [33]. It was an advantageous choice over transcriptome sequencing in this study because SGCD transcripts would have been absent in case 3 and possibly case 1 as well, necessitating additional sequencing of SGCD to identify the causative mutations. In dogs, WES has led to the identification of alleles underlying progressive retinal atrophy, primary angle closure glaucoma, and nemaline rod myopathy using small numbers of related cases [34,35,36,37,38] but is not ideal for detecting intergenic deletions or genomic rearrangements [39]. The development of improved WES enrichment kits for dogs [39, 40] will facilitate future detection of disease variants in canine models.
Conclusion
The identification of canine models of disease holds promise for new advances in the understanding and treatment of analogous human diseases. For example, the well-characterized Golden retriever model of Duchenne muscular dystrophy (DMD) has proven to be an invaluable resource for gene therapy and other trials [41, 42]. Here, we have clinically and genetically characterized the first large animal model of sarcoglycanopathy.
Abbreviations
- bp:
-
Base pair
- CAG:
-
Center for Animal Genetics, CAG GmbH
- CNL:
-
Comparative Neuromuscular Laboratory, University of California San Diego
- DMD:
-
Duchenne muscular dystrophy
- DNA:
-
Deoxyribonucleic acid
- H&E:
-
Hmatoxylin and eosin
- IBD:
-
Identical by descent
- IGV:
-
Integrative Genomics Viewer
- LGMD:
-
Limb-girdle muscular dystrophy
- PCR:
-
Polymerase chain reaction
- SGC:
-
Sarcoglycan complex
- SGCA:
-
α-sarcoglycan
- SGCB:
-
β-sarcoglycan
- SGCD:
-
δ-sarcoglycan
- SGCE:
-
ε-sarcoglycan
- SGCG:
-
γ-sarcoglycan
- SGCZ:
-
ζ-sarcoglycan
- UTR:
-
Untranslated region
- VCF:
-
Variant call format
- WES:
-
Whole exome sequencing
References
Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myologica. 2014;33:1-12.
Magri F, Brajkovic S, Brusa R, Comi GP, Govoni A. Revised genetic classification of limb girdle muscular dystrophies. Curr Mol Med. 2014;14:934-43.
Tarakci H, Berger J. The sarcoglycan complex in skeletal muscle. Front Biosci (Landmark Ed). 2016;21:744-56.
Shi W, Chen Z, Schottenfeld J, Stahl RC, Chan YM, Kunkel LM. Specific assembly pathway of sarcoglycans is dependent on beta- and delta-sarcoglycan. Muscle Nerve. 2004;29:409-19.
Trabelsi M, Kavian N, Daoud F, Commere V, Deburgrave N, Beugnet C, et al. Revised spectrum of mutations in sarcoglycanopathies. Eur J Hum Genet. 2008;16:793-803.
Pegoraro E, Hoffman EP. Limb-girdle muscular dystrophy overview. 2000 June 8 [Updated 2012 Aug 30]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1408.
Summers J, Diesel G, Asher L, McGreevy PD, Collins LM. Inherited defects in pedigree dogs. Part 2: disorders that are not related to breed standards. Vet J. 2010; 183:39–45.
Schatzberg SJ, Shelton GD. Newly identified neuromuscular disorders. Vet Clin N Am Small Anim Pract. 2004;34:1497-1520.
Shelton GD, Engvall E. Canine and feline models of human inherited muscle diseases. Neuromuscular Disord. 2005;15:127-38.
Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Nghiem P, Xiao X, et al. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome. 2012;23:85-108.
Atencia-Fernandez S, Shiel RE, Mooney CT, Nolan CM. Muscular dystrophy in the Japanese Spitz: an inversion disrupts the DMD and PGR genes. Anim Genet. 2015;46:175-84.
Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A, et al. A Duchenne muscular dystrophy gene hot spot in dystrophin-deficient Cavalier King Charles Spaniels is amenable to exon 51 skipping. PLoS ONE. 2010;5:e8647
Nghiem PP, Bello L, Balog-Alvarez C, Lopez SM, Bettis A, Barnett H, et al. Nghlem PP et al. Whole genome sequencing reveals a 7 base-pair deletion in DMD exon 42 in a dog with muscular dystrophy. Mamm Genome. 2017;28:106-13.
Deitz K, Morrison JA, Kline K, Guo L, Shelton G. Sarcoglycan-deficient muscular dystrophy in a Boston Terrier. J Vet Intern Med. 2008;22:476-80.
Dubowitz V, Sewry CA, Oldfors A. Muscle biopsy: a practical approach. 4th ed. Elsevier Health Sciences. 2013.
Roberds SL, Anderson RD, Ibraghimov-Beskrovnaya O, Campbell KP. Primary structure and muscle-specific expression of the 50-KDa dystrophin- associated glycoprotein (adhalin). J Biol Chem. 1993;368:23739-42.
Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461-71.
Ervasti JM, Campbell KP. Membrane stabilization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121-31.
Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, et al. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell. 2000;5:141-51.
Broad Institute. IGV Integrative Genomics Viewer. [Online]. Available from: http://software.broadinstitute.org/software/igv/. Accessed 1 May 2017.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078-9.
Bozeman MT. Golden Helix GenomeBrowse ® visualization tool (Version 2.1) [Software]. [Online]. Available from: http://www.goldenhelix.com.
Barresi R, Moore SA, Stolle CA, Mendell JR, Campbell KP. Expression of gamma-sarcoglycan in smooth muscle and its interaction with the smooth muscle sarcoglycan-sarcospan complex. J Biol Chem. 2000;275:38554-60.
Crosbie RH, Lebakken CS, Holt KH, Venzke DP, Straub V, Lee JC, et al. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol. 1999;145:153–65.
Holt KH, Campbell KP. Assembly of the sarcoglycan complex. Insights for muscular dystrophy. J Biol Chem. 1998;273:34667-70.
Fanin M, Duggan DJ, Mostacciuolo ML, Martinello F, Freda MP, Sorarù G, et al. Genetic epidemiology of muscular dystrophies resulting from sarcoglycan gene mutations. J Med Genet. 1997;34:973–7.
Ng R, Banks GB, Hall JK, Muir LA, Ramos JN, Wicki J, et al. Animal models of muscular dystrophy. Prog Mol Biol Transl. 2012;105:83-111.
Straub V, Duclos F, Venzke DP, Lee JC, Cutshall S, Leveille CJ, et al. Molecular pathogenesis of muscle degeneration in the δ-sarcoglycan-deficient hamster. Am J Pathol. 1998;153:1623–30.
Nigro V, Okazaki Y, Belsito A, Piluso G, Matsuda Y, Politano L, et al. Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet. 1997;6:601–7.
Ensembl.org. [Online].http://www.ensembl.org/Canis_familiaris/Transcript/Summary?db=core;g=ENSCAFG00000017592;r=4:53267777-53642810;t=ENSCAFT00000027892. Accessed 1 May 2017.
Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Ozawa E, Angelini C, et al. Mutations in the sarcoglycan genes in patients with myopathy. New Engl J Med. 1997;336:618-25.
Moreira ES, Vainzof M, Marie S, Nigro V, Zatz M, Passos-Bueno MR. A first missense mutation in the delta sarcoglycan gene associated with a severe phenotype and frequency of limb-girdle muscular dystrophy type 2F (LGMD2F) in Brazilian sarcoglycanopathies. J Med Genet. 1998;35:951–3.
Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. 2015;72:1424-32.
Ahonen S, Arumilli, Lohi H. A CNGB1 frameshift mutation in Papillon and Phalene dogs with progressive retinal atrophy. PLoS One. 2013;8:e72122.
Ahram DF, Collin RWJ, Kuehn MH, Grozdanic SD, Henkes A, Kecova H. Variants in nebulin (NEB) are linked to the development of familial primary angle closure glaucoma in basset hounds. PLoS One. 2015;10:e126660.
Evans M, Cox, Huska, Li F, Gaitero, Guo L, et al. Exome sequencing reveals a nebulin nonsense mutation in a dog model of nemaline myopathy. Mamm Genome. 2016;27:495-502.
Kropatsch R, Akkad DA, Frank M, Rosenhagen C, Altmüller J, Nürnberg P, et al. A large deletion in RPGR causes XLPRA in Weimaraner dogs. Canine Genet Epidemiol. 2016;Jul 8;3(1):7.
Forman OP, Pettitt L, Komáromy AM, Bedford P, Mellersh C. A novel genome-wide association study approach using genotyping by exome sequencing leads to the identification of a primary open angle glaucoma associated inversion disrupting ADAMTS17. PLoS One. 2015;10:e143546.
Broeckx BJ, Coopman F, Verhoeven GE, Bavegems V, De Keulenaer S, De Meester E, et al. Development and performance of a targeted whole exome sequencing enrichment kit for the dog (Canis Familiaris Build 3.1). Sci Rep. 2014;4:5597.
Broeckx BJ, Hitte C, Coopman F, Verhoeven GEC, De Keulenaer S, De Meester E, et al. Improved canine exome designs, featuring ncRNAs and increased coverage of protein coding genes. Sci Rep. 2015;5:12810.
Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev. 2015;26:57-69.
Yu X, Bao B, Echigoya Y, Yokota T. Dystrophin-deficient large animal models: translational research and exon skipping. Am J Transl Res. 2015;7:1314–31.
Acknowledgements
We are thankful to all the dog owners, breeders, and clinicians who have contributed samples to this study. The authors also thank Florian Battke, Sally Prouty, and Hammon Humphries for technical assistance.
Funding
Research reported in this publication was supported in part by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R15AR062868 (LAC) and the Clemson University Calhoun Honors College (AGD). HL was supported by the Jane and Aatos Erkko Foundation and the Academy of Finland. ES was supported by the Jenny and Antti Wihuri Foundation. This work was supported in part by a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant (1U54NS053672 to KPC). KPC is an investigator of the Howard Hughes Medical Institute.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Contributions
MC, JE, LAC, and GDS designed the research. MC, JE, LTG, AD, LG, JL, KC, AS-M, ES, and MH performed the experiments. MC, JE, KC, ES, MH, HL, LAC, and GDS analyzed the data. MC, JE, LAC, and GDS wrote the manuscript. All authors participated in editing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All dogs in this study were evaluated in a clinical veterinary practice by licensed veterinarians. The dogs in this study were examined and tissue biopsies collected with the written consent of their owners. Tissue studies were performed using protocols approved by the Institutional Animal Care and Use Committees (IACUC) of Clemson University, the University of California San Diego, and the University of Iowa, and the Animal Experiment Board in Finland (ESAVI/7482/04.10.07/2015), as well as the Baden-Württemberg veterinary office at the Landratsamt Tübingen Abt. 32: Veterinärwesen und Lebensmittelüberwachung, Tübingen, Germany (Registriernummer: DE 08 416 1038 21).
Consent for publication
Not applicable
Competing interests
MC is employed by CAG GmbH that performs canine DNA testing on a commercial basis. HL is a co-founder of Genoscoper Laboratories Oy that offers canine DNA testing on a commercial basis. All other authors declare that they have no competing interests.
Additional file
Additional file 1: Table S1.
Primers used to define and genotype SGCD mutations. (DOCX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cox, M.L., Evans, J.M., Davis, A.G. et al. Exome sequencing reveals independent SGCD deletions causing limb girdle muscular dystrophy in Boston terriers. Skeletal Muscle 7, 15 (2017). https://doi.org/10.1186/s13395-017-0131-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13395-017-0131-0